
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRealising the environmental benefits of metal–organic frameworks: recent advances in microwave synthesis
Ieuan
Thomas-Hillman†
 ,
Andrea
Laybourn†
*,
Chris
Dodds
and
Samuel W.
Kingman
,
Andrea
Laybourn†
*,
Chris
Dodds
and
Samuel W.
Kingman
Faculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: Andrea.Laybourn@nottingham.ac.uk
First published on 14th June 2018
Abstract
Metal–organic frameworks (MOFs) are a broad class of porous crystalline materials that show great potential for a wide-range of applications in areas such as energy and environmental sustainability. MOFs can show significant advantages in gas selectivity and separation over traditional adsorbents such as zeolites and activated carbons since they are tuneable both in terms of porosity and chemical functionality. The ability to control the pore environment of the MOF is one of their remarkable advantages and affords control over the structure and properties required for specific applications. Despite these advantages, the industrial adoption of MOFs is slow owing to the paucity of scalable, environmentally sustainable manufacturing methods and higher costs compared to zeolites. Microwave (MW) technology is an extremely promising method of MOF production owing to significantly reduced reaction times and subsequently lower process energy consumption, control over MOF properties, and the ability to produce MOFs and MOF-hybrids otherwise difficult to isolate or unobtainable through other synthetic routes. However, the ability to produce the multiple kilogram or even tonne quantities of MOFs required by industry using MW technology is yet to be achieved owing to little or no understanding of the interaction(s) of reactants and MOFs with the electric field, and crucially, how this informs the design of the scale up processes. This review aims to bridge this gap in knowledge by (1) highlighting recent advances in understanding of MW–MOF interactions and areas for future focus; (2) providing an up-to-date and comprehensive summary of literature on MW synthesis of MOFs, focusing on examples where MW heating has facilitated novel and unique results in the laboratory; and (3) emphasising the advantages, challenges and current steps and methodologies required towards industrial-scale MW production of MOFs.
 Ieuan Thomas-Hillman | Ieuan Thomas-Hillman received his Masters of Chemistry degree from Cardiff University in 2016. He is currently undertaking a PhD at the University of Nottingham on the synthesis of metal–organic frameworks using microwave technology. |
 Andrea Laybourn | Andrea Laybourn received her Masters of Chemistry and PhD from the University of Liverpool in 2008 and 2012, respectively. She currently holds a highly prestigious Nottingham Research Fellowship in the Faculty of Engineering at the University of Nottingham. Her research expertise is in the synthesis and characterisation of porous materials underpinned by her background in materials chemistry with a particular focus on the use of microwave heating for ultra-fast materials synthesis. |
 Chris Dodds | Chris Dodds is an Associate Professor at the University of Nottingham. He has fifteen years' experience in the design, scale-up and evaluation of industrial microwave processes and an in depth understanding of the interaction of microwave energy with a wide range of materials. He is an inventor on over 25 patents and an author of over 30 journal papers. Together with Prof. Kingman at the University of Nottingham, he has developed extensive experimental facilities for microwave processing research with many hundreds of kW of microwave power available coupled with unique metrology and applicator systems. |
 Samuel W. Kingman | Sam Kingman is the Associate Faculty Pro-Vice-Chancellor and Deputy Head of the Faculty of Engineering at the University of Nottingham. He was awarded a personal chair at Nottingham in 2006. In 2011 he was the recipient of the Bielby Medal and Prize award by the Society of Chemical Industry, the Royal Society of Chemistry and the Institute of Materials, Minerals and Mining. Professor Kingman has published over 150 journal papers, holds over 180 patents in the field of industrial microwave processing and has lead the development and construction of the world's largest industrial microwave heating processes. |
1. Introduction
Metal–organic frameworks (MOFs)2 are a specific subset of coordination polymers, comprised of metal nodes/clusters and organic linkers, which combine together forming porous supramolecular networks analogous to zeolites in structure. The field of MOFs first emerged in the 1990s with preliminary work performed by Hoskins and Robson.3,4 In 1995 Yaghi et al.5–7 used the term “metal–organic framework” for the first time, showing selective accommodation of guest species in the porous material. Since these early reports, an exponentially increasing number of frameworks have been synthesised and studied, leading to the development of one of the hottest areas of chemical research with over 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 papers in the field published to date, as shown in Fig. 1.8
000 papers in the field published to date, as shown in Fig. 1.8
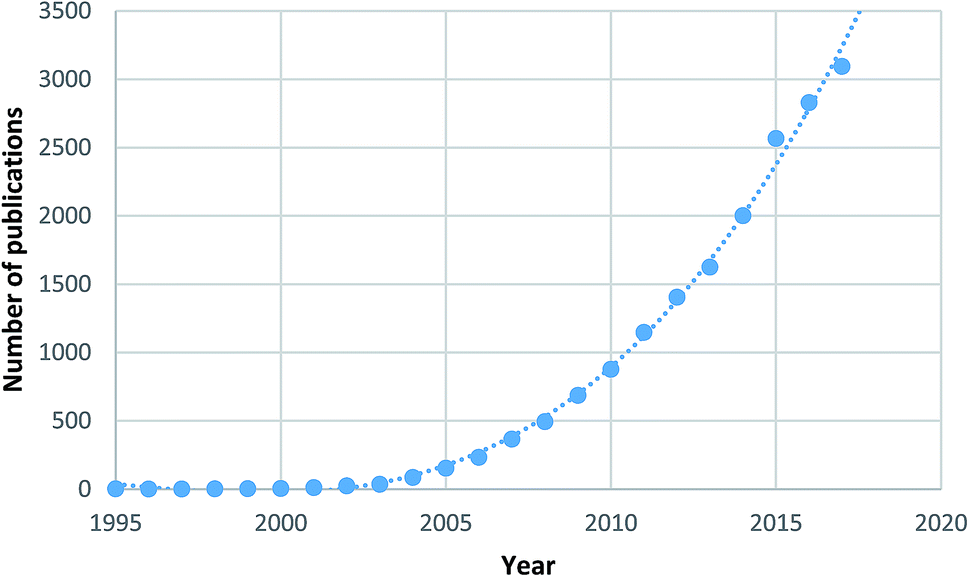 | ||
| Fig. 1 Number of MOF papers published by year highlighting the high level of research interest.8 | ||
MOFs have attracted extensive and continually increasing interest from both academia and, more recently, industry owing to their unprecedented porosity and structural and functional diversity. Proposed applications of MOFs include separations,9–11 gas storage,11 catalysis,11,12 sensing,11,13–15 as synthetic precursors to porous materials16,17 and as stimuli-responsive materials.18,19 A significant advantage of MOFs over traditional adsorbents such as zeolites and activated carbons is the ability to tailor the pore environment of the MOF which affords control over the structure and properties required for any specific application.20
Typically MOFs are prepared by solvothermal batch reactions whereby solutions of metal salt and organic linker are heated above the boiling point of the solvent and retained under autogenous pressure for up to one week.21 This synthetic route has inherent problems as it can produce MOFs of low quality (reduced crystallinity and porosity and non-uniform particle size and morphology, all of which are vital for processing to tailor practical applications). Additional problems include poor reproducibility between batches, long reaction times, large volumes of toxic solvent, and high cost of scale-up along with significant energy consumption.22 The development of technologies that reproducibly give high-quality MOFs whilst reducing the cost of manufacture in an efficient and sustainable way is a key enabling step in the transfer of MOF research from the laboratory to industry and the ultimate realisation of the environmental process benefits that MOFs offer.
Over the past two decades microwave technology has been investigated as an alternative heating method in the preparation of MOFs with a rapidly growing number of publications in this field (over 200 papers published to date).23 Microwave heating is a tremendously exciting route for MOF production as it offers the following benefits; considerably reduced synthesis times (from hours to seconds) and therefore significantly reduced energy consumptions24,25 and control over MOF properties (particle size,24,26,27 morphology25 and phase-selectivity28). Importantly, through rapid and selective heating, microwave technology has been used to prepare MOFs and MOF-hybrids otherwise difficult to isolate or unobtainable through other synthetic routes. Despite this hugely exciting research, the majority of published approaches have involved generic lab microwave systems and experiments that essentially use microwaves as a mechanism for simply rapid heating of solvent. The same is also true of attempts to process zeolites using microwave heating where a number of laboratory-scale studies have been reported using domestic microwave ovens.29 In order to facilitate future developments in microwave synthesis of MOFs (and zeolites) beyond the laboratory, greater understanding of the microwave parameters that are essential for scale-up is required. To this end, we present an up-to-date critical survey of literature relevant to MW synthesis of MOFs. This paper is organised as follows: first the theory of microwave heating and parameters that inform the design of scaled-up MW processes are discussed. Next we provide a comprehensive summary of MOF synthesis using microwave heating and compare materials prepared by this method to those obtained by conventionally heated solvothermal routes. Here we focus on reports of novel results exclusive to microwave heating as it is well-known that for economic impact MWs must be able to achieve outcomes that no other thermal processing method can due to high capital expenditure.1 Finally we discuss the challenges and current progress towards industrial-scale MW production of MOFs. This review aims to assist the progression of microwave-assisted MOF synthesis research by systematically organising previous knowledge in this area across the reported literature in an accessible and informative manner and by identifying areas for future growth.
2. The theory of microwave heating and its application to MOF synthesis
In conventional heating energy is delivered to the material (or reaction mixture in the case of MOF synthesis) by conductive, convective or radiative heat transfer. In dielectric or microwave heating, energy is delivered through interaction of the electronic structure of the material with the alternating electric field. The extent to which the material interacts with the electric field can be characterised by the dielectric properties of the mixture (specifically the dielectric constant, ε′, and dielectric loss, ε′′).30,31ε′ indicates the ability of the material to store energy through a form of polarisation, while ε′′ is the ability of the material to convert the stored energy to heat.30,31 At microwave frequencies, two polarisation or loss mechanisms occur, namely dipolar and ionic polarisation.30,32,33 Dipolar mechanisms occur for molecules that have a permanent dipole, such as water and N,N-dimethylformamide. These molecules experience torque which forces them to reorient in the direction of the applied electric field.32,33 Ionic mechanisms occur in solutions containing ions, such as dissolved salts; in this case the cations and anions are displaced by the oscillating electric field.30,32,33 In both dipolar and ionic polarisation, the species in solution experience frictional forces from neighbouring molecules which is lost as heat.30,32,33 It is important to note that the dielectric properties (ε′ and ε′′) greatly depend on a number of factors including, but not limited to, frequency of the electromagnetic field, temperature, physical state (solid, liquid, or gas), composition and concentration.30,32,33 Therefore, ε′ and ε′′ may vary considerably during a microwave heating process and determination of their values for each reaction is required for detailed understand of the interaction of the electric field with the material(s) of interest.31The dielectric properties of individual components within the bulk reaction mixture indicate which reactants interact more strongly with the electric field and thus heat more effectively. Generally, reactants with high values of ε′′ heat well and those with low ε′′ heat poorly. If there are large differences in the dielectric properties, then individual components will be selectively heated and become much hotter than the bulk.34 Through dielectric properties measurements, Laybourn et al.34 showed that aqueous metal(III) salts interact strongly with the electric field (ε′′ > 35), whereas terephthalic acid exhibits little interaction (ε′′ < 0.03). These results showed that the aqueous metal(III) salts will absorb more power and, therefore, heat more effectively than the ligand. This was the first indication of a selective heating process in MOF synthesis by microwaves. Further evidence of a selective heating mechanism in microwave synthesis of MOFs was also provided, for the first time, by Laybourn et al.25 Using a specifically designed single mode microwave applicator capable of housing a pressure vessel in a well-defined electric field. The authors showed an increase in yield of MIL-53(Al) (MIL = Materials Institute Lavoisier) with increasing average absorbed power at a constant total absorbed energy. Importantly, the reports by Laybourn et al. showed that while still often conducted in pressurised vessels and with identical reaction mixtures, microwave synthesis of MOFs proceeds via a different heating mechanism to conventionally heated reactions. Selective heating can be advantageous as it may give reaction products unobtainable through conventional heating and can result in lower production of unwanted side-products; this is discussed further in Section 3. Crucially it also gives rise for the potential to significantly reduce energy consumption in a process as the whole bulk of the reaction mixture does not need to be heated to the same overall temperature as in a conventional process.
