
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C2NxO1−x framework carbons with defined microporosity and Co-doped functional pores†
Zhihong
Tian
 *ab,
Nina
Fechler
b,
Martin
Oschatz
b,
Tobias
Heil
b,
Johannes
Schmidt
c,
Siguo
Yuan
a and
Markus
Antonietti
*b
*ab,
Nina
Fechler
b,
Martin
Oschatz
b,
Tobias
Heil
b,
Johannes
Schmidt
c,
Siguo
Yuan
a and
Markus
Antonietti
*b
aSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: zhihong.tian@mpikg.mpg.de
bDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Potsdam 14476, Germany. E-mail: office.cc@mpikg.mpg.de
cTechnical University of Berlin, Institute of Chemistry, Hardenberg str. 40, 10623 Berlin, Germany
First published on 10th September 2018
Abstract
Gallic acid and urea are used to produce C2NO materials with moderately defined micropores via direct condensation and ring closure. These materials show a unique heterocycle containing carbonaceous structure and feature an unusually high content of heteroatoms (nitrogen and oxygen) lining the insides of pores while having high specific surface areas. The multifunctional carbon materials demonstrate good performance for selective CO2 capture resulting from adjustable porosity and polarizability. In view of the simplicity of the salt flux synthetic method and the advantage of the available sustainable starting synthons, the C2NO framework has potential for use in diverse practical applications.
Introduction
Microporous carbon materials with high specific surface areas are key components in a wide range of fields including adsorption,1 gas separation,2 electrocatalysis,3 or in energy storage.4 Such “designer carbons” with well-defined aromatization, heteroatom content, and pore architectures are mostly synthesized by “carbonization”, i.e., the simple heating of organic matter to very high temperatures; this indicates the minimal extent of bottom-up control in structure and functionalization provided by this process.In the last few years, our group has published continued efforts to develop a more controlled, facile, sustainable, low-cost synthesis of carbonaceous materials while specifically controlling size and functionality of the pores. Especially, a “C2N”-species5 simultaneously described by the Baek group6 is discussed herein. The material is a porous, graphene-like structure in which 1/3 of all carbon atoms is replaced by pyrazinic nitrogen atoms (Scheme 1a), and it possesses a regular structure; it also exhibits mostly a 12-atom-sized circumvent, and the pores are lined tightly with basic nitrogen heteroatoms. Pores of related characteristics were shown in a recent paper to be “superhydrophilic”, i.e., filled with liquid water completely at relatively low partial pressures.7 Despite their high structural porosity and similarity to zeolites, these materials do not have unsaturated edge terminations; especially, there are no C–H bonds in the structure, justifying the understanding of these materials as “carbons” rather than as polymer frameworks. Interestingly, the synthesized species are thermodynamically stable, i.e., a whole range of carbonization species with very different educts (such as cyclohexanehexone, squaric acid/urea,5b thiooxamides,8 and dihydroxychinon/urea9) result in a similar final structure by spontaneous structural rearrangements. This motif is stable up to 700 °C, after which graphitization occurs. Carbons with dense and accessible hydrophilic micropores are desirable for a number of applications including ion adsorption,10 removal of polar gases,11 or electrocatalysis.12 Herein, we attempt to generalize previous synthesis protocols and describe a cheaper and flexible route towards similar systems based on naturally abundant or nature-derived starting materials. The “design rules” were recently described in a concept paper13 and involve monomer prealignment and directional “click-like” organic reactions that produce heteroatom-containing functional tectons with medium-high stability.
 | ||
| Scheme 1 (a) Idealized structure of 1,4 “dual-doped” carbons, i.e., pyrazine, oxazine, and dioxine containing frameworks. Please note that in this structural idealization, all heteroatoms are used in the most stable edge termination of a graphitic sheet; 33 mol% of all atoms are either N or O and all heteroatom substituents are in para position to each other. (b) Using urea, a pure “C2NxO1−x” structure was formed as expected. The structure depicts a reduced version as the products are electroactive. | ||
The intended generalizations concern two aspects:
- We want to replace elaborated synthetic educts with simpler, more available, and sustainable starting synthons; here, gallic acid and urea as a nitrogen source.
- We want to extend previous pyrazine functionalization to related, easy-to-bridge heterocycles, namely, oxazines and dibenzodioxines. The corresponding (idealized) frameworks with their heteroatom-lined pores are depicted in Scheme 1b.
