
Aromatic end-capped acceptor effects on molecular stacking and the photovoltaic performance of solution-processable small molecules†

Abstract
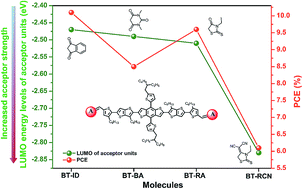
Aromatic end-capped acceptors are important in constructing donor materials and non-fullerene acceptors in organic solar cells. However, their features (such as electron-withdrawing ability and subtle change in planarity) and effects on molecular stacking and photovoltaic performance, lack a systematic study. This manuscript reports four molecules, namely, BT-RCN, BT-BA, BT-RA, and BT-ID, which are terminated by different acceptors in the same backbone. We quantify the molecular planarity through their dipole moment in the Z direction and investigate the effect of the degree of planarity on molecular properties and device performances. The four acceptors are classified into two groups based on acceptor strength: medium-strong and strong acceptors. Molecules based on medium-strong acceptors exhibit excellent efficiencies as follows: 10.1% for BT-ID, 9.6% for BT-RA, and 8.5% for BT-BA. Their decreased efficiency is quite consistent with their lowered hole mobility. Grazing incidence X-ray diffraction results demonstrate the positive relationship between the non-planarity of the acceptors and d-spacing distance in the π–π stacking direction, which are detrimental to mobility. BT-RCN, with a strong acceptor, obtains the lowest efficiency of 6.1%. These findings indicate the importance of matching the electron-donating ability of donor units and electron-withdrawing ability of acceptor units, and the subtle planarity change in molecular properties and aggregation.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