As well as governing the ability of the material/bulk reaction mixture to absorb and convert microwave energy to heat, the dielectric properties also directly affect the distribution of the electric field within the microwave cavity and the power density dissipation, as shown in eqn (1):
| Pd = 2πfε0ε′′E2 | (1) |
In addition, the temperature rise resulting from absorption of microwave energy by the material is related to the power density dissipation, as shown in eqn (2):
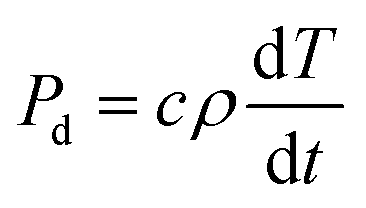 | (2) |
From eqn (1), it is evident that the power density dissipation varies proportionally to ε′′ and frequency, and by a power of two with the electric field. Consequently, significant increases in electric field strength, frequency and ε′′ will result in greatly increased power dissipation densities and thus heating rates (see eqn (2)). Therefore, it is essential to quantify the dielectric properties and electric field distribution when designing microwave cavities as this will control the power density in the heated phases of the material under treatment.
The final parameter crucial to the development of microwave heated systems is the penetration depth. The penetration depth is defined as the point at which the electric field reaches 37% of its original value; the amplitude of the electric field diminishes owing to absorption of the field by the material and conversion into heat. The penetration depth depends greatly on the dielectric properties of the material, as shown in eqn (3):
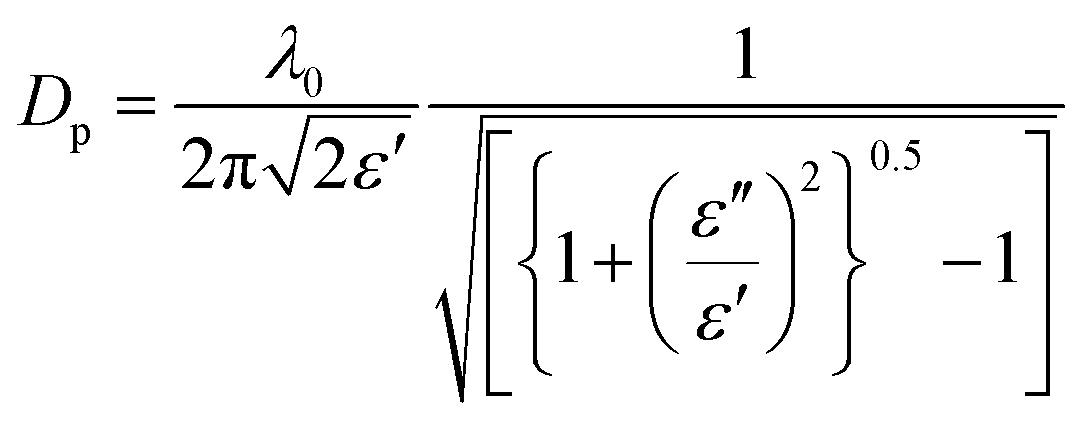 | (3) |
The penetration depth is an important parameter in microwave cavity design as it gives an indication of heat distribution within the material, i.e. whether heating is taking place primarily on the surface of the reaction mixture/material or volumetrically (heating the bulk). To put this into perspective, a typical MIL-53(Al) reaction mixture containing Al2(SO4)3·18H2O (0.77 g, 1.2 mmol), terephthalic acid (0.19 g, 1.2 mmol) and deionized water (6.7 mL) has a penetration depth of 0.38 cm.25
From this section, it should now be clear that the dielectric properties, power density, electric field distribution and penetration depth are all vital in microwave processing as these variables underpin the successful integration of microwave energy with chemical reactor systems capable of delivering the required materials, consistently at high quality and at the correct cost base and with minimal environmental impact. The literature presented in the following sections will be reviewed in this context.
3. Microwave synthesis of MOFs: a comparison of time with conventionally heated reactions
3.1. Background
With the prevalence of microwave heating in organic chemistry and the availability of ‘off the shelf’ microwave reactors, it is unsurprising that this technology has been increasingly applied to the synthesis of MOFs. Microwave technology has been used to prepare MOFs from transition metals35–41 p-block metals (namely lead and indium),42–44 alkali and alkaline earth metals,45–50 lanthanides and actinides;51–56 bimetallic MOFs;57 and mixed-linker MOFs.58–61 Additionally, microwave heating has been used in the synthesis of MOF linkers,62–66 MOF thin films,67–70 membranes,71–76 for surface deposition43,67,70,77–83 and for solvent-free synthesis of MOFs.84,85 Early reports typically used microwave heating as a faster route to known MOFs by adapting the conventionally heated solvothermal reaction conditions. Only recently has microwave synthesis been used independently for discovery of new MOFs facilitated by the ability to screen many reaction conditions in a shorter period of time compared to conventional heating routes.51,86–90The next sub-sections (3.2 to 3.6) of this paper summarise the effect of microwave heating on the reaction conditions and properties of MOFs produced. Where possible, information about microwave parameters such as power, treatment time, and type of cavity (or reactor) have been included and are discussed. However in all reported examples there is no information about the penetration depth and so it is not currently possible to determine whether the bulk synthesis mixture was heated by microwave energy or if the surface was heated by MW and the inside heated through conductive heat transfer. This is a major limitation in much of the literature currently published as it is impossible, without this knowledge, to determine if the systems were actually heated by microwave energy or a shell of the material heated by microwave energy with the majority of the reaction mixture being heated through conductive heat transfer from a microwave heated zone into the bulk.
3.2. Influence of microwave heating on MOF reaction time
The shorter reaction time enabled by microwave treatment has been noted across all synthetic chemistry and was readily apparent in the first report of microwave synthesis of a MOF in 2005.91 In this pioneering work Jhung et al. prepared MIL-100 via microwave and conventional routes by heating an aqueous mixture of metallic chromium, trimesic acid and hydrofluoric acid in a 1.0:0.67:2.0 molar ratio to 220 °C (the conditions for MIL-100 synthesis were previously established by Férey et al.92).91 The significance of microwave heating was demonstrated by a marked reduction in synthesis time without affecting the yield; 44% after 4 hours reaction in a Mars-5 CEM multimode microwave reactor (power not given) compared to 45% after 4 days in a conventionally heated oven.91 Thermo-gravimetric and X-ray diffraction analyses showed the MIL-100 products to be consistent regardless of heating method, however, a slightly reduced pore volume (from 1.16 to 0.97 cm3 g−1) was exhibited by the microwave-synthesised material.
Since 2005, microwave heating has continually been shown to facilitate a progressive reduction in MOF reaction time (in some cases the time is reduced by up to 99.8%, as presented in Table 1). Many papers show the ability to synthesise MOFs using microwave energy in the order of minutes.86 Two reports have also demonstrated the synthesis of MOFs in as little as 25 (1 mL solution heated in a model 520A microwave reactor from Resonance Instrument Inc.)24 and 4.3 seconds (6.7 mL solution heated in bespoke system with a cavity designed to enable precise control of the power and energy input to the reaction mixture);25,34 with the latter representing the fastest reported synthesis of a MOF on the tens of milligram scale to date.25,34
| MOF (trivial name) | Systematic name | Reaction time (minutes) | Reduction in synthesis timeb (%) | Change in MOF yield (%) | Change in crystal size (%) | Change in surface area (%) | Microwave conditions | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH | MW | Cavity type | Powerc (W) | Temp. (°C) | |||||||
| a All percentages calculated from (MW/CH) × 100 = x, x – 100. b Reaction time of microwave synthesis (MW) relative to conventional heating (CH) expressed as a percentage to two decimal places. c Power set on the microwave control unit; this will likely have changed once the target temperature was reached and does not reflect the actual power absorbed by the reaction mixture. d Quantitative data not reported in the original reference. e Single mode cavities. f Multi-mode cavities. g No data available, every minute the authors removed the vessel, stirred and re-heated to avoid overpressure. | |||||||||||
| HKUST-1 | Cu3(BTC)2 | 1440 | 60 | 95.83 | −5 | −50 | +21.35 | CEM MARS-5f | 1200 | 140 | 93 |
| 1440 | 30 | 97.92 | +2 | +5.51 | CEM discovere | 200 | 180 | 94 | |||
| 1200 | 4 | 99.67 | +22.36 | Domestic Galanz, PT021TP-6f | 700 | 95 | |||||
| Ln-MOFs | [Ln(TTTPC)(NO2)2(Cl)]·(H2O)10 | 2880 | 5 | 99.83 | Biotage initiator 8EXPe | 400 | 85 | 56 | |||
| MIL-101 | [Cr3OH(H2O)2(BDC)3] | 1080 | 15 | 98.61 | −9 | −21.21 | +15.72 | CEM discovere | 300 | 220 | 96 |
| MIL-53 | [Fe(OH)BDC] | 4320 | 150 | 96.53 | CEM MARS-5f | 1200 | 70 | 97 | |||
| MOF-5 | [Zn4O(BDC)3] | 1440 | 30 | 97.92 | −95 | −6.0 | CEM MDS-2100e | 1000 | 95–165 | 98 | |
| CPO-27 | Co2(DHTP)(H2O)2·8H2O | 510 | 47 | 90.78 | +12.64 | CEM MARS-5f | 400 | 70 | 99 | ||
| No trivial name given | Fe(1,4-naphthalene dicarboxylate) | 4320 | 2 | 99.95 | −70 | +40.6 | PreeKem Scientific Instruments WF-4000e | 1500 | 100 | ||
| UiO-66 | [Zr6O4(OH)4(BDC)6] | 1440 | 18 | 98.75 | −3 | +100 | −10.5 | CEM MARS-5f | Step 1 200 step 2 80 | 101 | |
3.3. Influence of microwave heating on MOF crystal size