It is well known that the introduction of other elements such as nitrogen or oxygen into the carbon lattice greatly enhances material properties in terms of chemical and thermal stability, band positions, and catalytic efficiency; also, it significantly improves oxidation stability14 and contributes to more specific interactions with liquids, solvated ions, or gases.15
In this study, we show that the resulting heteroatom-doped carbon materials (ideally with about 33 mol% heteroatom doping, i.e., every second ring has to host 2 heteroatoms, which is energetically preferred in para positions to each other) are unusually polar, electrochemically active. They constitute a new type of carbonaceous sorbent with a high sorption strength, e.g., to CO2 and thus exhibit high capacity under relevant conditions. Due to the predominant para-character of substitution patterns, all doping centers can easily electronically communicate with each other; the novelty to be expected as compared to that of classical heteroatom-doped carbons comes from the fact that the heteroatoms are in significant desirable mutual arrangement and thus, carbon properties are altered by the concerted action of at least two doping atoms.
Results and discussion
Gallic acid is used as the primary starting product, as shown in Scheme S1.† It is a well-known natural resource found in gallnuts, witch hazel, tea leaves, sumac, oak bark, and other plants. Gallic acid is known to decarboxylate at higher temperatures to yield pyrogallol,16i.e., the carboxylic acid unit acts as a “protecting/leaving group” to enable controlled substitution chemistry to realize high structural definition.As an initial scheme for more effective functionalization, we use the addition of urea and three well-known chemical reactions, which were found to occur simultaneously throughout monomer condensation:
- Urea decomposes at around 130 °C under the formation of ammonia and isocyanic acid, both of which act as linkers.17 Isocyanic acid undergoes spontaneous isocyanate formation with oxygen nucleophiles such as phenolic groups.
- Aromatic –OH groups can be conveniently substituted with amines to exhibit –NH2 substitution patterns using Lewis acid catalysis.18 Urea serves as a convenient and sustainable ammonia source at elevated temperatures (first point and Scheme S1†).
- Gallic acid condenses with aromatic amines to oxazoles in a highly defined fashion, e.g., in the well-known synthesis of gallocyanines.19 After the formation of the central oxazole ring, the structure decarboxylates even more easily, allowing further substitution on the pyrogallol ring. This simple heterocyclic reaction coupled with consecutive liberation of carboxylate “protection group” from the 4 position makes gallates favorable starting products in our opinion extensively for 2-d para-heteroatom-doped carbons via a polycondensation scheme. Interestingly, gallocyanines as the related, low-molecular-weight dyes are black in the solid state, i.e., they have an appropriate electronic structure of a narrow bandgap semiconductor in their molecular crystals.
Condensation occurs as expected: while forming a liquid eutectic mixture of gallic acid, urea, and molten ZnCl2 as a catalyst and solvent, spontaneous foaming via excessive CO2 formation and no release of ammonia occur at around 100–120 °C. The observed mass loss agrees with the decarboxylation of gallic acid with the release of a large extent of CO2 from urea by transamidation, isocyanate formation and hydrolysis due to the added Lewis acid ZnCl2. In this stage, the reaction rapidly increases solution viscosity due to the ongoing polymerization processes. Next, the reaction mixture gels, which clearly proves the ability of the monomers to participate in branching reactions and finally, the gel turns resin-like due to high extent of multidimensional polymerization and cross-linking. This occurs well before any onset of carbonization and resembles a typical urethane foam formation. ZnCl2 fluxes act as catalyst and solvent to promote the condensation reaction and later as porogens throughout the condensation processes; thus, the obtained material is mechanically stabilized against shrinkage. The obtained carbons are referred to as GU11-Y, GU12-Y and GU13-Y, with 11–13 signifying the molar ratios of gallic acid to urea, and Y is the condensation temperature. G–Y is the urea-free carbon obtained solely by condensation of gallic acid.
This condensation process is based on the elimination of carboxyl, hydroxyl, carbonyl and amine groups in the carbons. We used FT-IR measurements to probe the functional groups on these carbons (Fig. S1†). FTIR spectra of both the 350 °C and 500 °C carbons show rather broad signals with low intensity, confirming the high degree of condensation at a comparably low temperature.20 The carbonaceous nature is also supported by Raman spectra of GU13-350 and GU13-500 (Fig. S2†). A sharp G band is observed at 1591 cm−1, and a D band (usually assigned to defects) is found at 1359 cm−1, which can be generally ascribed to a disordered graphitic lattice. We confirm that both the carbons obtained at 350 °C and 500 °C are very stable by performing thermogravimetric analysis (TGA) on the samples in air (Fig. S3†). The samples without urea tend to decompose earlier, manifesting that nitrogen doping further enhances oxidation resistance. The 500 °C series (burn off maximum: 500–600 °C) show higher thermal stability than the 350 °C series (burn off maximum: 400–550 °C). This is due to the well-defined framework that is fully developed in the 500 °C samples.21 Compared with standard activated carbons,22 these dual-doped carbons have higher thermal stability, i.e., they oxidize at higher temperatures due to the large amount of O and N inside their structures.23 The samples decompose/oxidize, leaving almost no residual mass at 700 °C. Such a high temperature needed for complete decomposition is very unusual for other carbonized materials but is typical for regular, thermodynamically stable materials, where clear decomposition chemistry can be assigned.14 The complete mass loss also indicates the absence of Zn residuals, which is independent of the synthesis temperature and the urea content.
