
A heterojunction strategy to improve the visible light sensitive water splitting performance of photocatalytic materials

Abstract
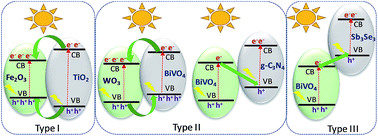
Photocatalytic water splitting is a promising path for generating hydrogen which will decrease the dependency on conventional fossil fuels to generate power and provides an environmentally benign way to store solar energy. Designing photocatalysts is one of the key challenges in making photocatalytic water splitting efficient and economically viable. Heterojunction formation using different functional materials in a single photo-catalyst has the potential of broadening light harvesting properties, improving chemical stability, enhancing the photoexcited charge separation, and thus boosting water splitting efficiency. This article gives an overview on the heterojunction strategy for improving photocatalytic water splitting performance. Recent developments in different visible light-responsive heterojunction systems are discussed for metal oxide–metal oxide and metal oxide–non-metal oxide heterojunctions. Along with experimental observation and the proposed charge separation mechanism, synthesis techniques of heterojunction materials are also included in this article. In addition to recent progress in the heterojunction-based photocatalytic system, this review article also provides future directions for photocatalytic water splitting.
- This article is part of the themed collections: Recent Review Articles and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