A consequence of the shorter reaction times afforded by microwave heating is the reduction in crystal and/or particle size of MOFs compared to those produced using conventionally heated solvothermal routes. In some cases a 50% reduction in crystal size is observed (see Table 1 above).93 Unlike conventionally heated routes, a narrower crystal size distribution is also exhibited by MOFs produced using microwave-assisted synthesis.25 Many research groups have highlighted the importance of particle size and distribution for environmental applications such as chromatographic separations,102 adsorption,42,103 catalysis104–106 as well as for making multiferroics.107 These results (small crystals and narrow size distribution) have been ascribed to faster kinetics of crystal nucleation and growth owing to hot-spots (or selective heating of components) in the reaction mixture.97 It has been hypothesised that the hot-spots give rise to a high concentration of nucleation points and thus a large nuclei to reactant ratio. As the crystals grow on these nuclei, remaining reactant is rapidly consumed, resulting in crystal size reduction.108 In attempts to quantify the effect of heating source on the crystallisation of MOFs, Haque et al. followed a two-step synthesis of MIL-53(Fe) prepared by ultrasound, conventional, and microwave heating methods using ex situ powder X-ray diffraction (PXRD).97 The authors reported increased acceleration factors (calculated from the pre-exponential factor in the Arrhenius equation) of 14.8 and up to 53.1 for crystal nucleation and growth, respectively, under microwave conditions (CEM Mars 5 reactor, up to 1200 W, 70 °C) compared to conventional heating (70 °C).97 Ultrasound (VCX 750 ultrasonic generator from Sonics & Materials, Inc; power varied at fixed reaction temperatures) was also found to have increased acceleration factors (48.7 and 284.2 for the nucleation and crystal growth steps, respectively) compared to conventional heating.97 However, in the case of ultrasound synthesis, the mode of acceleration is ascribed to cavitation whereby formation, growth, and instantaneous collapse of bubbles in the reaction mixture generates local hot spots.109 Further kinetic studies by ex situ PXRD have also shown significant acceleration of crystal nucleation and growth by microwave heating for a range of MOFs.99,110 However, all kinetic studies conducted thus far carry a level of inherent inaccuracy. No consideration is given to the influence of power absorbed by the reaction mixture in the microwave or ultrasound experiments. These approaches also do not take into account the effect of localised microwave heating and simply measure average bulk temperature arising from the differential heating of individual components, coupled with subsequent heat transfer.In addition to a reduction in crystal size and distribution, many groups have demonstrated the ability to control the crystal size during microwave synthesis. Ni and Masel were able to fine-tune the crystals of IRMOF-1, -2 and -3 on the sub-micrometre scale by altering the concentration of the reaction mixtures, more dilute mixtures being preferable.24 Bag et al. showed control over crystal size for a series of isostructural lanthanide MOFs by careful manipulation of the reaction time; 5 minute reactions yielded micro-crystals and longer times (>30 minutes) gave larger crystals similar in size to the conventionally heated solvothermal route.56 Similarly, Li et al. reported control over IRMOF-3 crystal size by varying the synthesis time.111 Bunzen et al.112 and Liu et al.113 demonstrated control over particle size for MFU-4 and a cyclodextrin MOF, respectively, by variation of reaction time and addition of chemical modulators to the reaction mixture. Reactions involving addition of a modulator with the same chemical functionality as the linker to the reaction mixture in varying amounts is referred to as coordination modulation.114 Finally, Laybourn et al. tuned the particle size and distribution of MIL-53(Al) during microwave synthesis by altering the absorbed power at a fixed energy (250–4000 W; 56 kJ mol−1, based on water as the solvent) showing smaller crystals and narrower size distributions with increasing absorbed power.25
3.4. Influence of microwave heating on the morphology of MOFs
In most instances of microwave synthesis of MOFs, the morphology (i.e. crystal or particle shape) of products is the same as those produced using conventionally heated solvothermal methods. However, few examples of differences in morphology have been reported. Taylor et al. showed a change from spiral rods (1–2 μm) to smaller (300 nm) block-like particles for the room temperature and microwave-assisted syntheses, respectively.115 Using microwave-assisted coordination modulation (for further explanation see Section 3.3), Sakata et al. altered the morphology of Zn2(ndc)2(dabco) (ndc: 1,4-naphthalenedicarboxylate; dabco: 1,4-diazabicyclo[2,2,2]octane) from large micron-sized cubic crystals to nano-sized rods by changing the quantity of modulator added to the reaction mixture.114 Guo et al. demonstrated morphological control over MIL-53(Fe), and the resultant iron oxide nanostructures produced from the calcined MOF, by varying the reaction time in the MOF synthesis step.116 Reaction times of 0.5 and 2 hours gave 1.5 μm long spindles and shorter, fatter spindles, for the iron oxide materials respectively (see Fig. 2).116 At longer times, the microwave reaction led to the production of yolk–shell octahedra with increasing size until 12 hours where large rod structures were observed (Fig. 2).116 In all cases PXRD confirmed production of MIL-53(Fe) in the microwave-synthesis step116 except for the 12 hour reaction which produced a different, though previously reported phase.117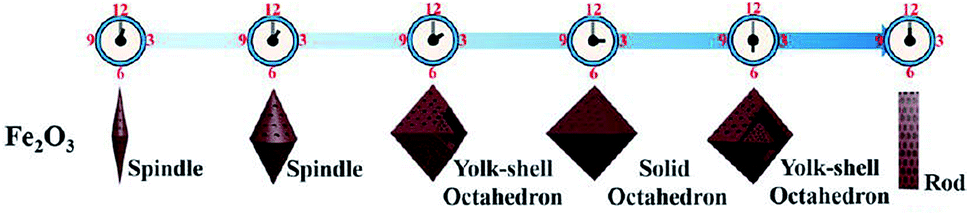 | ||
| Fig. 2 Morphological progression of iron oxide produced from MIL-53(Fe) in a two-step synthesis. Step 1 involved microwave synthesis of the MOF. In step 2, the MOF is calcined. Control of morphology was demonstrated by altering the reaction time in step 1. Reprinted (adapted) with permission from (W. Guo, W. Sun, L.-P. Lv, S. Kong and Y. Wang, ACS Nano, 2017, 11, 4198–4205). Copyright (2017) American Chemical Society. | ||
Finally, Chouhan et al. reported aggregated rhomboidal crystals and thin needle crystals for a MOF based on 2-pyridyltetrazole and zinc produced by conventional and microwave heating methods, respectively;118 and rhomboidal (Fig. 3a) and bundled fibrous (Fig. 3b) nano-sized crystals for the conventional and microwave synthesis of the isostructural cadmium analogue, respectively.119
 | ||
| Fig. 3 Morphological differences between microwave (a) and conventionally heated (b) syntheses of a 2-pyridyltetrazole cadmium MOF.119 Reprinted from Chinese Journal of Chemistry, A. Chouhan, G. Pilet, S. Daniele, et al., Shape Controllable Preparation of Submicronic Cadmium Tetrazole-Based Metal–Organic Frameworks via Solvothermal or Microwave-Assisted Methods and Their Photocatalytic Studies, with permission from John Wiley and Sons. Copyright 2017 SIOC, CAS, Shanghai & WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Interestingly, although PXRD analyses of the zinc and cadmium 2-pyridyltetrazole MOFs indicated identical crystal structures, the materials produced via microwave and conventional methods exhibited subtly different properties (BET surface areas, UV-vis absorption, and luminescence).119 As a result of these differences, the microwave products performed better in the studies of photo-catalytic degradation of methylene blue, which the authors attributed to the higher surface area and therefore more accessible reactive sites in the microwave-synthesised MOFs.119 Although changes in morphology as a result of microwave heating have been reported, the effect of microwave heating on the growth mechanism is yet to be established.
3.5. Influence of microwave heating on the surface area of MOFs
The surface area varies largely for any given MOF. For example, MIL-53(Al) from commercial sources (tradename: Basolite® A100) is supplied with a surface area ranging from 1100 to 1500 m2 g−1.120 Many factors are known to affect the surface area, including solvent choice, quantity of solvent in the reaction mixture, ratio of metal to linker, choice of metal salt and reaction temperature.121–123 Therefore, in order to make a fair assessment of the effect of heating method (conventional solvothermal vs. microwave) on the surface area, comparison between reactions involving the same chemical conditions are required. In these cases, microwave synthesis of MOFs has generally been shown to give products with higher surface areas than those produced via conventional heating routes (see Table 1 and Fig. 4).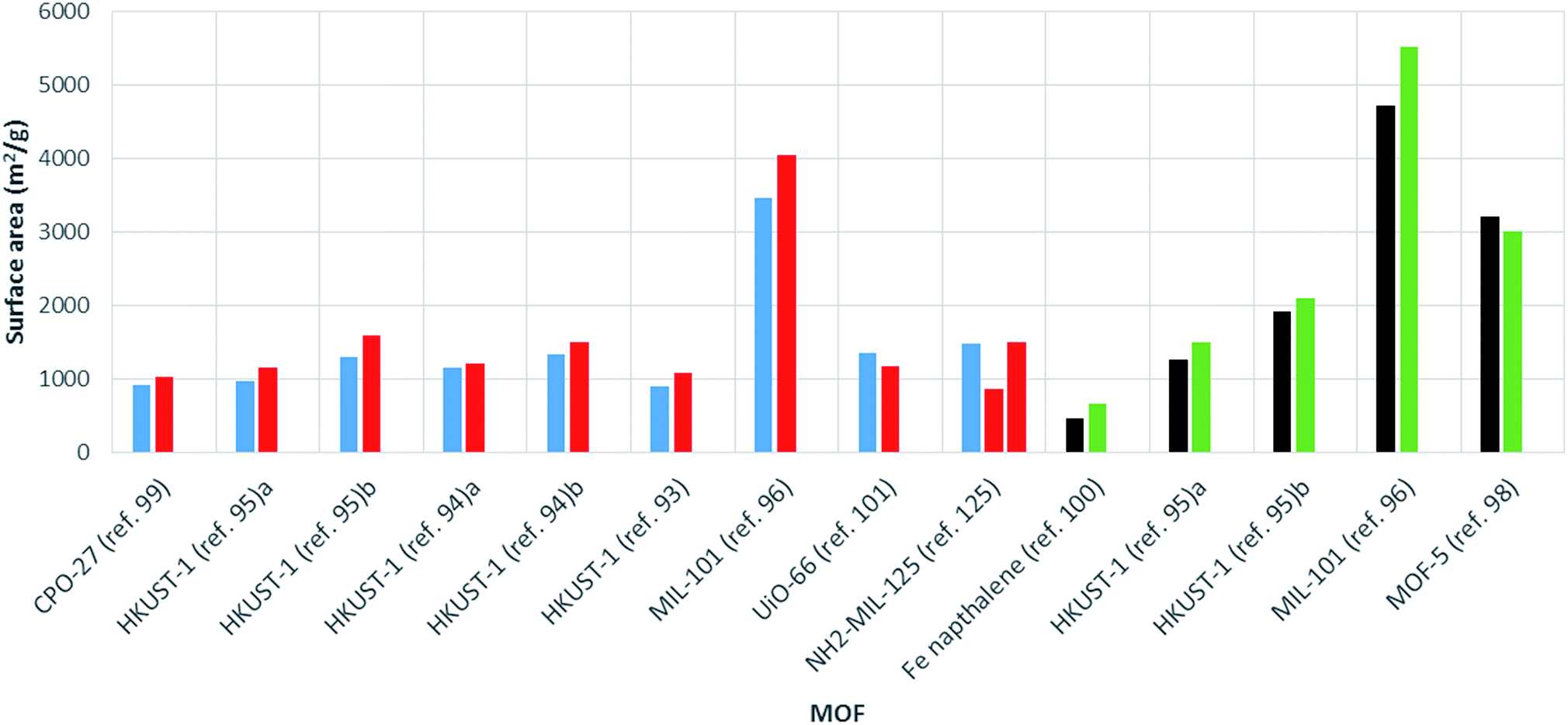 | ||
| Fig. 4 Bar chart showing comparative surface areas between MOFs prepared using microwave (red, BET; green, Langmuir) and conventionally heated solvothermal (blue, BET; black, Langmuir) synthesis. Ref. 95a MOF activated by dissolution in CHCl3. Ref. 95b MOF activated using supercritical carbon dioxide. Synthesis conducted in ethanol and water (ref 94a) and DMF (ref 94b). | ||
In a comparative study, Khan and Jhung found HKUST-1 synthesised using microwave heating to have a larger surface area and pore volume than the conventionally heated solvothermal product (1080 m2 g−1 and 0.39 cm3 g−1 compared to 890 m2 g−1, 0.32 cm3 g−1, respectively, see Table 1).93 Increased surface areas and pore volumes for MOFs produced by microwave heating compared to conventional heating have also been reported for MIL-10196 and further examples of HKUST-194,95 values are given in Table 1. The largest increase in surface area with microwave heating to date was reported by Lou et al. for a 1,4-naphthalene di-carboxylate based iron MOF synthesised using a WF-4000 microwave reactor (PreeKem Scientific Instruments).100 In this case a surface area increase of around 40% was observed (see Table 1).100
An increase in surface area for microwave-synthesised MOFs has been attributed to the production of smaller crystals compared to those resulting from conventional heating methods.99 There are many cases where nano-sized crystals have been found to exhibit greater surface areas than larger MOF crystals.124 However, few examples of reduced surface areas for MOFs produced by microwave heating have been reported. These include MOF-5,98 UiO-66101 and MIL-53(Al)25 where the reduction in surface area was attributed to trapping of oxidised reaction solvent in the pores of the MOF,98 a reduction in linker defects,101 and thermally-induced deterioration of crystal quality at high absorbed powers,25 respectively. Finally, Kim et al. reported both larger and smaller surface areas for MIL-125 and NH2-MIL-125 materials produced using microwave heating (power and reaction time varied) compared to conventionally heated reactions with no clear trend.125 This result suggests that any effect microwave heating has on surface area is dependent on the specifics of the particular synthesis (reactant ratios, solvent systems).