The elemental compositions of the condensation products, as determined by combustion elemental analysis, are summarized in Table S1.† Especially, while performing the reaction with gallic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea in a 1:3 mole ratio in the presence of excess salt, the system is indeed composed of 2/3 carbon atoms and 1/3 doped heteroatoms; for example, the structure of GU13-350 is C2O0.7N0.2, whereas that of GU13-500 is C2O0.5N0.2. Thus, we observe a minor extent of C–C condensation in addition to the targeted reaction cascades. At 500 °C condensation, we still found a similar material but with less hydrogen and slight excess of carbon when compared to nitrogen and oxygen, i.e., the doping atoms get eliminated during a typical side reaction.
:urea in a 1:3 mole ratio in the presence of excess salt, the system is indeed composed of 2/3 carbon atoms and 1/3 doped heteroatoms; for example, the structure of GU13-350 is C2O0.7N0.2, whereas that of GU13-500 is C2O0.5N0.2. Thus, we observe a minor extent of C–C condensation in addition to the targeted reaction cascades. At 500 °C condensation, we still found a similar material but with less hydrogen and slight excess of carbon when compared to nitrogen and oxygen, i.e., the doping atoms get eliminated during a typical side reaction.
As is typical for such materials, further increase in condensation temperature to 800 °C and 900 °C under inert gas leads to more extensive carbonization within the structure and the destruction of the “para-dual doping motif”. Consequently, the N content decreases with the increase in temperature. However, the amount of N and O remaining after condensation at 800 °C and 900 °C is still in between 24-25 and 20–21 wt%. Based on our studies, we assign the thermal stability limit of heteroatom-lined pores that is similar to those presented in Scheme 1 at 700–750 °C.
The morphologies of the samples were first analyzed with SEM (Fig. 1). All powders have defined nanostructures with a primary grain size of 50–200 nm, i.e., very high contributions of an external surface area to interface properties are not to be expected. These powders are also well packed at the secondary hierarchy level, and we can specifically exclude one- or two-dimensional characters of the primary particles, which would lead to a different texture.
 | ||
| Fig. 1 SEM images of (a) G-350, (b) GU11-350, (c) GU12-350, (d) GU13-350, (e) G-500, (f) GU11-500, (g) GU12-500 and (h) GU13-500. | ||
Nitrogen physisorption was employed to study the inner specific surface area and pore size of the obtained condensation products (Fig. 2a, b and S4†); the corresponding quantitative data are summarized in Table 1. Urea-containing samples condensed at 350 °C have only minor porosity accessible for nitrogen, and the low values are related to the outer texture seen in Fig. 1. The sample without urea possesses the highest porosity and is predominantly microporous. At 500 °C, high porosity and specific surface area (SSA) are developed. Higher contents of urea result in a relatively stronger contribution, well-defined microporosity and less mesopores, which can be ascribed to higher fluidity of the reaction mixtures and more heteroatom-lined micropores. To further understand the process of pore formation, GU13-400 and GU13-450 were prepared by the method used for GU13-350 and GU13-500 but at 400 and 450 °C, respectively. The amount of nitrogen adsorbed at 77 K shows a gradual increase as the condensation temperature changes from 350 to 500 °C (Fig. S5†). This is accompanied with a growth of the micropore volume, and the sorption curves remain similar, i.e., more and more micropores become accessible for the adsorbate molecules. We attribute this process to the removal of pendant functional groups and the formation of phenazines, oxazine or dioxine rings through ring closure during the controlled condensation process. Specifically, the polymeric aromatic precursor systems stack together before 350 °C, but the relevant organic rearrangement and the opening and stacking of closed 12-atom-lined pores begin at 400 °C and are completed at 500 °C. Therefore, the porosity of the well-defined 1,4 dual doped carbons mainly comes from the formation of holes surrounded by completed and closed aromatic rings in the materials' skeleton, i.e., the pores are tectonic and not due to bonding or packing defects.
