
Molecular blends of methylated-poly(ethylenimine) and amorphous porous organic cages for SO2 adsorption†

Abstract
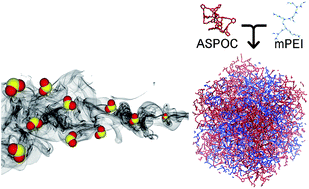
Porous organic cages (POCs) are emerging porous materials that exhibit intriguing properties in the areas of self-assembly, host–guest interaction, and solution processability. In this work, we explore the applicability of POCs as molecular porous supports for polymeric amines. We find that primary amines in poly(ethylenimine) (PEI) can undergo metathesis with the imine bonds present in POCs, resulting in non-porous products. This problem can be overcome by transforming the primary amines in PEI to tertiary amines via methylation. The methylated PEI (mPEI) forms homogeneous composites with amorphous scrambled porous organic cages (ASPOCs) without undesired reactions or phase separation. The microscopic structure of the composites is studied using molecular dynamics simulations. These composite materials are evaluated as adsorbents for low concentration SO2 (200 ppm) adsorption and show good thermal and cyclic stability.
- This article is part of the themed collection: Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