3.6. Summary
In Section 3 we have shown that microwave synthesis is an extremely promising route for MOFs as it potentially offers shorter reaction times, and therefore, lower energy consumptions, control over crystallite size and morphology, and can also produce MOFs with higher surface areas than those produced by conventional solvothermal methods. However, despite the huge activity in this area, gaps in fundamental understanding are still prevalent. For example, the underlying effect of microwave heating on morphology, crystal growth and surface area are still not fully understood. Additionally, many of the reported studies use the average temperature of the reaction mixture as a key indicator for microwave heating. In order for the area to move forward the effect of microwave parameters such as electric field strength and distribution and the dielectric properties must be considered in order to determine if the bulk synthesis mixture was heated by microwave energy.4. Microwave synthesis of new MOFs and MOF-hybrids
4.1. Background
Microwave heating is advantageous for MOF discovery owing to the ability to screen many reaction conditions in a short time-frame. Additionally, selective heating offers the potential to produce new MOF structures through kinetically driven reactions. This section discusses the use of microwave technology in the preparation of new MOFs and MOF-hybrids.4.2. Phase-selective synthesis of MOFs
MOFs can exhibit multiple phases; that is different arrangements of the same metal nodes and linkers, resulting in fundamentally different crystal structures. In some instances up to 5 separate structural phases have been produced from the same reactants.126,127 Different MOF phases often exhibit different properties, notably surface area, therefore production of specific phases is essential for targeting precise properties.128 Synthesis of specific MOF phases can be achieved through control of pH and temperature, by templating and by microwave heating.21 For example, Khan and Jhung28 investigated phase selective synthesis of two MOFs, namely MIL-53(Cr) and MIL-101(Cr), from aqueous reaction mixtures of chromium chloride hexahydrate and terephthalic acid (H2BDC).28 The reaction mixtures were heated to 210 °C under autogenous pressure using either conventional or microwave heating for specific time intervals (daily and hourly, respectively) and the ratio of phases was determined by ex situ PXRD analysis.28 In the early stages of reaction, <1 day for conventional and <3 hours for microwave heating, the kinetically favourable MIL-101(Cr) product was dominant.28 Longer reaction times (>3 hours) resulted in greater quantities of the thermodynamically favourable MIL-53(Cr) product.28 The authors proposed a mechanism for the formation of these two phases; MIL-101 forms first, disassembles, re-assembling to form the MIL-53 phase, as shown in Fig. 5.28,129 Exclusive synthesis of the MIL-101(Cr) phase (termed ‘phase-pure’) was achieved using microwave heating with reaction times of 1 hour.28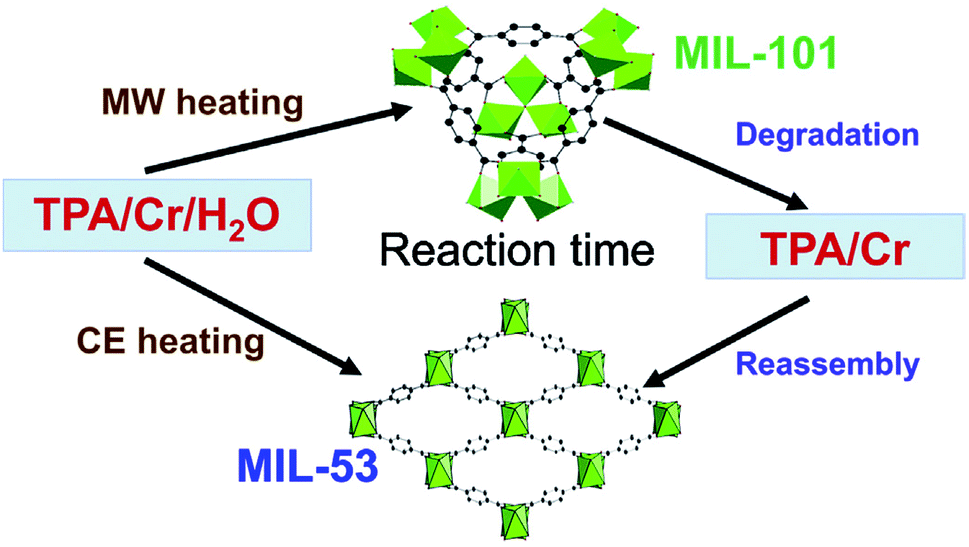 | ||
| Fig. 5 Schematic representation for the formation of two MOF phases (MIL-101 and MIL-53) from reaction between CrCl3·6H2O and H2BDC in water using microwave and conventional heating. Reprinted from Coordination Chemistry Reviews, 285, N. A. Khan, S. H. Jhung, Synthesis of metal–organic frameworks (MOFs) with microwave or ultrasound: rapid reaction, phase-selectivity, and size reduction, 11–23, Copyright (2015), with permission from Elsevier. | ||
Further work by Khan et al. focused on a series of three frameworks based on aluminium metal nodes and trimesic acid, namely MIL-96(Al), MIL-100(Al) and MIL-110(Al).128 Alteration of the reaction time and pH during microwave heating enabled isolation of the separate phases; MIL-110(Al) < 2 h, 2.3 M equivalents of base; MIL-100(Al) < 2 h, 2.3 M equivalents of nitric acid and reactions conducted above 2 hours afforded the thermodynamically stable MIL-96(Al) phase.128 Interestingly, the authors identified a downward trend in porosity with BET surface areas of 1056, 639 and 216 m2 g−1, for MIL-100, MIL-110 and MIL-96, respectively, indicating that more porous structures are inherently less thermodynamically stable.128 Finally, during the microwave synthesis of MIL-77 (a cubic nickel glutarate MOF, previously prepared by Guillou et al.130), Jhung and co-workers discovered a new tetragonal phase.26 Alteration of the reaction conditions showed that the cubic phase preferentially forms at low pH, low temperature and especially under conventional heating whereas the tetragonal phase is obtained favourably at high pH, high temperature and particularly using microwave heating (Mars-5 CEM multimode reactor, power input was varied at different stages of the reaction).26 This result was unexpected as previous reports had shown the denser tetragonal phase to be thermodynamically favourable and the less dense cubic phase to be kinetically favourable and so one would expect the cubic and tetragonal phases to dominate in microwave and conventionally heated reactions, respectively.26 The authors hypothesised the formation of the denser tetragonal phase under microwave heating to be the result of changes in crystallisation rates for the two phases.26
4.3. “One pot” synthesis of MOF hybrids
In chemistry, a one-pot synthesis involves carrying out successive chemical reactions in one reactor. This strategy is often employed to improve the efficiency of reactions as multiple work-up steps are avoided. In MOF chemistry, one-pot synthesis is often used to prepare MOF-hybrids or MOFs with additional functionality as the porous structure can be further functionalised by the secondary reaction. Examples of one-pot microwave-assisted syntheses of MOF hybrids include; MOF–silica131,132 and MOF–alumina131 composites; a photochromic indium trimesate (trivial MOF name, JUC-120) hybrid film produced by entrapping guest molecules within the framework;133 a catalyst for alcoholysis of styrene oxide produced by doping MIL-101(Cr) with phosphotungstic acid;134 and a heterogeneous catalyst for microwave heated organic coupling reactions comprising of IRMOF-3 doped with silver nanoparticles synthesised from the MOF precursor solution and silver nitrate.111 In the latter example the sizes of silver nanoparticles were controlled by altering the reaction time.111 All of these reports are summarised in Table 2.| Classification of material produced | Details | Ref. |
|---|---|---|
| Core–shell hybrid | MIL-53-Cr/V | 135 |
| Inorganic composites | Silica/HKUST-1 | 131 |
| Alumina/HKUST-1 | 131 | |
| Silica (MCM-41)/HKUST-1 | 132 | |
| Spiropyran incorporation in thin film JUC-120 | 133 | |
| Entrapped molecules/‘doped’ hybrid MOFs | Keggin phosphotungic acid doped MIL-101 | 134 |
| IRMOF-3 doped with silver nanoparticles | 111 | |
| Cu-BDC/reduced graphene oxide composite as a precursor for hybrid metal oxide | 136 |
Recently Depauw et al. used microwave heating to prepare a mixed metal MIL-53 MOF via a one-pot reaction containing a mixture of chromium and vanadium salts and H2BDC linker.135 Under conventionally heated solvothermal conditions a homogenous framework was produced, however microwave heating led to the formation of an “egg yolk” structure comprising of a Cr/V core surrounded by a pure chromium MIL-53 shell as determined by energy-dispersive X-ray (EDX) spectroscopy mapping shown in Fig. 6.135
 | ||
| Fig. 6 EDX spectroscopy mapping of a homogenous (A and B) and “egg yolk” (C and D) mixed metal MIL-53(Cr/V) MOF synthesised by conventional and microwave heating, respectively.135 Reproduced from ref. 135 with permission from the Royal Society of Chemistry. | ||
4.4. Post-synthetic modification of MOFs
Microwave heating has also been used for the post-synthetic modification (PSM) of MOFs, whereby a pre-formed MOF is altered. PSM is a particularly important technique as it enables further enhancement and fine-tuning of MOF properties for a wide range of applications through inclusion of alternative chemical species/functionalities.137–139 Post synthetic modification can be categorised into several types, including modification of the linkers (without changing the structure of the MOF) or metal nodes, incorporation of additional chemical species (also called doping, often into the pores of the MOF), or modification of the whole MOF structure. Examples of PSM facilitated by microwave heating are now discussed and a summary is presented in Table 3.| Type of PSM | Details | Comment | Ref. |
|---|---|---|---|
| Incorporation of catalytic nanoparticles | MIL-101@Pd, MIL-101@Cu and MIL-101@Pd/Cu (bimetallic nanoparticles incorporated) | Catalytic oxidation of CO | 140 |
| ScBTC@Pd | Cross coupling reactions | 141 | |
| MIL-101@Cu | Catalytic reduction of nitro-aromatics | 142 | |
| Incorporation of catalytically active nanoparticles | UiO-66@Pd | Cross coupling reactions | 143 |
| MIL-101@Cu and MIL-101@Ni | Augmented CO2 storage | 144 | |
| MIL-101@Fe2O3 | Catalytic oxidation of alcohols and alkenes | 145 | |
| Incorporation of graphene oxide | ZIF-8 derived ZnO with incorporated graphene oxide | Photo-catalytic degradation of methylene blue | 146 |
| Linker functionality modification | UiO-66-Br → UiO-66-CN | 90% yield, route to otherwise unobtainable linker functionality | 147 |
| De-protection of an amine functionalised with tert-butoxycarbonyl (Boc) group IRMOF-74 (Mg) | 148 | ||
| MIL-101-NH2 → MIL-101-peptide | Up to tetra-amino acids grafting onto the linkers without racemization of the amino acids | 149 | |
| MIL-68-NH2 → MIL-68-peptide | |||
| UiO-66-NH2 → UiO-66-peptide | |||
| MIL-53-NH2 functionalised with an alkyl halide | Catalytic synthesis of cyclic carbonates | 150 | |
| IRMOF-74-III | Multivariate in pore tri-peptide functionality | 151 | |
| De-protection of IRMOF-74-III-Boc to give the primary amine | Carbon capture | 152 | |
| MOF–polymer monoliths | MIL-101 | Chromatography | 153 |
| MIL-101(Fe), MIL-101(Al), MIL-100(Cr), UiO-66, MIL-88B(Cr), MIL-53(Al) | Solid phase extraction of penicillin | 154 | |
| MIL-53 | 155 | ||
| Cation exchange | Ni-BTC → Ni/Cu-BTC | Base for production of hybrid metal oxides | 156 |
| UiO-66 → UiO-66(Zr/Ti) | Photocatalysis | 157 | |
| Ni-BTC to mixed metal Ni/Sn-BTC | Base for production of hybrid metal oxides | 158 | |
| Synthetic precursor to hybrid metal oxide | MIL-100 → magnetic Fe2O3/C composite | Ionothermal synthesis with doped zinc chloride | 159 |
| MIL-53 → magnetic Fe2O3/C composite | Ionothermal synthesis with doped zinc chloride, for dye removal and degradation | 160 | |