| Sample | S BET (m2 g−1) | S mic (m2 g−1) | V t (cm3 g−1) | V mic (cm3 g−1) |
|---|---|---|---|---|
| a S BET calculated from nitrogen adsorption isotherms (77 K) by the Brunauer–Emmett–Teller method. Vt: total pore volume at P/P0 = 0.995. Vmic: cumulative DFT pore volume (<2 nm). | ||||
| G-350 | 627 | 448 | 0.49 | 0.19 |
| GU11-350 | 11 | 2.9 | 0.03 | 0.002 |
| GU12-350 | 4 | 0 | 0.03 | 0 |
| GU13-350 | 17 | 0 | 0.1 | 0 |
| G-500 | 1699 | 678 | 1.57 | 0.37 |
| GU11-500 | 1532 | 669 | 1.22 | 0.35 |
| GU12-500 | 1339 | 742 | 1.09 | 0.36 |
| GU13-500 | 1227 | 791 | 1.02 | 0.35 |
Brunauer–Emmett–Teller (BET) specific surface areas (hereafter simply referred to as surface area) at 500 °C are between 1200 and 1700 m2 g−1, depending on urea content. These values exceed those of zeolites and even those of most hierarchical zeolites.24 This is expected as carbon with similar pore sizes and wall thicknesses to those of silica has a lower mass density. Please note that the nitrogen surface areas of all 350 °C samples are significantly lower than those of the samples at 500 °C, which we attribute to poorer accessibility to the internal pore volume (as shown below with CO2-sorption experiments). With increasing condensation temperature from 350 to 500 °C (Table S2†), the surface area increases from 17, 46, 595 to 1227 m2 g−1 for samples GU13-350, GU13-400, GU13-450 and GU13-500, respectively. The pore volume follows a similar trend, expanding from 0.1 cm3 g−1 for GU13-350 to 1.02 cm3 g−1 for GU13-500. The surface area decreases again with higher temperatures (650–900 °C), which can be due to the disintegration of the oxygen-rich framework. A condensation temperature of 850 °C yields GU13-850, and the specific surface area decreases to 1167 m2 g−1 (Fig. S6†). In general, further increase in temperature leads to pore collapse (with the interface energy as the driving force) and formation of carbons with lower surface areas as the walls become thicker; this slowly transitions the system towards more extended graphene units and more pronounced aromatic stacking in the walls.
Low pressure nitrogen physisorption measurements in polar materials can suffer from high quadrupole moment of nitrogen, which results in large errors during the determination of micropore size. Thus, we carried out argon physisorption at 87 K for sample GU13-500 to determine the micropore size (Fig. S7†). The shape of the isotherm is comparable to the measurement from nitrogen adsorption, and the average size of micropores for GU13-500 is 1.45 nm.
Water vapor adsorption at 298 K (Fig. S8†) was conducted for GU13-350 and GU13-500 samples to investigate the strength of water adsorption (a particular feature of zeolites). Both isotherms show a significant uptake of water even at very low relative pressures. Such a strong interaction of the carbon pores with water does not occur in classical porous carbons even if they contain very narrow micropores. These measurements indicate the presence of strong water binding sites in our carbons, which is very similar to that in zeolites. In accordance with the nitrogen physisorption experiments, GU13-500 has an overall higher water uptake as compared to GU13-350 with the former having a higher pore volume.
The powder XRD patterns of 350 °C carbon showed a broad feature centered at 2θ ≈ 26° (Fig. S9a†), which is usually ascribed to the stacking of aromatic systems.25 However, the degree of stacking is not pronounced. Interestingly, this peak nearly vanishes as the condensation temperature is increased to 500 °C (Fig. S9b†), which indicates that negligible layer packing is present in all the samples. Aromatic systems with dioxine bridges are known to bend easily and can even form stable transversal rings. The absence of graphitic packing is supported by high resolution transmission electron microscopy (HR-TEM). As shown in Fig. 2d, no clear graphitic domains are observed, but rectangles were found to cross each other, which was similar to that in a fiber fabric.
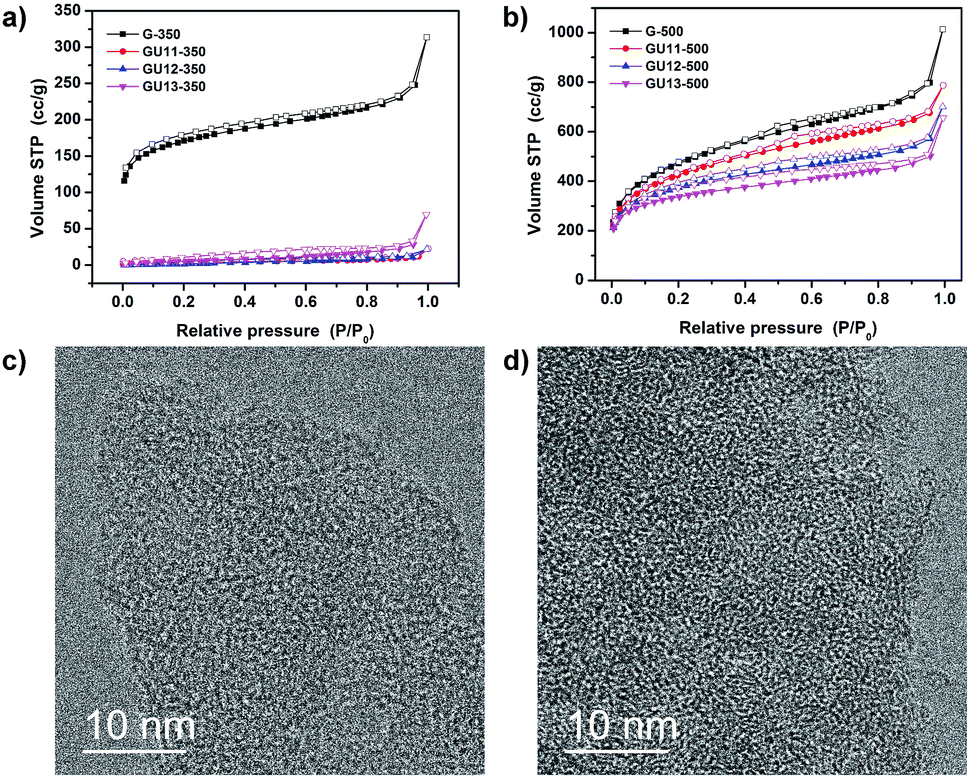 | ||
| Fig. 2 N2 adsorption (solid symbols) and desorption (open symbols) isotherms at 77 K for the C2NO frameworks obtained at (a) 350 °C and (b) 500 °C. HR-TEM of (c) GU11-350 and (d) GU11-500. | ||
XPS measurements were employed to identify the chemical composition in different materials (Fig. S10†). In agreement with the combustion analysis (in which some adsorbed water may contribute to the detected oxygen, especially in urea-rich samples), XPS elemental analysis confirms the high heteroatom content in the materials (Table S3†). C 1s spectrum (Fig. 3a) shows mainly three types of C. The primary peak at 284.3 eV corresponds to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H species, followed by a peak at 285.7 eV for C–N and C–O. Moreover, the peak at 288.7 eV (usually ascribed to highly oxidized carbon or C+) is formed throughout the process of condensation, and it becomes larger with more urea. We cannot exclude the presence of carboxylates or even very strongly adsorbed CO2, but such electron-poor carbons also exist in multiple heteroatom-doped aromatic systems. The N 1s region of the spectra can be deconvoluted into two peaks (Fig. 3b): the binding energy at 398 eV is generally assigned to a secondary amine in the para position of oxygen; the other one with a higher binding energy of 399.5 eV can be ascribed to nitrogen in aromatic rings, i.e., pyridines, pyrazines, and oxazines. The high intensity of the latter peak suggests high proportion of N in six-membered aromatic rings. Together with the very high heteroatom content of about 30 mol%, these results indicate that N is integrated into the carbon framework most presumably in the para position to a second heteroatom. The peaks for O 1s are found for all samples at 531.4 and 532.5 eV corresponding to CO and C–O–C, respectively (Fig. 3c). Interestingly, the peak that corresponds to O+ for O 1s was observed at 533.3 eV. Similar to many natural dyes, e.g., methylene blue and anthocyanin, our synthesis scheme based on gallic acid and urea gives aromatic systems conjugated through the oxygen atom, localizing a formal (+) charge at this heteroatom position.
C and C–H species, followed by a peak at 285.7 eV for C–N and C–O. Moreover, the peak at 288.7 eV (usually ascribed to highly oxidized carbon or C+) is formed throughout the process of condensation, and it becomes larger with more urea. We cannot exclude the presence of carboxylates or even very strongly adsorbed CO2, but such electron-poor carbons also exist in multiple heteroatom-doped aromatic systems. The N 1s region of the spectra can be deconvoluted into two peaks (Fig. 3b): the binding energy at 398 eV is generally assigned to a secondary amine in the para position of oxygen; the other one with a higher binding energy of 399.5 eV can be ascribed to nitrogen in aromatic rings, i.e., pyridines, pyrazines, and oxazines. The high intensity of the latter peak suggests high proportion of N in six-membered aromatic rings. Together with the very high heteroatom content of about 30 mol%, these results indicate that N is integrated into the carbon framework most presumably in the para position to a second heteroatom. The peaks for O 1s are found for all samples at 531.4 and 532.5 eV corresponding to CO and C–O–C, respectively (Fig. 3c). Interestingly, the peak that corresponds to O+ for O 1s was observed at 533.3 eV. Similar to many natural dyes, e.g., methylene blue and anthocyanin, our synthesis scheme based on gallic acid and urea gives aromatic systems conjugated through the oxygen atom, localizing a formal (+) charge at this heteroatom position.
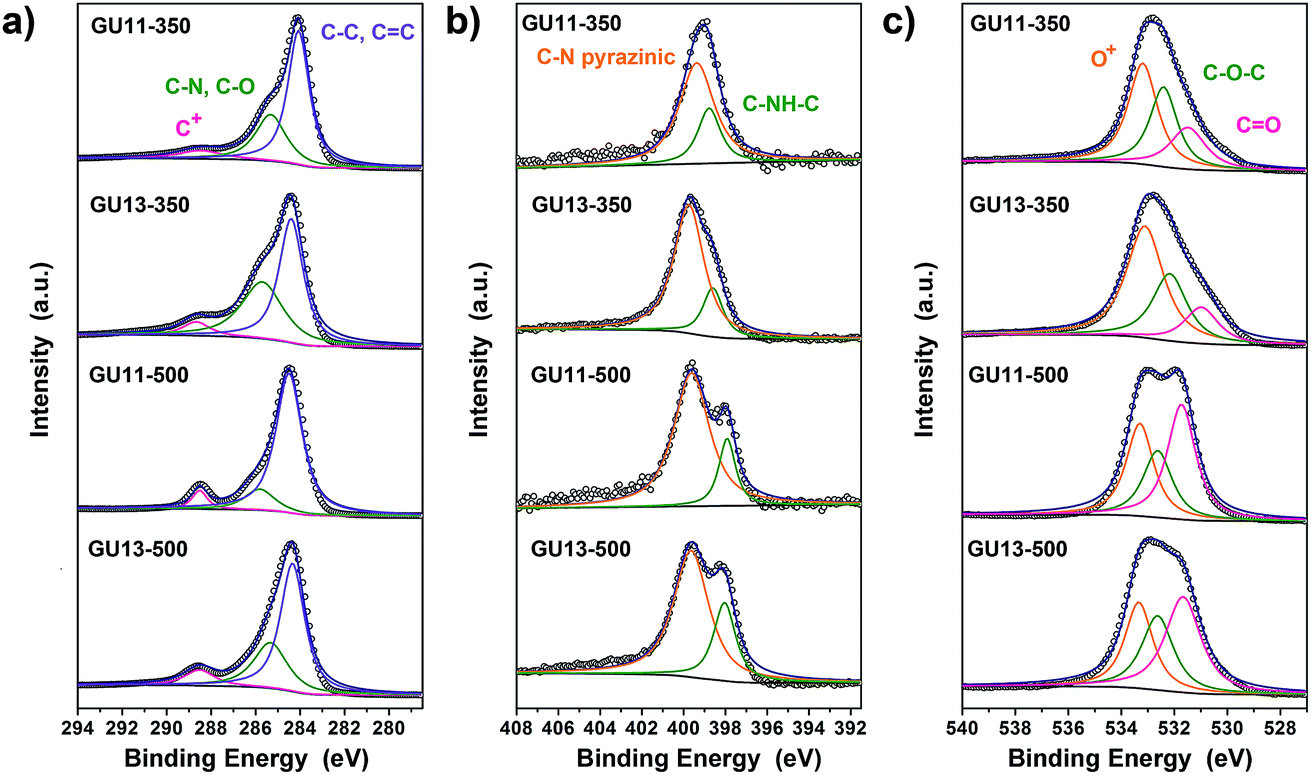 | ||
| Fig. 3 Deconvoluted XPS spectra of the carbons: (a) C 1s, (b) N 1s, (c) O 1s. | ||
Considering the combination of high heteroatom content, predominant 1,4-para dual heteroatom doping and a well-defined high specific surface area induced by the employed unique chemical condensation motifs, the samples were tested for CO2 sorption to determine their selectivity. As we pointed out in a recent review article, such materials will be very attractive for selective CO2 capture at low pressure because a strongly polarizing surface is more important for carbon dioxide uptake at 1 bar than a high nitrogen surface area.2a An ideal material for this purpose would combine notable CO2 adsorption with kinetic exclusion of nitrogen. The data are summarized in Fig. 4 and S11†
 | ||
| Fig. 4 CO2 adsorption (solid symbols) and desorption (open symbols) isotherms at 273 K for materials prepared at (a) 350 °C and (b) 500 °C. (c) CO2/N2 selectivity as calculated by the IAST method for a CO2:N2 = 0.15:0.85 gas mixture at 273 K. (d) Thermal response measurements of CO2 adsorption/desorption (1 bar, 298 K) of the materials prepared at 500 °C. | ||
Ordinary activated microporous carbons are known to possess low IAST CO2/N2 selectivity in the range of 5–20 as the polarizable conjugated graphitic carbons bind many molecules based on the rather non-selective polarization resulting from van der Waals interactions.2a As a result, this is how our highly porous but non-nitrogen-doped sample G-500 performs (Fig. 4c); compared to this, the sorption behavior of N,O-dual doped carbons only has little in common with ordinary carbonaceous materials. The selectivities are rather high for both 350 and 500 °C samples prepared with urea. It is widely accepted that (even rather random) heteroatom doping as such can significantly enhance CO2/N2 selectivity by introducing specific binding sites, inducing strong polarization of CO2 (typical values for N-doped carbons with comparable nitrogen contents are in the range of 20–40);11a,26 however, the polarization of CO2 is apparently stronger in our well-defined 1,4 dual-doped materials and their selectivities of 67.5 (GU13-350) and 50 (GU13-500) indeed approach the values typical for performance separation products.
CO2 adsorption kinetics of the 350 °C samples are poor, and a weak hysteresis is observed. Nevertheless, CO2 at room temperature can reach some of the internal pores but N2 cannot. Taking this into consideration, it can be concluded that the “real” (equilibrium) selectivity of these samples might be higher, and such polymer structures do offer the possibility of CO2 molecular sieving. Due to the presence of larger pores, 500 °C samples show rapid equilibration as well as sufficient selectivity reaching 50 for GU13-500, making them promising for CO2 capture under relevant conditions (e.g., for removal of CO2 from flue gas). Interestingly, while nitrogen sorption at liquid nitrogen temperatures decreases with increasing urea content, an opposite trend was observed for CO2 sorption at 273 K; the higher urea content during synthesis promoted the formation of smaller micropores with dense heteroatom lining and induced specific adsorption sites for CO2. The strength of the interactions between adsorbed CO2 molecules and the surface of our materials can be estimated by calculating the isosteric heat of CO2 adsorption (Qst) using the Clausius–Clapeyron equation. We calculated apparent Qst values for 500 °C samples (assuming kinetic equilibrium for all cases) from CO2 adsorption isotherms measured at 273 K (Fig. 4b) and 300 K (Fig. S12†). The plot of Qst as a function of CO2 uptake is shown in Fig. S13.† Among the 500 °C samples, the most nitrogen-rich structure of GU13-500 has the highest apparent Qst, which ranges from 47 to 42 kJ mol−1 for the strongest binding sites. The Qst values reported herein are higher than those of most previously reported porous carbon materials, especially activated carbons, even with nitrogen heteroatoms present.27 High Qst is clearly due to the pore size, and we assume that it is also because the 1,4-para dual heteroatom doping plays a crucial role. Slightly higher Qst values for CO2 adsorption are reported only for some strongly polarizing metal–organic frameworks, especially for modified zeolites or nitrogen-doped microporous carbons functionalized with extra alkali metal cations.2a,11a,28
InfraSORP29 measurements with CO2 as the test gas at constant flow of 80 mL min−1 further indicate the structural properties of 500 °C samples and their kinetic adsorption behavior (Fig. 4d). Notably, CO2 diffusion in 500 °C samples is very fast, as indicated by the nearly symmetric adsorption and desorption signals as well as large and sharp temperature increase throughout cycling. In spite of the predominantly microporous structure, adsorption and desorption times are between 20 and 40 s. Accordingly, thermal response measurements of the 500 °C carbons show mass-related thermal response peak areas between 5.4 and 9.6 mg−1 (Fig. S14†). This further indicates that the most structured GU13-500 sample has very strong affinity and a high binding enthalpy for CO2, especially despite its relatively lower nitrogen surface area. The surface modifications described here thereby provide high CO2 adsorption capacity, high CO2/N2 selectivity, high adsorption strength, and favorable adsorption kinetics.
Conclusions
In summary, we have developed a synthesis scheme based on gallic acid and urea to generate an assembled framework of heterocyclic rings, forming a layered carbonaceous structure with about 30 wt% of heteroatoms through directed condensation and ring closure. The unusually high content of heteroatoms (oxygen and nitrogen) can only be meaningfully assigned and included as phenazines, oxazine, or dioxine rings, which inherit the material high functionality and redox activity. Such a structure when coupled with salt flux synthesis and templating can form a framework with high specific surface area with nearly complete heteroatom lining of the micropores. This method is simple, straightforward, and sustainable due to its starting products and is highly promising for scale up in an industrial environment. We believe that the newly described oxazoles and dioxine frameworks can contribute to a growing library of functional tectons for carbon-based materials with a wider range of properties; for example, they can be used for selective adsorption, as catalytic supports, or in electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Max Planck Society is gratefully acknowledged for financial support. MA wants to thank the German Excellence UNICAT for continued support. We thank Regina Rothe for technical assistance and Ralf Walczak for the Ar and H2O vapour physisorption tests. M. O. acknowledges financial support by a Liebig Fellowship of the German Chemical Industry Fund. Zhihong Tian sincerely acknowledges the financial support provided by China Scholarship Council (CSC) of the Ministry of Education, P. R. China (No. 201707040019). Open Access funding provided by the Max Planck Society.Notes and references
-
(a) X. Yu, Z. Tang, D. Sun, L. Ouyang and M. Zhu, Prog. Mater. Sci., 2017, 88, 1–48 CrossRef
; (b) P. Puthiaraj, Y.-R. Lee and W.-S. Ahn, Chem. Eng. J., 2017, 319, 65–74 CrossRef CAS
-
(a) M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC
-
(a) A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef
-
(a) X. Chen, R. Paul and L. Dai, Natl. Sci. Rev., 2017, 4, 453–489 CrossRef
-
(a) N. Fechler, N. P. Zussblatt, R. Rothe, R. Schlögl, M. G. Willinger, B. F. Chmelka and M. Antonietti, Adv. Mater., 2016, 28, 1287–1294 CrossRef CAS PubMed
-
(a) J. Mahmood, S.-M. Jung, S.-J. Kim, J. Park, J.-W. Yoo and J.-B. Baek, Chem. Mater., 2015, 27, 4860–4864 CrossRef CAS
- G. P. Hao, G. Mondin, Z. Zheng, T. Biemelt, S. Klosz, R. Schubel, A. Eychmüller and S. Kaskel, Angew. Chem., Int. Ed., 2015, 54, 1941–1945 CrossRef CAS PubMed
- J. Xu, J. Zhu, X. Yang, S. Cao, J. Yu, M. Shalom and M. Antonietti, Adv. Mater., 2016, 28, 6727–6733 CrossRef CAS PubMed
- L. Li, Y. Zhao, M. Antonietti and M. Shalom, Small, 2016, 12, 6090–6097 CrossRef CAS PubMed
-
(a) J. Zhao, H. Lai, Z. Lyu, Y. Jiang, K. Xie, X. Wang, Q. Wu, L. Yang, Z. Jin and Y. Ma, Adv. Mater., 2015, 27, 3541–3545 CrossRef CAS PubMed
-
(a) Y. Zhao, X. Liu, K. X. Yao, L. Zhao and Y. Han, Chem. Mater., 2012, 24, 4725–4734 CrossRef CAS
-
(a) W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed
- N. Fechler and M. Antonietti, Nano Today, 2015, 10, 593–614 CrossRef CAS
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 CrossRef PubMed
-
(a) J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC
- Z. Xia, A. Singh, W. Kiratitanavit, R. Mosurkal, J. Kumar and R. Nagarajan, Thermochim. Acta, 2015, 605, 77–85 CrossRef CAS
-
(a) L. Stradella and M. Argentero, Thermochim. Acta, 1993, 219, 315–323 CrossRef CAS
-
(a) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed
-
(a)
T. Posner, Lehrbuch der synthetischen Methoden der organischen Chemie, Рипол Классик, 1903 Search PubMed
- Y. Chang, M. Antonietti and T. P. Fellinger, Angew. Chem., Int. Ed., 2015, 54, 5507–5512 CrossRef CAS PubMed
-
(a) S. Gadipelli, T. T. Zhao, S. A. Shevlin and Z. X. Guo, Energy Environ. Sci., 2016, 9, 1661–1667 RSC
- J. Lim, S. Y. Ryu, J. Kim and Y. Jun, Nanoscale Res. Lett., 2013, 8, 227 CrossRef PubMed
- S. Zhang, S. Tsuzuki, K. Ueno, K. Dokko and M. Watanabe, Angew. Chem., Int. Ed., 2015, 54, 1302–1306 CrossRef CAS PubMed
-
(a) A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed
- K. P. Gierszal, M. Jaroniec, T.-W. Kim, J. Kim and R. Ryoo, New J. Chem., 2008, 32, 981–993 RSC
- J. W. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W.-G. Bae, L. Pan and J. B.-H. Tok, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed
- X. Ren, H. Li, J. Chen, L. Wei, A. Modak, H. Yang and Q. Yang, Carbon, 2017, 114, 473–481 CrossRef CAS
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed
- M. Oschatz, M. Leistner, W. Nickel and S. Kaskel, Langmuir, 2015, 31, 4040–4047 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta03213k |
| This journal is © The Royal Society of Chemistry 2018 |