| Prussian blue: Zn(CO3) | Precursor for a porous ZnO/ZnFe2O4 composite | 161 |
An example of PSM by doping was reported by El-Shall et al.140 In their work a catalyst for the oxidation of CO gas was prepared by doping MIL-101 with metallic and bimetallic nanocrystals.140 During the PSM step, metal salts of palladium and copper where allowed to diffuse into the pores of MIL-101 before addition of a hydrazine hydrate reducing agent.140 This was followed by a 2 minute microwave reaction to yield the final MOF@nanoparticle materials.140 Several examples of PSM involving the MOF linker have been reported, in this case an exposed pendant functional group on the linker undergoes chemical reaction. For instance, Bonnefoy et al. successfully grafted oligopeptides onto the linker of 3 MOFs using microwave heating.149 The authors highlighted the diversity of peptide chemistry as a potential route to a large library of MOF-oligopeptide materials with potential applications in sensing, catalysis and separation.149 It is important to note that the ability to produce MOFs with numerous different pendant functional groups on the linker is particularly powerful as it is conceivably possible to perform countless reactions to augment MOFs using this method of PSM. A final notable example of PSM by microwave heating involves preparation of MOF–polymer monoliths reported by Lin et al.154 The various MOFs (see Table 3 ‘MOF–polymer monoliths’ for exact details) were suspended in a monomer mixture, loaded into a column and then polymerised by microwave heating to give the MOF–polymer monoliths.154 The monoliths were tested for their ability to extract and recover penicillin-type compounds in solid phase extraction, with excellent results.162
4.5. Summary
From the many examples discussed in Section 4 and summarised in Tables 2 and 3, it is clear that microwave heating is an effective method for the production of new MOFs and MOF hybrids by phase-selective or one-pot synthesis or through PSM. However, as identified in Section 3 of this paper, there is little understanding of the mechanisms of microwave heating. In particular the effect of microwave energy on MOF crystallisation is yet to be determined.5. Towards industrial scale microwave synthesis of MOFs
5.1. Background
The diverse and tuneable properties of MOFs offer immense opportunities for economic and environmental impact; for example in the field of ambient pressure CO2 capture,163,164 an industry which is estimated to be worth between $128 billion and $221 billion by 2030.165 However, current state of the art in industrial manufacture has restricted widespread adoption of MOFs for commercial applications, as at scale they deliver poor quality materials, whilst incurring high energy and capital costs, meaning that many applications are not economically viable. The production of MOFs at large scales is hindered by a combination of chemical, environmental and engineering challenges including; dependence on high temperatures and pressures to give solvothermal synthesis conditions; use of large quantities of toxic, corrosive and highly flammable chemicals; production of acidic by-products, long times required for crystal growth; cost and availability of large scale reaction rigs; and energy requirements.22 Additional challenges also exist in materials handling, such as; heterogeneous reaction conditions, mixing requirements, reactions that form precipitates; and separation of products after synthesis.In order to address these challenges, microwave reactions under continuous flow conditions have recently been developed. This production route has several advantages; the combination of rapid microwave heating and high surface area-to-volume ratio for a reaction mixture in a flow reactor leads to improvements in heat and mass transfer and thus significant reduction in synthesis time; typically less solvent is required; process intensification leads to less energy consumption and safer implementation of harsh reaction conditions; improved control over the synthesis parameters allows faster optimisation of reactions to give reproducible MOF products of high quality; and continuous flow reactors are more scalable and often greener compared to batch systems.22
This section focuses on recent research efforts involving the production of MOFs beyond the laboratory scale using microwave technology. Current microwave systems for MOF synthesis on the gram to kilogram scale are presented and their advantages and limitations are discussed. Additionally, the successful development of industrial scale microwave reactors in other sectors is described in order to highlight important considerations and learnings transferrable to the development of an industrial scale microwave process for the synthesis of MOFs. Finally, the economic and environmental implications of industrial scale microwave reactors are discussed.
5.2. Current efforts in scaling-up MOF synthesis using microwave heating
The first continuous flow microwave reactor for the synthesis of MOFs was reported in 2015.166 The pioneering work by Albuquerque et al. involved laboratory scale preparation of MOF-74(Ni) using a gas liquid segmented flow reactor.166 Gas liquid segmented flow was used to improve mixing and to prevent blockages in the reactor.166 The reactor comprises a 1/16 inch tube fed through two sections; a microwave heated zone (see Table 4 for specific details) used for nucleation, and a conventionally heated oil bath zone for crystal growth, as shown in Fig. 7.166 Both sections were found to be necessary for MOF production as broad peaks were observed in the PXRD patterns of MOF-74(Ni) prepared without the oil bath zone.166 The authors reported high conversion of reagents (ca. 96.5%) and space time yields (STY) of (∼90 g h−1 L−1 or 720 kg m−3 d−1, where 1 day is equivalent to 8 hours),166 a significant improvement on the conventional heated batch process (STY of 28.5 g L−1 h−1).167| Name of continuous flow system | MOFs prepared using the reactor | Reaction conditions | Microwave conditions | Production rate (g h−1) | Space time yield (kg m−3 d−1)d | Ref. | |
|---|---|---|---|---|---|---|---|
| Cavity type | Power (W) | ||||||
| a DMF = N,N-dimethylformamide. b Acetic acid was used as a modulator. c Based on a 53 mL microwave heated zone and production rate in g h−1 (in the adjacent column). d Number of hours equivalent to 1 day is variable, depending on the source, and not formally given. e STY not stated, 80–90 g L−1 h−1 given instead. | |||||||
| Segmented flow reactor | MOF-74 (Ni) | 2.5 bar, DMFa | Teflon tube with 1/16 inch inner diameter fed through a WR340 waveguide (single mode) | <3000 | 4.5 | —e | 166 |
| Plug flow reactor | MIL-53(Al) | 6 bar, DMF,a H2O | PTFE tube with 4.35 mm inner diameter fed through the cavity of a CEM MARS 5 multimode synthesizer. Reactor volume altered by changing the number of coils in the tube | 200 | 7.1 | 3168c | 168 |
| UiO-66 | 3 bar, DMF,a H2O, acetic acidb | 200 | 14.4 | 7204c | |||
| HKUST-1 | 6 bar, DMFa | 360 | 79.4 | 64800c |
|||
| Continuous tubular microwave reactor | HKUST-1 | Ethanol, 3% solids | PTFE tube (1 mm internal diameter) fed through the side vents of a of a domestic microwave oven (Logik L20MS10) | ca. 80 | Not given | 80000 |
169 |
| HKUST-1 | Ethanol, 15% solids | 400000 |
|||||
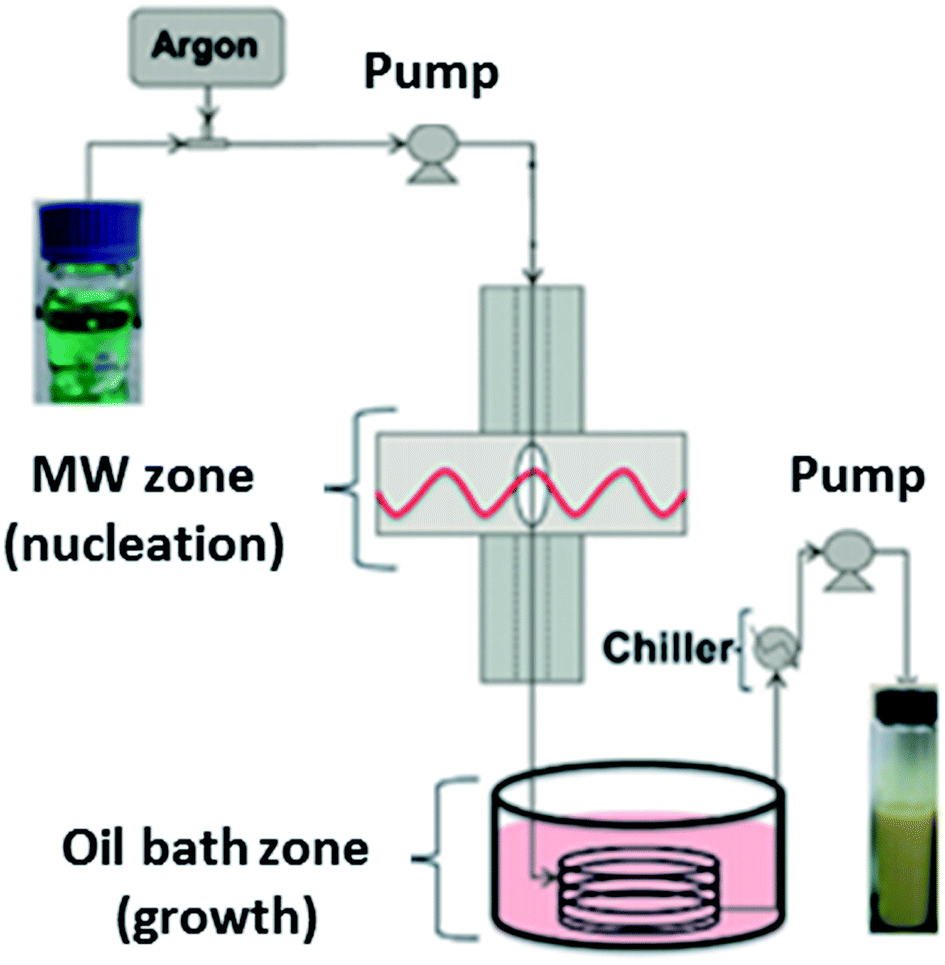 | ||
| Fig. 7 Schematic diagram of the gas liquid segmented continuous flow reactor for synthesis of MOF-74(Ni) developed by Albuquerque et al.166 Reproduced from ref. 166 with permission from the Royal Society of Chemistry. | ||
Building upon their batch process involving simultaneous heating of multiple discrete vessels containing MOF reaction in a multimode microwave reactor (CEM MARS 5 synthesizer),101 Taddei et al. investigated the continuous flow microwave synthesis of three MOFs, namely UiO-66, HKUST-1 and MIL-53.168 The reactor comprises a coiled PTFE tube fed through a CEM MARS 5 synthesizer.168 The volume of the microwave section of the reactor was altered by changing the number of coils in the tube.168 Downstream of the microwave heated section, the tube is introduced into a custom-built pressurised ‘collector’ made of glass, as shown in Fig. 8.168 The collector negates the need to flow slurries containing reactants and products through the back pressure regulator, preventing blockages in the reactor.168 Specific reaction conditions (pressures, solvent systems) and microwave parameters are given in Table 4, above. STYs of 3618, 7204, 64800 kg m−3 d−1 for MIL-53, UiO-66, and HKUST-1, respectively were achieved.168
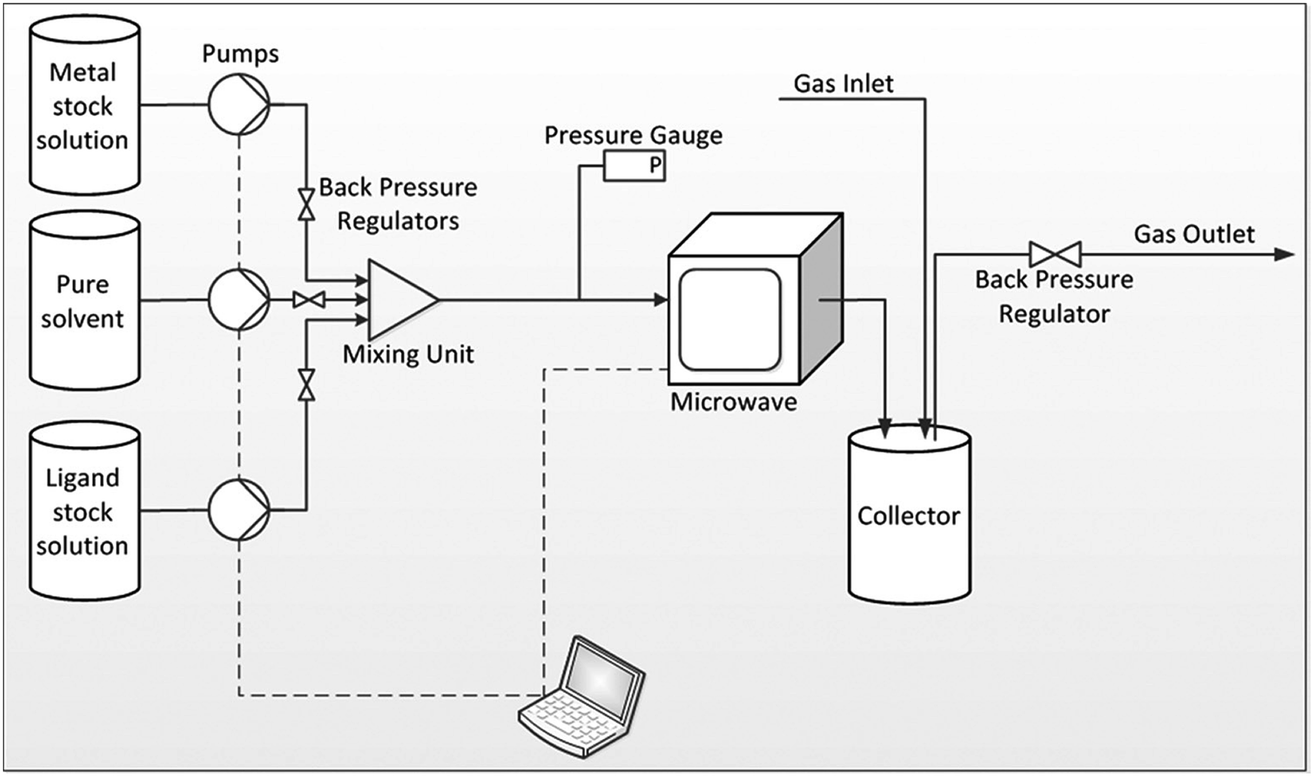 | ||
| Fig. 8 Schematic diagram of the plug flow microwave reactor developed by Taddei et al.168 Reprinted from Chemistry A European Journal, M. Taddei, D. A. Steitz, J. A. van Bokhoven, et al. Continuous-Flow Microwave Synthesis of Metal–Organic Frameworks: A Highly Efficient Method for Large-Scale Production, with permission from John Wiley and Sons. Copyright 2016 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Using the same reactor (Fig. 8),168 Taddei et al. investigated the crystallisation of UiO-66 using high-resolution powder X-ray diffraction by placing the outlet tube (coming out of the microwave heated section) in the focus of the X-ray beam.170 This experimental set-up enabled in situ monitoring of the reaction after microwave heating had taken place. The effect of varying quantities of water and acetic acid modulator in the reaction mixture on the yield and crystallite size of UiO-66 was investigated over a range of residence times. No consideration was given to the effect of absorbed microwave energy on the reaction. A constant power of 280 W was applied in all experiments, therefore the reaction mixtures were subjected to varying amounts of microwave energy depending upon the flow rate. Results showed the rate of product formation, yield and crystallite size to be highly dependent on the water/acetic acid ratio and age of metal salt stock solution.170
Most recently, McKinstry et al. reported the continuous flow microwave synthesis of HKUST-1 using a PTFE tube (1 mm internal diameter) fed through the side vents of a domestic microwave oven (Logik L20MS10, 800 W).169 The authors highlighted that alterations to the microwave cavity casing were avoided for safety reasons,169 however, it is extremely important to note that microwave leakage may still occur even without modification. As electromagnetic wave is attenuated by the reaction mixture (i.e. the amplitude of the electric field diminishes owing to absorption of the field by the reaction mixture, see Section 2 for further details) it is possible for the wave to be small enough to pass along the tube and out of the microwave cavity. Consideration and mitigation of microwave leakage is discussed further in Section 5.3. Using their continuous flow microwave system, McKinstry et al. investigated the effect of varying concentration (reported as % of solids) and residence times on the yield and surface area of HKUST-1 with a constant power input of 80 W; a summary of these results is given in Table 4.169 As the production rate was found to greatly affect the surface area (a key indicator of MOF quality), the authors used surface area production rates (SAPRs, the amount of surface area of MOF produced per reactor volume and time, m2 per m3 per day, where a day is equivalent to 24 hours)169 to compare between experiments and other reported methods of production. A residence time of 6 minutes with 3% solids gave an STY of 2700 kg m−3 d−1 with an SAPR of 5.2 × 106 m2 m−3 d−1 (BET surface area of 1930 m2 g−1). A residence time of 13 seconds and solids of 3 and 15% gave STYs of 80000 and 400000 kg m−3 d−1, respectively.169 However, in both cases a decrease in surface area was observed; 1550 and 600 m2 g−1 (corresponding to an SAPR of 240000 × 106 m2 m−3 d−1) for 3 and 15% reaction solids, respectively.169 Although an STY of 400000 kg m−3 d−1 is, to the best of our knowledge, the highest value for any MOF produced to-date via any synthesis method, a significant reduction in surface area indicates production of poor quality MOF. The trade-off between product quality and rapid, scalable bulk production of MOFs is an important consideration for industrial manufacture.
5.3. Development of industrial scale microwave reactors; important factors to consider
Scaling-up microwave systems from the laboratory to industry requires a multi-disciplinary approach and many factors must be considered from a materials processing, microwave design chemical, environmental and economic perspective. It is beneficial to transfer key learning outputs from successful development of industrial scale microwave reactors in other processing sectors. An example of this is the microwave treatment of ores for the mining industry, work which has led to the largest mass throughput microwave processing systems ever developed. In their work, Buttress et al.171 and Batchelor et al.172 demonstrated the necessity for understanding the fundamental parameters required for designing microwave systems capable of a stable and reliable treatment that also meets with occupational health and safety (OHS) and electromagnetic compatibility (EMC) regulations. These parameters (discussed in this paper in Section 2) include (i) dielectric properties, defining the efficiency of power coupling and distribution of the electric field within the heating cavity; (ii) penetration depth and relationship with reactor design and specification; (iii) power density in the heated phase, which is a direct function of the dielectric loss, the applied frequency and the electric field strength. All of these variables underpinned the design of microwave cavities capable of delivering the power density distribution (energy per unit volume over the treatment time) needed to produce the required material effect, consistently at high quality and with maximum treatment efficiency. Buttress et al. also noted the importance of using single mode cavities as the interaction between the applied microwave energy and the material is very difficult to characterise in multi-mode systems owing to the complexity of the electric field pattern.171 Integration of the microwave cavity with a materials handling system based on flow requirements (e.g. mixing), and chemical compatibility was also essential. For continuous flow reactors, the design of choking structures to confine the electric field within the microwave system is paramount in order to achieve compliance with safety standards. All of these factors support the successful integration of microwave energy with chemical reactor systems capable of delivering the economic large-scale manufacturing processes at the correct cost and minimal environmental impact.Current state of the art in industrial manufacture of MOFs has restricted widespread adoption of MOFs for commercial applications, as at scale they deliver poor quality materials, whilst incurring high energy and capital costs, meaning that many applications are not economically viable. Microwave technology can address these challenges through rapid and selective heating, which can increase production rates by orders of magnitude, give controllable product quality and at a reduced energy and potentially capital cost. Furthermore, the cost of MOF production varies considerably depending on the type of MOF, reagents, synthesis temperature, energy and recyclability of reagents; to ensure that the MOFs and reactors being developed are sustainable on a life cycle basis, it is necessary to evaluate their environmental and economic implications from an early stage of development using life cycle assessment (LCA) and techno-economic analysis (TEA). At present, life cycle, environmental and commercial data in the literature are scarce for conventionally heated well-established MOFs and are non-existent for MOFs synthesised using microwave heating and so further work in this area is much needed. In order for the breakthroughs in research to occur it is our view that there is a requirement to integrate expertise in microwave technology, process engineering design, techno-economic and environmental analyses, and MOF chemistry to deliver a fundamental understanding of the interactions between microwave energy and commercially and scientifically important MOF systems. This knowledge could then be used to deliver the highest quality scientific and engineering breakthroughs to underpin the development of novel scalable and sustainable chemical reactor manufacturing technologies based upon integration of advanced electromagnetic design and process intensification techniques. For successful delivery, this work requires innovation at the boundaries of process assessment and engineering, microwave and materials interaction, process design and materials chemistry. If it can be delivered, we propose that it will commercially unlock the use of MOFs for a number of high impact energy and environmental applications.
6. Conclusions
MOFs are an incredibly large and diverse class of materials. The potentially high commercial value of MOFs arises from their high porosity and tuneable pore environment which gives them an extremely high capacity and high selectivity for gas and hydrocarbon applications. Their proposed applications address challenges in energy, environmental sustainability and innovative healthcare. However, challenges in scaling-up MOF production and their high product and environmental cost compared to traditional sorbents has restricted widespread adoption in industry. Therefore, the development of technologies that reduce the cost of manufacture in an efficient and sustainable way is a key enabling step in the transfer of MOF research from the laboratory to industry.Microwave (MW) technology shows great promise for scale up of MOF synthesis as it offers benefits over other methods including significantly reduced reaction times (from hours to seconds), high space time yields, and improved energy efficiency. However the most noteworthy advantage of microwave heating is the high level of control over MOF properties such as morphology, particle size and phase; achieved by altering the applied power and treatment time. Further control is realised by altering the reaction chemistry through addition of modulators and/or co-reactants and by varying the solvent. Additionally, microwave heating has been shown to facilitate MOF discovery and has the ability to produce new MOFs and MOF-hybrids that are otherwise difficult to prepare or inaccessible by other synthetic approaches. These advantages demonstrate the applicability and value of microwave heating both at laboratory and industrial scales, through acceleration of MOF design and discovery and as a route to large scale production. However, research in the field of microwave synthesis of MOFs is dominated by the use of commercially available multi-mode microwave systems and a lack of understanding of the interactions of reactants and MOFs with the electric field, and crucially, how this informs the design of the scale up processes. MOFs are potential “step-change” materials, but failure to determine this fundamental knowledge thus far explains the general failure in scale-up from the laboratory and to the non-realization of the potential benefits offered.
Conflicts of interest
There are no conflicts of interest to declare.List of abbreviations for linkers and MOFs
| DHTP | 1,5-Dihydroxyterephthalic acid |
| TTTPC | 1,1′,1′′-Tris(2,4,6-trimethylbenzene-1,3,5-triyl)-tris(methylene)-tris(pyridine-4-carboxylic acid) |
| BTC | Trimesic acid (1,3,5-benzenetricarboxylic acid) |
| BDC | Terephthalic acid (1,4-benzenedicarboxylic acid) |
| HKUST | Hong Kong University of Science and Technology |
| Ln MOF | Lanthanide metal–organic framework |
| MIL | Materials of Institut Lavoisier |
| IRMOF | Isoreticular MOF |
| JUC | Jilin University, Changchun |
| UiO | University of Oslo |
Acknowledgements
Andrea Laybourn acknowledges the University of Nottingham for the award of a Nottingham Research Fellowship.References
- R. J. Meredith, Engineers' handbook of industrial microwave heating, IET, UK, 1998 Search PubMed.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, S. M. Paik and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef.
- O. M. Yaghi and G. Li, Angew. Chem., Int. Ed. Engl., 1995, 34, 207–209 CrossRef.
- Scopus database search for “Metal–Organic Framework” or “Metal Organic Framework” – Total results 17759-retrieved 01/12/17.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef PubMed.
- S. Qiu and G. Zhu, Coord. Chem. Rev., 2009, 253, 2891–2911 CrossRef.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef PubMed.
- D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef PubMed.
- P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef.
- L. Zhang, H. B. Wu and X. W. (David) Lou, J. Am. Chem. Soc., 2013, 135, 10664–10672 CrossRef PubMed.
- K. J. Lee, J. H. Lee, S. Jeoung and H. R. Moon, Acc. Chem. Res., 2017, 50, 2684–2692 CrossRef PubMed.
- C. L. Jones, A. J. Tansell and T. Easun, J. Mater. Chem. A, 2016, 4, 6714–6723 Search PubMed.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef PubMed.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- Scopus database search for “Metal–Organic Framework” or “Metal Organic Framework” and “Microwave” – Total results 284-retrieved 20/03/18.
- Z. Ni and R. I. Masel, J. Am. Chem. Soc., 2006, 128, 12394–12395 CrossRef PubMed.
- A. Laybourn, J. Katrib, R. S. Ferrari-John, C. G. Morris, S. Yang, O. Udoudo, T. L. Easun, C. Dodds, N. R. Champness, S. W. Kingman and M. Schröder, J. Mater. Chem. A, 2017, 5, 7333–7338 Search PubMed.
- S. H. Jhung, J.-H. Lee, P. M. Forster, G. Férey, A. K. Cheetham and J.-S. Chang, Chem.–Eur. J., 2006, 12, 7899–7905 CrossRef PubMed.
- Y. K. Hwang, J.-S. Chang, S.-E. Park, D. S. Kim, Y.-U. Kwon, S. H. Jhung, J.-S. Hwang and M. S. Park, Angew. Chem., Int. Ed., 2005, 44, 556–560 CrossRef PubMed.
- N. A. Khan and S. H. Jhung, Cryst. Growth Des., 2010, 10, 1860–1865 Search PubMed.
- R. Xu, W. Pang and Q. Huo, Modern Inorganic Synthetic Chemistry, Elsevier, 2011 Search PubMed.
- J. B. Hasted, Aqueous dielectrics, Chapman and Hall, 1973 Search PubMed.
- A. Metaxis and R. J. Meredith, Industrial Microwave Heating, IET, UK, 1983 Search PubMed.
- G. B. Dudley, R. Richert and A. E. Stiegman, Chem. Sci., 2015, 6, 2144–2152 RSC.
- U. Kaatze and R. Behrends, Tech. Mess., 2009, 69, 5 Search PubMed.
- A. Laybourn, J. Katrib, P. A. Palade, T. L. Easun, N. R. Champness, M. Schröder and S. W. Kingman, Phys. Chem. Chem. Phys., 2016, 18, 5419–5431 RSC.
- W. Liu, L. Ye, X. Liu, L. Yuan, X. Lu and J. Jiang, Inorg. Chem. Commun., 2008, 11, 1250–1252 CrossRef.
- S. Biswas, M. Grzywa, H. P. Nayek, S. Dehnen, I. Senkovska, S. Kaskel and D. Volkmer, Dalton Trans., 2009, 6487–6495 RSC.
- A. Centrone, T. Harada, S. Speakman and T. A. Hatton, Small, 2010, 6, 1598–1602 CrossRef PubMed.
- J. L. Crane, K. E. Anderson and S. G. Conway, J. Chem. Educ., 2015, 92, 373–377 CrossRef.
- M. Ma, A. Bétard, I. Weber, N. S. Al-Hokbany, R. A. Fischer and N. Metzler-Nolte, Cryst. Growth Des., 2013, 13, 2286–2291 Search PubMed.
- G. Y. Yoo, W. R. Lee, H. Jo, J. Park, J. H. Song, K. S. Lim, D. Moon, H. Jung, J. Lim, S. S. Han, Y. Jung and C. S. Hong, Chem.–Eur. J., 2016, 22, 7444–7451 CrossRef PubMed.
- Y.-Y. Liu, S. Couck, M. Vandichel, M. Grzywa, K. Leus, S. Biswas, D. Volkmer, J. Gascon, F. Kapteijn, J. F. M. Denayer, M. Waroquier, V. Van Speybroeck and P. Van Der Voort, Inorg. Chem., 2013, 52, 113–120 CrossRef PubMed.
- L. Hashemi and A. Morsali, CrystEngComm, 2012, 14, 779–781 RSC.
- S. Khanjani and A. Morsali, CrystEngComm, 2012, 14, 8137–8142 RSC.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, Chem.–Eur. J., 2016, 22, 6654–6665 CrossRef PubMed.
- H.-K. Liu, T.-H. Tsao, Y.-T. Zhang and C.-H. Lin, CrystEngComm, 2009, 11, 1462–1468 RSC.
- P.-C. Liang, H.-K. Liu, C.-T. Yeh, C.-H. Lin and V. Zima, Cryst. Growth Des., 2011, 11, 699–708 Search PubMed.
- M. Mazaj, V. Kaucic, A. Golobic and N. Zabukovec Logar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, m4–m6 Search PubMed.
- C.-T. Yeh, W.-C. Lin, S.-H. Lo, C.-C. Kao, C.-H. Lin and C.-C. Yang, CrystEngComm, 2012, 14, 1219–1222 RSC.
- C.-Y. Wu, D. S. Raja, C.-C. Yang, C.-T. Yeh, Y.-R. Chen, C.-Y. Li, B.-T. Ko and C.-H. Lin, CrystEngComm, 2014, 16, 9308–9319 RSC.
- C. P. Cabello, C. O. Arean, J. B. Parra, C. O. Ania, P. Rumori and G. T. Palomino, Dalton Trans., 2015, 44, 9955–9963 RSC.
- P. Silva, F. Vieira, A. C. Gomes, D. Ananias, J. A. Fernandes, S. M. Bruno, R. Soares, A. A. Valente, J. Rocha and F. A. A. Paz, J. Am. Chem. Soc., 2011, 133, 15120–15138 CrossRef PubMed.
- Z.-J. Lin, Z. Yang, T.-F. Liu, Y.-B. Huang and R. Cao, Inorg. Chem., 2012, 51, 1813–1820 CrossRef PubMed.
- S. M. F. Vilela, D. Ananias, A. C. Gomes, A. A. Valente, L. D. Carlos, J. A. S. Cavaleiro, J. Rocha, J. P. C. Tomé and F. A. A. Paz, J. Mater. Chem., 2012, 22, 18354–18371 RSC.
- P. Silva, D. Ananias, S. M. Bruno, A. A. Valente, L. D. Carlos, J. Rocha and F. A. Almeida Paz, Eur. J. Inorg. Chem., 2013, 2013, 5576–5591 CrossRef.
- R. F. Mendes and F. A. Almeida Paz, Inorg. Chim. Acta, 2017, 460, 99–107 CrossRef.
- P. P. Bag, X.-S. Wang and R. Cao, Dalton Trans., 2015, 44, 11954–11962 RSC.
- H. Li, M. Liang, W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 1098–1103 CrossRef.
- Y.-S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592–8598 CrossRef PubMed.
- S. Bernt, M. Feyand, A. Modrow, J. Wack, J. Senker and N. Stock, Eur. J. Inorg. Chem., 2011, 2011, 5378–5383 CrossRef.
- A. C. Kathalikkattil, R. Roshan, J. Tharun, H.-G. Soek, H.-S. Ryu and D.-W. Park, ChemCatChem, 2014, 6, 284–292 CrossRef.
- A. C. Kathalikkattil, D.-W. Kim, J. Tharun, H.-G. Soek, R. Roshan and D.-W. Park, Green Chem., 2014, 16, 1607–1616 RSC.
- S. Mukhopadhyay, B. G. Mukhopadhyay, M. F. C. G. da Silva, J. Lasri, M. A. J. Charmier and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 11334–11341 CrossRef PubMed.
- W. Karuehanon, W. Fanfuenha, A. Rujiwatra and M. Pattarawarapan, Tetrahedron Lett., 2012, 53, 3486–3489 CrossRef.
- W. Funfuenha, W. Phakhodee and M. Pattarawarapan, Tetrahedron, 2014, 70, 5415–5419 CrossRef.
- L. Martins, R. Nasani, M. Saha, S. Mobin, S. Mukhopadhyay and A. Pombeiro, Molecules, 2015, 20, 19203–19220 CrossRef PubMed.
- A. D. Martin, T. L. Easun, S. P. Argent, W. Lewis, A. J. Blake and M. Schröder, CrystEngComm, 2017, 19, 603–607 RSC.
- Y. Yoo and H.-K. Jeong, Chem. Commun., 2008, 0, 2441–2443 RSC.
- Z.-Q. Li, M. Zhang, B. Liu, C.-Y. Guo and M. Zhou, Inorg. Chem. Commun., 2013, 36, 241–244 CrossRef.
- Y. Abdollahian, J. L. Hauser, I. R. Colinas, C. Agustin, A. S. Ichimura and S. R. J. Oliver, Cryst. Growth Des., 2014, 14, 1506–1509 Search PubMed.
- W.-J. Li, J.-F. Feng, Z.-J. Lin, Y.-L. Yang, Y. Yang, X.-S. Wang, S.-Y. Gao and R. Cao, Chem. Commun., 2016, 52, 3951–3954 RSC.
- H. Bux, F. Liang, Y. Li, J. Cravillon, M. Wiebcke and J. Caro, J. Am. Chem. Soc., 2009, 131, 16000–16001 CrossRef PubMed.
- Y. Yoo, Z. Lai and H.-K. Jeong, Microporous Mesoporous Mater., 2009, 123, 100–106 CrossRef.
- J. Caro, Curr. Opin. Chem. Eng., 2011, 1, 77–83 CrossRef.
- H. T. Kwon and H.-K. Jeong, Chem. Commun., 2013, 49, 3854–3856 RSC.
- Y. Zhu, Q. Liu, J. Caro and A. Huang, Sep. Purif. Technol., 2015, 146, 68–74 CrossRef.
- S. Shahid, K. Nijmeijer, S. Nehache, I. Vankelecom, A. Deratani and D. Quemener, J. Membr. Sci., 2015, 492, 21–31 CrossRef.
- Y.-S. Li, F.-Y. Liang, H. Bux, A. Feldhoff, W.-S. Yang and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 548–551 CrossRef PubMed.
- A. Centrone, Y. Yang, S. Speakman, L. Bromberg, G. C. Rutledge and T. A. Hatton, J. Am. Chem. Soc., 2010, 132, 15687–15691 CrossRef PubMed.
- S. Xiao, P. Liu, W. Zhu, G. Li, D. Zhang and H. Li, Nano Lett., 2015, 15, 4853–4858 CrossRef PubMed.
- M. Bechelany, M. Drobek, C. Vallicari, A. A. Chaaya, A. Julbe and P. Miele, Nanoscale, 2015, 7, 5794–5802 RSC.
- C. Liu, Y. Wu, C. Morlay, Y. Gu, B. Gebremariam, X. Yuan and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 2552–2561 Search PubMed.
- N. E. Tari, A. Tadjarodi, J. Tamnanloo and S. Fatemi, J. CO2 Util., 2016, 14, 126–134 CrossRef.
- R. D. Arrua, A. Peristyy, P. N. Nesterenko, A. Das, D. M. D'Alessandro and E. F. Hilder, Analyst, 2017, 142, 517–524 RSC.
- H. Zhang, J. Zhong, G. Zhou, J. Wu, Z. Yang and X. Shi, J. Nanomater., 2016, 2016, 9648386 Search PubMed.
- H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef PubMed.
- J. Klinowski, F. A. Almeida Paz, P. Silva and J. Rocha, Dalton Trans., 2011, 40, 321–330 RSC.
- P. Maniam and N. Stock, Inorg. Chem., 2011, 50, 5085–5097 CrossRef PubMed.
- T. Ahnfeldt, J. Moellmer, V. Guillerm, R. Staudt, C. Serre and N. Stock, Chem.–Eur. J., 2011, 17, 6462–6468 CrossRef PubMed.
- H. Reinsch and N. Stock, Microporous Mesoporous Mater., 2013, 171, 156–165 CrossRef.
- S. Halis, N. Reimer, A. Klinkebiel, U. Lüning and N. Stock, Microporous Mesoporous Mater., 2015, 216, 13–19 CrossRef.
- S. H. Jhung, J.-H. Lee and J.-S. Chang, Bull. Korean Chem. Soc., 2005, 26, 880 CrossRef.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- N. A. Khan and S. H. Jhung, Bull. Korean Chem. Soc., 2009, 30, 2921–2926 CrossRef.
- M. Schlesinger, S. Schulze, M. Hietschold and M. Mehring, Microporous Mesoporous Mater., 2010, 132, 121–127 CrossRef.
- Z. Xiang, D. Cao, X. Shao, W. Wang, J. Zhang and W. Wu, Chem. Eng. Sci., 2010, 65, 3140–3146 CrossRef.
- L. Bromberg, Y. Diao, H. Wu, S. A. Speakman and T. A. Hatton, Chem. Mater., 2012, 24, 1664–1675 CrossRef.
- E. Haque, N. A. Khan, J. H. Park and S. H. Jhung, Chem.–Eur. J., 2010, 16, 1046–1052 CrossRef PubMed.
- J.-S. Choi, W.-J. Son, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 116, 727–731 CrossRef.
- E. Haque and S. H. Jhung, Chem. Eng. J., 2011, 173, 866–872 CrossRef.
- Y. Lou, J. Chen, J. Jiang and Q. Bao, Dalton Trans., 2013, 43, 1261–1266 RSC.
- M. Taddei, P. V. Dau, S. M. Cohen, M. Ranocchiari, J. A. van Bokhoven, F. Costantino, S. Sabatini and R. Vivani, Dalton Trans., 2015, 44, 14019–14026 RSC.
- D. Cabooter, A. Fanigliulo, G. Bellazzi, B. Allieri, A. Rottigni and G. Desmet, J. Chromatogr. A, 2010, 1217, 7074–7081 CrossRef PubMed.
- H. Uehara, S. Diring, S. Furukawa, Z. Kalay, M. Tsotsalas, M. Nakahama, K. Hirai, M. Kondo, O. Sakata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 11932–11935 CrossRef PubMed.
- M. Tonigold, Y. Lu, B. Bredenkötter, B. Rieger, S. Bahnmüller, J. Hitzbleck, G. Langstein and D. Volkmer, Angew. Chem., Int. Ed., 2009, 48, 7546–7550 CrossRef PubMed.
- A. Dhakshinamoorthy, M. Alvaro, Y. K. Hwang, Y.-K. Seo, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 10719–10724 RSC.
- K. Hindelang, A. Kronast, S. I. Vagin and B. Rieger, Chem.–Eur. J., 2013, 19, 8244–8252 CrossRef PubMed.
- N. Abhyankar, M. Lee, M. Foley, E. S. Choi, G. Strouse, H. W. Kroto and N. S. Dalal, Phys. Status Solidi RRL, 2016, 10, 600–605 CrossRef.
- C. Carlucci, H. Xu, B. Federica Scremin, C. Giannini, D. Altamura, E. Carlino, V. Videtta, F. Conciauro, G. Gigli and G. Ciccarella, CrystEngComm, 2014, 16, 1817–1824 RSC.
- M. Yaser Masoomi, A. Morsali and P. C. Junk, RSC Adv., 2014, 4, 47894–47898 RSC.
- E. Haque, N. A. Khan, C. M. Kim and S. H. Jhung, Cryst. Growth Des., 2011, 11, 4413–4421 Search PubMed.
- D. Li, X. Dai, X. Zhang, H. Zhuo, Y. Jiang, Y.-B. Yu, P. Zhang, X. Huang and H. Wang, J. Catal., 2017, 348, 276–281 CrossRef.
- H. Bunzen, M. Grzywa, M. Hambach, S. Spirkl and D. Volkmer, Cryst. Growth Des., 2016, 16, 3190–3197 Search PubMed.
- B. Liu, Y. He, L. Han, V. Singh, X. Xu, T. Guo, F. Meng, X. Xu, P. York, Z. Liu and J. Zhang, Cryst. Growth Des., 2017, 17, 1654–1660 Search PubMed.
- Y. Sakata, S. Furukawa, C. Kim and S. Kitagawa, Chem. Lett., 2012, 41, 1436–1438 CrossRef.
- K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358–14359 CrossRef PubMed.
- W. Guo, W. Sun, L.-P. Lv, S. Kong and Y. Wang, ACS Nano, 2017, 11, 4198–4205 CrossRef PubMed.
- C. Zhang, L. Ai and J. Jiang, J. Mater. Chem. A, 2015, 3, 3074–3081 Search PubMed.
- A. Chouhan, P. Mayer and A. Pandey, Indian J. Chem., 2015, 54A, 851–857 Search PubMed.
- A. Chouhan, G. Pilet, S. Daniele and A. Pandey, Chin. J. Chem., 2017, 35, 209–216 CrossRef.
- Calculated from the advertised surface area range of Basolite MOFs on Sigma Aldrich, retrieved 18/09/17.
- R. Sabouni, H. Kazemian and S. Rohani, Chem. Eng. Technol., 2012, 35, 1085–1092 CrossRef.
- M. Märcz, R. E. Johnsen, P. D. C. Dietzel and H. Fjellvåg, Microporous Mesoporous Mater., 2012, 157, 62–74 CrossRef.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef.
- A. Carné, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291–305 RSC.
- S.-N. Kim, J. Kim, H.-Y. Kim, H.-Y. Cho and W.-S. Ahn, Catal. Today, 2013, 204, 85–93 CrossRef.
- F.-L. Yuan, Y.-Q. Yuan, M.-Y. Chao, D. J. Young, W.-H. Zhang and J.-P. Lang, Inorg. Chem., 2017, 56, 6522–6531 CrossRef PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Inorg. Chem., 2008, 47, 7751–7756 CrossRef PubMed.
- N. A. Khan, J. S. Lee, J. Jeon, C.-H. Jun and S. H. Jhung, Microporous Mesoporous Mater., 2012, 152, 235–239 CrossRef.
- N. A. Khan and S. H. Jhung, Coord. Chem. Rev., 2015, 285, 11–23 CrossRef.
- N. Guillou, C. Livage, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 5314–5317 CrossRef PubMed.
- J. Górka, P. F. Fulvio, S. Pikus and M. Jaroniec, Chem. Commun., 2010, 46, 6798–6800 RSC.
- N. E. Tari, A. Tadjarodi, J. Tamnanloo and S. Fatemi, Microporous Mesoporous Mater., 2016, 231, 154–162 CrossRef.
- F. Zhang, X. Zou, W. Feng, X. Zhao, X. Jing, F. Sun, H. Ren and G. Zhu, J. Mater. Chem., 2012, 22, 25019–25026 RSC.
- L. H. Wee, F. Bonino, C. Lamberti, S. Bordiga and J. A. Martens, Green Chem., 2014, 16, 1351–1357 RSC.
- H. Depauw, I. Nevjestic, J. D. Winne, G. Wang, K. Haustraete, K. Leus, A. A. Verberckmoes, C. Detavernier, F. Callens, E. D. Canck, H. M. Vrielinck and P. V. D. Voort, Chem. Commun., 2017, 53, 8478–8481 RSC.
- D. Li, D. Yan, X. Zhang, J. Li, T. Lu and L. Pan, J. Colloid Interface Sci., 2017, 497, 350–358 CrossRef PubMed.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef PubMed.
- M. S. El-Shall, V. Abdelsayed, A. E. R. S. Khder, H. M. A. Hassan, H. M. El-Kaderi and T. E. Reich, J. Mater. Chem., 2009, 19, 7625–7631 RSC.
- L. Zhang, Z. Su, F. Jiang, Y. Zhou, W. Xu and M. Hong, Tetrahedron, 2013, 69, 9237–9244 CrossRef.
- F. Wu, L.-G. Qiu, F. Ke and X. Jiang, Inorg. Chem. Commun., 2013, 32, 5–8 CrossRef.
- W. Dong, C. Feng, L. Zhang, N. Shang, S. Gao, C. Wang and Z. Wang, Catal. Lett., 2016, 146, 117–125 CrossRef.
- M. Montazerolghaem, S. F. Aghamiri, S. Tangestaninejad and M. R. Talaie, RSC Adv., 2015, 6, 632–640 RSC.
- A. M. Balu, C. S. K. Lin, H. Liu, Y. Li, C. Vargas and R. Luque, Appl. Catal., A, 2013, 455, 261–266 CrossRef.
- G. Zhu, X. Li, H. Wang and L. Zhang, Catal. Commun., 2017, 88, 5–8 CrossRef.
- M. Kim, S. J. Garibay and S. M. Cohen, Inorg. Chem., 2011, 50, 729–731 CrossRef PubMed.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef PubMed.
- J. Bonnefoy, A. Legrand, E. A. Quadrelli, J. Canivet and D. Farrusseng, J. Am. Chem. Soc., 2015, 137, 9409–9416 CrossRef PubMed.
- H.-G. Seok, D.-W. Kim, J.-G. Yang, M.-I. Kim and D.-W. Park, J. Nanosci. Nanotechnol., 2016, 16, 4612–4619 CrossRef PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef PubMed.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef PubMed.
- H.-Y. Huang, C.-L. Lin, C.-Y. Wu, Y.-J. Cheng and C.-H. Lin, Anal. Chim. Acta, 2013, 779, 96–103 CrossRef PubMed.
- C.-L. Lin, S. Lirio, Y.-T. Chen, C.-H. Lin and H.-Y. Huang, Chem.–Eur. J., 2014, 20, 3317–3321 CrossRef PubMed.
- S. Lirio, W.-L. Liu, C.-L. Lin, C.-H. Lin and H.-Y. Huang, J. Chromatogr. A, 2016, 1428, 236–245 CrossRef PubMed.
- W. Guo, W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef PubMed.
- J. Tu, X. Zeng, F. Xu, X. Wu, Y. Tian, X. Hou and Z. Long, Chem. Commun., 2017, 53, 3361–3364 RSC.
- R. Dai, W. Sun, L.-P. Lv, M. Wu, H. Liu, G. Wang and Y. Wang, Small, 2017, 13, 1700521 CrossRef PubMed.
- J.-D. Xiao, L.-G. Qiu, X. Jiang, Y.-J. Zhu, S. Ye and X. Jiang, Carbon, 2013, 59, 372–382 CrossRef.
- C. Zhang, F. Ye, S. Shen, Y. Xiong, L. Su and S. Zhao, RSC Adv., 2015, 5, 8228–8235 RSC.
- X. Yang, H. Xue, Q. Yang, R. Yuan, W. Kang and C.-S. Lee, Chem. Eng. J., 2017, 308, 340–346 CrossRef.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 Search PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef.
- Carbon Capture and Sequestration Market Could Reach $128 Billion by 2030, https://www.navigantresearch.com/newsroom/carbon-capture-and-sequestration-market-could-reach-128-billion-by-2030, accessed May 16, 2017.
- G. H. Albuquerque, R. C. Fitzmorris, M. Ahmadi, N. Wannenmacher, P. K. Thallapally, B. P. McGrail and G. S. Herman, CrystEngComm, 2015, 17, 5502–5510 RSC.
- S. Cadot, L. Veyre, D. Luneau, D. Farrusseng and E. A. Quadrelli, J. Mater. Chem. A, 2014, 2, 17757–17763 Search PubMed.
- M. Taddei, D. A. Steitz, J. A. van Bokhoven and M. Ranocchiari, Chem.–Eur. J., 2016, 22, 3245–3249 CrossRef PubMed.
- C. McKinstry, E. J. Cussen, A. J. Fletcher, S. V. Patwardhan and J. Sefcik, Chem. Eng. J., 2017, 326, 570–577 CrossRef.
- M. Taddei, N. Casati, D. A. Steitz, K. C. Dümbgen, J. van Bokhoven and M. Ranocchiari, CrystEngComm, 2017, 19, 3206–3214 RSC.
- A. J. Buttress, J. Katrib, D. A. Jones, A. R. Batchelor, D. A. Craig, T. A. Royal, C. Dodds and S. W. Kingman, Miner. Eng., 2017, 109, 169–183 CrossRef.
- A. R. Batchelor, A. J. Buttress, D. A. Jones, J. Katrib, D. Way, T. Chenje, D. Stoll, C. Dodds and S. W. Kingman, Miner. Eng., 2017, 111, 5–24 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |