DOI:
10.1039/C8TA02622J
(Paper)
J. Mater. Chem. A, 2018,
6, 14449-14463
Transition metal speciation as a degradation mechanism with the formation of a solid-electrolyte interphase (SEI) in Ni-rich transition metal oxide cathodes†
Received
21st March 2018
, Accepted 25th June 2018
First published on 27th June 2018
Abstract
An ever-growing demand for high-energy density and high-power Li-ion batteries has driven active research for electrode materials with superior capacity. Recent years have seen the development of Ni-rich transition metal oxide cathode materials due to their high reversible capacity and lower cost. To achieve full capacity from the charge compensation process, a high voltage (>4.4 V) charging is required. However, the battery operation at higher voltages eventually results in dramatic capacity fading and voltage decay with a rapid decomposition of the electrolyte upon further charge–discharge. While previous studies have reported the degradation mechanism within the electrode surface, there have been few empirical investigations into the solid-electrolyte interphase (SEI) formation on Ni-rich cathodes. In the current work, we visualize the different nature of the electrode–electrolyte interphase at various cut-off voltages (2.0–4.2 V, 2.0–4.5 V, and 2.0–4.8 V). We correlate the key properties of the SEI layer with the capacity fading mechanism in the high capacity battery system. The speciation of transition metal elements (Ni, Mn, and Co) into various oxidation and spin states has been identified as the dominant process of the capacity degradation.
1. Introduction
The widespread adoption of lithium-ion batteries in energy storage systems for electric transportation, e.g. hybrid electric vehicles (HEV), battery electric vehicles (BEV), and rover vehicles, has heightened the need for developing high-energy density and high power batteries with enhanced stability.1–5 The successful commercialization of layered LiCoO2 rechargeable batteries has revolutionized mobile electronics during the past few decades. There are, however, inherent limitations for the layered LiCoO2 cathode such as low operating capacity (∼140 mA h g−1), low reversible capacity, instability in the deintercalated state, Co toxicity, and limited availability of Co resources.6–8 Much effort has been put into tailoring the layered LiCoO2 cathode, with particular attention to overcoming its technical hurdles to achieve high-energy density with improved stability. LiNi1/3Co1/3Mn1/3O2 with a layered α-NaFeO2 structure, generally represented as NCM111, is of much interest because of its synergetic advantages over a single transition metal. The replacement of Co with Mn not only offers benefits with respect to ecological aspects, but it also provides merits for stabilizing the framework for the electrode structure, retaining the initial oxidation state of Mn.9 On the other hand, Ni is the key element that offers a large amount of cell capacity by a two-stage redox reaction between Ni2+/Ni3+ and Ni3+/Ni4+.10–12 The battery charge can also be achieved by the redox reaction of Co3+/Co4+, which usually takes place between 67% and 100% state of charge (SOC).13 In addition, the presence of Co3+ can suppress some structural distortions resulting from the Ni3+ Jahn–Teller effect and cation disorder.14–17 It is therefore the benefits offered by Co that lead to the improvement in the rate capability of lithium-ion batteries.12,17 In spite of the advantages attributed to the balanced ternary combination between Mn, Co, and Ni, recent development has been directed towards enhancing the specific discharge capacity by increasing the Ni content in the transition metal oxide cathode.18,19 The major benefit of Ni-rich cathodes is the high discharge capacity that can be practically achieved (200–220 mA h g−1) with lower material cost.20–22 However, achieving high capacity from Ni-rich cathodes requires high operating voltage (4.4–4.6 V), which in turn results in a critical capacity degradation stemming from the electrolyte decomposition and/or surface modifications of the electrode.23–26 A large amount of Li+ (de)intercalation from the electrode at higher voltage exerts strong influence on the layered structure of the transition metal oxide.27 Furthermore, the occurrence of unstable Ni4+ during Li+ deintercalation leads to the reduction of LiMO2, where M = Ni1−x−yCoxMny (1 − x − y ≥ 0.5), to the inactive NiO phase, thereby reducing the battery cycle life.18,24,27 The Ni4+ would also be reduced to Ni3+ at elevated temperature followed by oxygen release, causing structural instability in the electrode.19,28 A previous study by Cherkashinin et al. also reported the reduction of Ni3+ to Ni2+ with oxygen loss in a Ni-based layered cathode material.29 The aforementioned processes together result in inherent limitations of Ni-rich layered cathode batteries, namely, the surface reconstruction from a layered structure into a spinel/rock-salt structure is a major degradation mechanism during charge–discharge, leading to poor coulombic efficiency, mechanical strain, and capacity decline.18,23,27,30,31 Jung et al. investigated the fading mechanism of the LiNi0.5Co0.2Mn0.3O2 cathode material, and reported the occurrence of the cubic NiO phase on the surface of the electrode.23 The formation of the reconstructed surface by NiO is likely to be different at different cut-off voltage levels. While the NiO cubic phase appears sporadically in low voltage cycling (3.0–4.5 V), the NiO cubic phase occurs uniformly on the active material surface at high voltage (3.0–4.8 V).23 It has also been studied that the cell cycled at high voltage (2.0–4.7 V) is more likely to undergo transition metal reduction than the cell cycled at low voltage (2.0–4.3 V), because of the lattice disruption during charge–discharge.31 While some research has been conducted on the understanding of the surface reconstruction for Ni-rich cathodes, surprisingly, there has been considerably less discussion about the formation of a solid-electrolyte interphase (SEI). The electrolyte interactions with the electrode surface result in the formation of the SEI layer, which is strongly associated with the charge–transfer reaction between the electrolyte and the electrode bulk.32 It is likely that the condition of the modified surface at voltage levels determines the nature of the SEI layer.33
The present study reports a detailed examination of the voltage-dependent properties of the SEI layer formed on a Ni-rich transition metal oxide cathode (LiNi0.5Co0.2Mn0.3O2). The electrode–electrolyte interphase has been visualized on the basis of secondary-ion mass spectroscopy (SIMS) measurement to correctly understand the nature of the SEI at different cut-off voltages. Particular attention has been devoted to the investigation on the capacity fading behaviour with the SEI formation. The state of Li+ trapping in the interphase layer is likely to be one key factor that determines the cell performance, but the local-atomic environment of the transition metals (Ni, Mn, and Co) has stronger influence on the cell stability. It has been believed that the transition metal dissolution is not related to the degradation of the Ni-rich cathode at high voltage.34 However, our study reveals the transition metal speciation into various oxidation and spin states at higher cut-off voltages. The precipitation of the modified transition metals as SEI species would be suggested as the dominant process of the capacity fading and voltage decay mechanism.
2. Experimental
Electrode preparation
The Li(Ni0.5Mn0.3Co0.2)O2 electrode (labelled as NMC5) was prepared by blending the Li(Ni0.5Mn0.3Co0.2)O2 active material (MTI, Japan) with conductive carbon (Super C65) and the polyvinylidene fluoride (PVDF) binder in a N-methyl-2-pyrrolidone (NMP, Sigma Aldrich) solution. The weight ratio of the slurry was 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 (=active material:carbon black:PVDF). After overnight mixing, the Ni-rich electrode slurry was introduced onto an aluminium current collector to make the NMC5 electrode with a doctor blade. This coated film was dried with an integrated dryer inside a coater (MTI, Japan) at 60 °C overnight. Following that, it was placed into a vacuum oven and dried at 120 °C for 12 h. The casted NMC5 on the Al-current collector was roll-pressed with a thickness of 10 μm.
:10:10 (=active material:carbon black:PVDF). After overnight mixing, the Ni-rich electrode slurry was introduced onto an aluminium current collector to make the NMC5 electrode with a doctor blade. This coated film was dried with an integrated dryer inside a coater (MTI, Japan) at 60 °C overnight. Following that, it was placed into a vacuum oven and dried at 120 °C for 12 h. The casted NMC5 on the Al-current collector was roll-pressed with a thickness of 10 μm.
Cell assembling and electrochemical measurement
The cell assembly was conducted in an Ar-glove box (H2O < 1 ppm and O2 < 1 ppm). The electrodes were cut into discs with a diameter of 16 mm, and they were assembled with Li metal (anode), a polypropylene based membrane as a separator (CELGARD Inc.), a stainless-steel spacer and a steel spring, in order to make CR2032 coin cells. A hydraulic crimping machine (MTI, MSK-110) was utilized to seal the cell parts together. 1 M lithium hexafluorophosphate (LiPF6) in ethylcarbonate (EC), diethylcarbonate (DC), and dimethylcarbonate (DMC) was used as the electrolyte (EC:DC:DMC = 1:1:1 in volume). The pristine material is labelled as REF, indicating that no current was applied. The assembled coin cells were charged and discharged with a current rate of 0.4C in the voltage windows of 2.0–4.2 V (NMC5 4.2 V), 2.0–4.5 V (NMC5 4.5 V), and 2.0–4.8 V (NMC5 4.8 V) at room temperature with 100 cycles. A 8-channel battery tester (NEWARE, Battery Test Station) was used to conduct constant-current cycling for the coin cells. 1 C-rate was calculated to be 160 mA h g−1. The loading of the active materials was 6.60 mg cm−2. Electrochemical impedance spectroscopy (EIS, Autolab PGSTAT204 with an FRA32 module) measurement was performed at the initial charge state and the cycled discharge state of the coin cells in the frequency range of 0.1 Hz to 100 kHz with an AC amplitude of 5 mV.
Material characterization
The cycled coin cells at the different cut-off voltages were disassembled inside an Ar-glove box. The electrodes were cleaned with DMC and dried overnight prior to the material characterization. Magnetic measurement on the electrode material was conducted using a physical property measurement system (Quantum Design PPMS Dynacool). The reciprocal molar magnetization was examined with a 1000 Oe magnetic field in the temperature range between 5 K and 300 K. The magnetic susceptibility of the electrode was examined under a magnetic field of 30 kOe at 5 K. Secondary-ion mass spectrometry (SIMS) visualization was performed on the basis of a positive ion detection mode (PID) of a SIMS (Kratos Axis ULTRA) equipped with a quadrupole mass spectrometer (HAL 7, Hiden Analytical) and ion sputter gun (IG20, Hiden Analytical). The SIMS system employs quadrupole mass spectrometry (QMS). A more detailed description of the QMS detection system can be found in ref. 35. A 1 keV Ar+ primary beam with a 50 nA current and a diameter of 100 μm was employed for the SIMS measurements with sputtering. The Ar+ bombardment time of the SIMS visualization was 600 s for each electrode. The beam angle was 45° with regards to the sample surface. The chamber pressure was 10−9 mbar, and it increased to 10−6 mbar during SIMS operation. The morphology of the electrode at different cut-off voltages was compared using scanning electron microscopy (SEM, FEI Quanta 250 FEG). The powder diffraction pattern was measured by using a Bruker D8 X-ray diffractometer (X-ray source: Cu Kα radiation = 8 keV) by step scanning with 0.02° increments in the range of 2θ = 10–80°. Rietveld refinement was carried out based on the XRD spectra using the MAUD application for the phase analysis. X-ray photoelectron spectroscopy (XPS, AXIS Ultra HSA KRATOS) measurement was performed to investigate the surface chemistry of the electrode (X-ray source: Al Kα = 1486.6 eV). The measurement conditions were 15 kV with an applied power of 150 W under ultra-high vacuum (10−9 torr). The background treatment for the XPS spectra was performed with a Shirley-type function. Data fitting was carried out with a Gaussian–Lorentzian function using CasaXPS software. To evaluate the work function changes of the electrodes, ultraviolet photoelectron spectroscopy (UPS) measurements were also conducted under an ultra-high vacuum environment (10−9 mbar) with an emission source of helium gas (He I: 21.22 eV). The calibration of the binding energy (eV) for UPS was conducted by measuring the Fermi edge (EF = 0 eV) on an Au surface. The energy resolution of USP is estimated to be 0.14 eV. The applied sample bias voltage was −9 V. The electrodes from the coin cells were extracted after the electrochemical characterization. To summarize the sequence, the morphology of the electrodes was firstly observed by SEM, and then XRD was conducted. Followed by the electrode splitting, magnetic measurements and XPS/SIMS were carried out on each sample. While a piece was taken from the electrode for the XPS measurement (e.g. NMC5 4.2 V-XPS), another piece was also taken from an identical electrode (e.g. NMC5 4.2 V-SIMS) for the SIMS measurement, independently.
3. Results and discussion
Fig. 1 compares the electrochemical properties of Li(Ni0.5Mn0.3Co0.2)O2 (NMC5) cathodes at different cut-off voltages, i.e., 2.0–4.2 V, 2.0–4.5 V, and 2.0–4.8 V. All samples were cycled up to 100 times with a constant current of 0.4 C-rate. Both NMC5 4.5 V and NMC5 4.8 V have delivered a very high specific capacity of 225 mA g h−1 during the initial cycle. On the other hand, NMC5 4.2 V revealed a charge capacity of 168 mA g h−1, as shown in Fig. 1a. The achievement of the larger capacity at the high voltage levels can be attributed to the location of the O:2p band and the Ni3+/4+:eg band in the energy vs. density of states diagram, with a high Madelung energy that makes higher oxidation states accessible.5,36 At lower voltage levels (<4.3 V), Ni2+/3+:eg is likely to be the major contributor to the charge compensation mechanism. Upon further charging, electrons can be extracted from both Co3+/4+:t2g and Ni3+/4+:eg bands, providing higher charge capacity. However, a deeper Li+ (de)intercalation with excessive voltages can lead to a significant loss of electrons from the O2−:2p band, resulting in irreversible oxygen removal and causing rapid capacity fading. This inherent limitation originated from the electrode design is one of the key mechanisms that lead to the dramatic capacity fading of the NMC5 4.8 V cell. In spite of the lowest charge capacity during 50 cycles, the NMC5 cell charged at 2.0–4.2 V showed the best battery capacity after 100 cycles (Fig. 1a). The level of the cut-off voltage is a vital factor that affects the capacity fading behavior, as well as the voltage decay process. It appears that an over-voltage cycling can cause a higher voltage drop, as can be seen in Fig. 1b–d. The differential capacity (dQ/dV) plots for each cut-off voltage are presented in Fig. 1e–g. A major peak was observed upon both charge and discharge in the voltage range between 3.7 V and 4.6 V. The peak is mainly associated with the redox reaction between Ni2+/Ni3+ and Ni3+/Ni4+. Particular interest has been devoted to the phase transition at different voltage levels. It is apparent from the dQ/dV plot that higher cut-off voltages result in dynamic phase transitions, as evidenced from the spectral shifts. Especially, the reduction peak at around 3.6–3.7 V is likely to be associated with Mn4+ in the layered LiTmO2 (Tm = Ni, Mn, Co), and therefore the spectral shift of the peak indicates the phase transition from a layered structure to a spinel structure.37–39
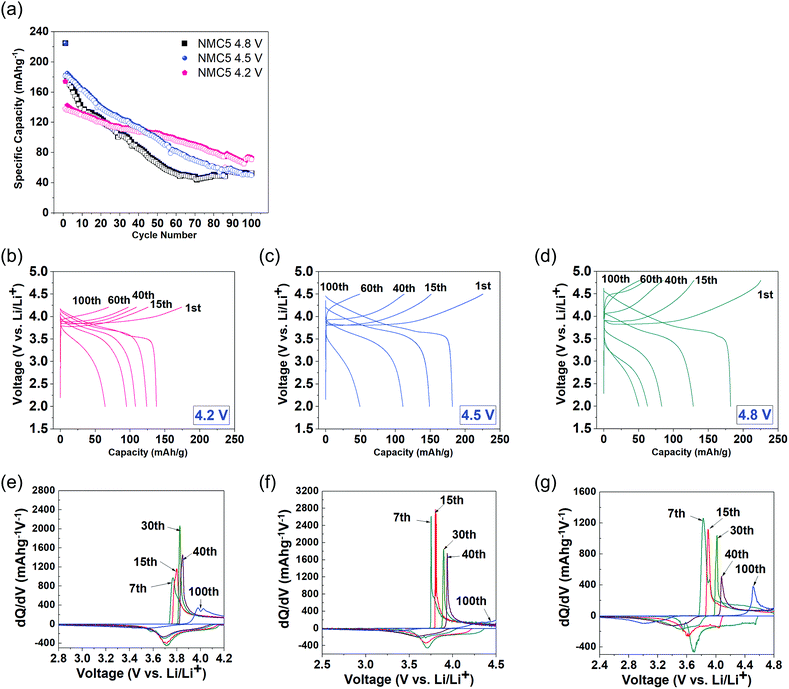 |
| | Fig. 1 Electrochemical properties of Li(Ni0.5Mn0.3Co0.2)O2 (NMC5) cells at different cut-off voltage levels: (a) comparison of charge and discharge capacities for 100 cycles and the galvanostatic profiles at (b) 4.2 V cut-off voltage, (c) 4.5 V cut-off voltage, and (d) 4.8 V cut-off voltage with 0.4 C-rate current applied. The corresponding dQ/dV plots of (e) 4.2 V, (f) 4.5 V, and (g) 4.8 V cut-off voltages for the 7th, 15th, 30th, 40th, and 100th cycles. | |
The distribution of some key elements that constitute the SEI layer was visualized at different cut-off voltages, based on a log scale of the SIMS depth profiling (Fig. 2a). The Li (or Li compounds) showed the dominant signal in the electrode or SEI, as can be seen in Fig. 2a and b. A major advantage of the present elemental visualization is the detailed information on the local-atomic environment of the electrode interphase. As shown in Fig. 2a, the cell charged at higher voltages displayed diverse elemental bands with higher concentrations. This suggests the increment in the number of the SEI species at higher cut-off voltages. The chemical reactions of lithium with the electrolyte can result in the formation of Li2O, Li2CO3, and LiF (with LiPF6 electrolyte) at the solid-electrolyte interphase, after continuous charging and discharging.33,40 On the other hand, alkyl carbonates (e.g. ROCO2Li) were reported as the major products at the electrolyte side.41 The distinct changes in the atomic mass unit between 50.0 and 60.0 amu at different voltages are likely to be attributed to the surface reconstruction (NiO, MnOn, and CoOn) and/or the accumulation of the dissolved transition metal ions onto the electrode (e.g. NiF2, NiF3, MnF2, LiF2, and CoF3).33,42–44 Also, the dissolution of the C5OF bond between the carbon black and PVDF binder possibly contributes to the formation of dissimilar SEI environments at different voltage levels. The components at approximately 47.0 amu and 52.0 amu can be assigned to the decomposed products from the electrolyte, i.e., P–O and OF2, respectively. This will be further discussed in a later section. It is likely that the precipitation of metallic species determines the properties of the SEI layer, thereby leading to a dissimilar electrochemical behaviour upon cycling. The trapped Li+ is another key aspect that is strongly associated with the properties of the SEI layer (Fig. 2b). The major process of the Li+ trapping can be explained by the following electron reduction schemes:45
| | | 2(CH2O)2CO (EC) + 2e− + 2Li+ → (CH2OCO2Li)2 + C2H4 | (1) |
| | | (CH3CH2)2OCO2 (DEC) + e− + Li+ → CH3CH2OCO2Li + CH3CH2 | (2) |
| | | (CH2O)2CO + 2e− + 2Li+ → Li2CO3 + C2H4 | (3) |
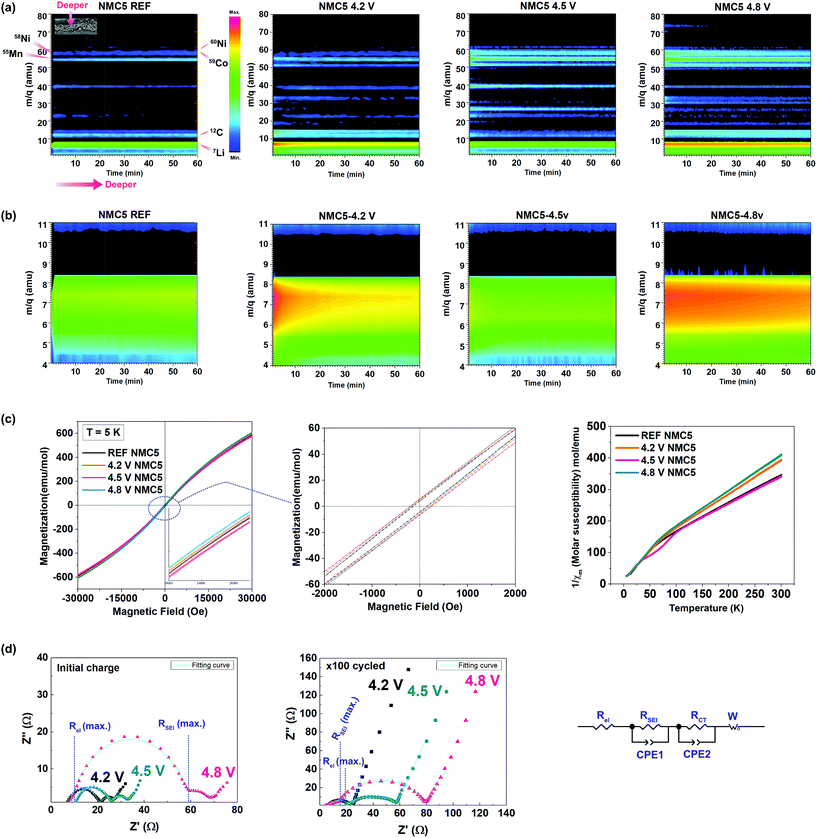 |
| | Fig. 2 Elemental visualization of the key SEI species based on the SIMS depth profiles of lithium, carbon, oxygen, manganese, nickel, and cobalt of Li(Ni0.5Mn0.3Co0.2)O2 at (a) the reference state where no current was applied (REF), and after 2.0–4.2 V cycling, 2.0–4.5 V cycling, and 2.0–4.8 V cycling with a current rate of 0.4 C-rate 100 times. (b) The elemental visualization of the trapped Li+ for each electrode. (c) Hysteresis loop of magnetization (at 5 K) of the Li(Ni0.5Mn0.3Co0.2)O2 cathodes cycled at 2.0–4.2 V (4.2 V NMC5), 2.0–4.5 V (4.5 V NMC5), and 2.0–4.8 V (4.8 V NMC5), and their reciprocal magnetic susceptibility between 5 K and 300 K under a magnetic field of 1000 Oe. (d) EIS measurements. Nyquist plots (with its equivalent circuit) and the fitting curves for the electrode after the initial cycle and after 100 cycles for NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V cells. | |
Li+ trapping was identified at the electrodes cycled in the voltage ranges of 2.0–4.2 V and 2.0–4.8 V. It is somewhat surprising that there had been no trapped Li+ in the electrode cycled to 4.5 V. To understand this interesting observation, the cycled cathodes were also characterized by magnetization measurement, EIS, and XRD with Rietveld refinement. The magnetic properties of the NMC5 cathodes are shown in Fig. 2c. The local-atomic states for each transition metal element and their spin states could be evaluated from the magnetization (5 K) and reciprocal magnetic susceptibility measurements. There were no saturations in the magnetization curves as presented in Fig. 2c, indicating antiferromagnetic interactions for all samples. On the other hand, the NMC5 cathodes showed a paramagnetic behavior between 150 K and 300 K (Fig. 2c). It is therefore possible to apply the Curie–Weiss law of Xm = C (T − θ), where C is the Curie constant (C = Nμ2/3kB, N: Avogadro number, μ: effective moment and kB: Boltzmann constant) and θ is the Weiss constant, for the linear fitting. The fitting results are exhibited in Table S1.† The Curie–Weiss temperature was −95.43 K, −66.42 K, −95.88 K and −63.46 K for the reference, NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V cathodes, respectively. The dominant antiferromagnetic interaction in the cathode samples can be further supported by the negative value of the Curie–Weiss temperatures. The antiferromagnetic interaction is likely to be attributed to the interactions between Ni2+ in the Li-layer and in the transition metal layer of the Li(Ni0.5Mn0.3Co0.2)O2 electrode.46,47 A small hysteresis loop was found in the magnetization curve (Fig. 2c). The remnant magnetization was 4.67 emu mol−1 for the reference NMC5 cathode. It increased to 5.66 emu mol−1, 5.92 emu mol−1, and 6.53 emu mol−1 for NMC5 4.8 V, NMC5 4.5 V, and NMC5 4.3 V, respectively, after the electrochemical cycles. The transition metal migration into the Li-layer appears to be slightly more active for the NMC5 cathode cycled at a lower voltage level, as evidenced by the higher remnant magnetization.48,49 The molar magnetization was relatively high for NMC5 4.8 V, and this can be explained by the precipitation of the transition metal based SEI species on the electrode surface. The effective magnetic moments were measured to be 3.01 μB, 2.72 μB, 3.03 μB, and 2.65 μB for NMC5 reference, NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V samples, respectively (Table S1†). The measurements of the effective magnetic moments enable us to identify transition metals with particular oxidation and spin states. Mn4+, Co3+, and Ni2+ are the major elements that constitute the cathode composite, but we could also find Mn3+ and Ni3+ in the pristine state of the Li(Ni0.5Mn0.3Co0.2)O2 composite. While the coexistence of Mn4+ and Mn3+ has been widely studied, there is relatively less information about the presence of Ni3+ in the pristine material. A couple of recent studies have reported the mixed oxidation state between Ni2+ and Ni3+ in the layered transition metal oxide cathode.24,50 The information about the oxidation states obtained from the magnetic measurements (Table S1†) is in good agreement with previous studies, and it is further supported by the XPS investigation (Fig. 4). One of the more crucial findings of the present study is the identification of Ni3+ with different spin states in relation to the cut-off voltage levels. While high-spin Ni3+ was observed in the pristine Li(Ni0.5Mn0.3Co0.2)O2 (REF) and the NMC5 4.5 V cathodes, low-spin Ni3+ was detected in the NMC5 4.2 V and NMC5 4.8 V cathodes (Table S1†). In addition, we could also observe similarities in the local atomic environment between these sample groups. The molar ratio and the spin states of the transition metals of the Li(Ni0.5Mn0.3Co0.2)O2 (NMC5) cathode cycled at 2.0–4.5 V are comparable to those of pristine NMC5. On the other hand, the atomic environment of the Li(Ni0.5Mn0.3Co0.2)O2 (NMC5) cathode cycled at 2.0–4.2 V is similar to that of the NMC5 cathode cycled in the voltage range of 2.0–4.8 V. Electrochemical impedance spectroscopy (EIS) was used to compare the resistances resulting from the formation of the SEI layer (RSEI) and the charge transfer between the electrode and electrolyte (RCT), at different cut-off voltage levels (Fig. 2d). Also, the lithium-ion diffusivity could be deduced from the Warburg impedance of the EIS measurements. The EIS fitting results are summarized in Table S3.† After the initial charge, there has been a large SEI impedance (RSEI = 52 Ω) for the cathode cycled at 2.0–4.8 V. The SEI resistances of NMC5 4.2 V and NMC5 4.5 V were notably smaller than that of the NMC5 4.8 V sample. After 100 cycles, NMC5 4.8 V also exhibited the highest SEI impedance, but the impedance from the electrolyte (Rel = 19.39 Ω) steeply increased for the cathode cycled to 4.5 V cut-off voltage. On the other hand, the charge transfer resistance was 16.08 Ω, 34.33 Ω, and 60.68 Ω for NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V, respectively. These discrepancies could be attributed to the dissimilar surface conditions of the SEI layer at the cut-off voltages, which in turn invokes the formation of different organic and metallic mixtures at the interphase. For instance, the alkyl carbonate compounds (e.g. ROCO2Li) are likely to remain in the electrolyte, when the cell is cycled in the voltage range of 2.0–4.5 V, and thus cause a higher resistance in the electrolyte. The lithium diffusion coefficient (DLi) was evaluated to be 5.78 × 10−10 cm2 s−1, 1.41 × 10−11 cm2 s−1, 9.44 × 10−11 cm2 s−1, and 3.56 × 10−11 cm2 s−1 for NMC-REF, NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V, respectively. Among the cycled samples, NMC5 with 4.5 V cut-off voltage revealed the best lithium-ion diffusivity, which agrees with the lithium-ion depth profile visualized in Fig. 2b. The powder diffraction peaks obtained from the XRD measurements and the SEM observations are presented in Fig. 3. The morphology of the SEI layer with the precipitation of the decomposed products varied at the different cut-off voltages, as exhibited in Fig. 3d, e, g, h, j and k. The thickness of the surface layer, which includes the active material, carbon black, and PVDF binder, was relatively thin, when the cathode was cycled at 2.0–4.5 V (Fig. 3h) due to the dissolution of the electrode in the electrolyte including the SEI layer formed on top. To understand the crystallography phase transition of the electrode, which is likely to be linked to the dissolution of the transition metals, Rietveld refinement was carried out on the basis of the powder diffractions (Fig. 3f, i and l). Although our XRD system employed is considered to be optimum for thin-film type samples, SEI layers of nanometer scales would not be detectable with X-ray diffractometers. However, as the powder diffraction provides an averaged structure, some information on the surface phase should remain in the diffraction pattern. The occurrence of the spinel (like) phase on the surface is a well-known degradation process with SEI formation, particularly for Ni-rich transition metal oxide cathodes.18,23 While the bulk retains the layered structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), the surface of Ni-rich cathodes transforms into a spinel phase and further into a cubic phase forming NiO upon charge–discharge.18,23 Therefore, it is likely that the modifications in the surface phase of the Ni-rich electrode can notably contribute to the occurrence of the cubic spinel (Fdm) in the crystallography information fitted by Rietveld refinement. The phase transition could be further supported by the magnetism study conducted at the different cut-off voltages. The fitting result is presented in Table S2.† The phase transition from a trigonal structure (space group: Rm) to a cubic structure (space group: Fdm) has been detected when the cells are cycled with the cut-off voltages of 4.2 V and 4.8
V. Especially, the NMC5 4.8 V cathode showed a significant phase transition during charge–discharge. By contrast, there has been no spinel phase in the NMC5 4.5 cathode, but we can see notable modifications in the lattice parameters of the trigonal structure (see Table S2†).
m), the surface of Ni-rich cathodes transforms into a spinel phase and further into a cubic phase forming NiO upon charge–discharge.18,23 Therefore, it is likely that the modifications in the surface phase of the Ni-rich electrode can notably contribute to the occurrence of the cubic spinel (Fdm) in the crystallography information fitted by Rietveld refinement. The phase transition could be further supported by the magnetism study conducted at the different cut-off voltages. The fitting result is presented in Table S2.† The phase transition from a trigonal structure (space group: Rm) to a cubic structure (space group: Fdm) has been detected when the cells are cycled with the cut-off voltages of 4.2 V and 4.8
V. Especially, the NMC5 4.8 V cathode showed a significant phase transition during charge–discharge. By contrast, there has been no spinel phase in the NMC5 4.5 cathode, but we can see notable modifications in the lattice parameters of the trigonal structure (see Table S2†).
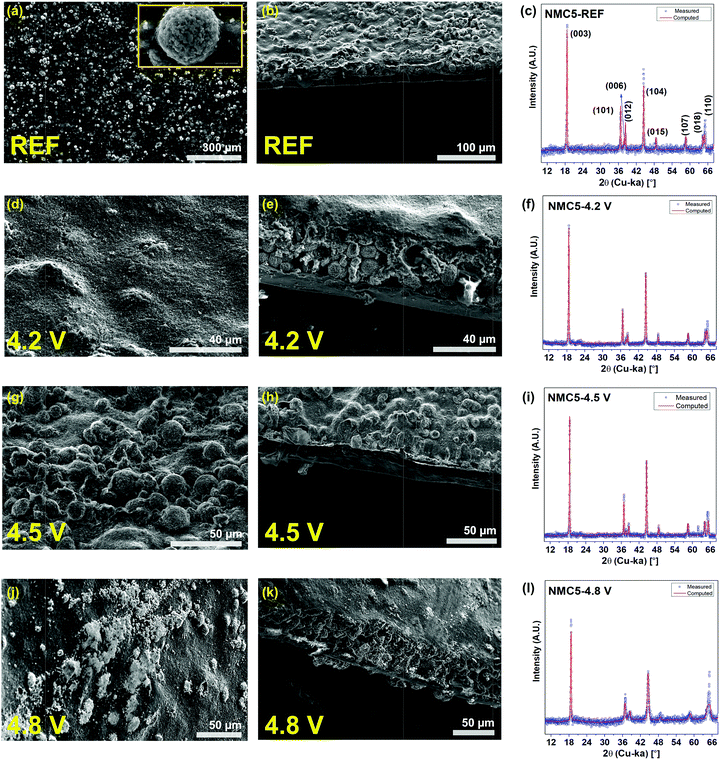 |
| | Fig. 3 SEM images and XRD analyses. Powder diffraction is fitted with the red curve (computed) for Rietveld refinement. (a) Top view SEM of NMC5 REF. (b) Cross sectional view SEM of NMC5 REF. (c) Powder diffraction of NMC5 REF. (d) Top view SEM of NMC5 4.2 V. (e) Cross sectional view SEM of NMC5 4.2 V. (f) Powder diffraction of NMC5 4.2 V. (g) Top view SEM of NMC5 4.5 V. (h) Cross sectional view SEM of NMC5 4.5 V. (i) Powder diffraction of NMC5 4.5 V. (j) Top view SEM of NMC5 4.8 V. (k) Cross sectional view SEM of NMC5 4.8 V. (l) Powder diffraction of NMC5 4.8 V. The cells have been cycled 100 times at 2.0–4.2 V (NMC5 4.2 V), 2.0–4.5 V (NMC5 4.5 V), and 2.0–4.8 V (NMC5 4.8 V) with a current rate of 0.4C. | |
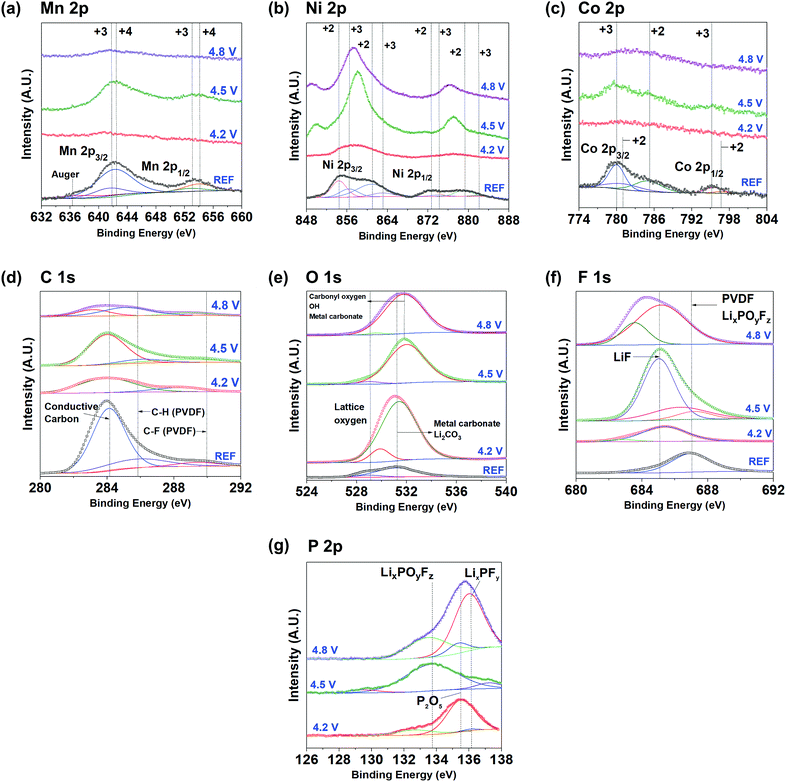 |
| | Fig. 4
Ex situ XPS spectra of NMC5 REF, NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V electrodes after 100 cycles with a constant current of 0.4C. (a) Mn 2p, (b) Ni 2p, (c) Co 2p, (d) C 1s, (e) O 1s, (f) F 1s, and (g) P 2p. | |
Ex situ XPS measurements were conducted to examine the surface chemistry of the electrode materials at different voltage levels. The XPS spectra of Mn 2p, Ni 2p, Co 2p, C 1s, O 1s, F 1s, and P 2p for the reference and the cycled cathodes are shown in Fig. 4. The binding energy of Mn (2p3/2) and Mn (2p1/2) was measured to be 642.5 eV and 654.1 eV, respectively, at the pristine state of Li(Ni0.5Mn0.3Co0.2)O2 (REF). Mn4+ is likely to be dominant in the Mn 2p spectrum, but there have also been small peaks at around 641.6 eV and 653.0 eV, signifying the presence of some Mn3+ in the pristine cathode.51 The occurrence of Mn3+ could be explained by the electron transfer between Mn4+ and Ni2+, as well as low-spin Mn3+ in the pristine material.52–54 The appearance of low-spin Mn3+ was confirmed by the magnetic measurements (Fig. 2c and S1†). However, the intensity of these peaks dramatically decreased, after the electrochemical cycles in the voltage range between 2.0 V and 4.2 V (Fig. 4a). By contrast, the XPS spectrum of the NMC5 cathode cycled at 2.0–4.5 V was very similar to that of the pristine sample (REF). This Mn 2p spectrum disappeared again, when it was charged to 4.8 V. The trend of the Mn 2p XPS at different cut-off voltages is comparable to that of Co 2p, as presented in Fig. 4c. The two major Co (2p3/2) and Co (2p1/2) peaks were observed at 780.0 eV and 795.1 eV, respectively, indicating the presence of Co3+ in the pristine material (REF). The broad satellite peak at 785.2 eV indicates the presence of a small amount of Co2+ with Co3+ in the pristine material.55,56 Similar to Mn 2p, there has been a sharp decrease in the Co 2p XPS peaks, after the electrochemical cycles up to 4.2 V. The major peaks reoccurred after charge–discharge in the voltage range of 2.0–4.5 V, as can be seen from Fig. 4c. These peaks disappeared again, when the cell was cycled between 2.0 and 4.8 V. The surface conditions varied at the cut-off voltages tend to determine the nature of the electrode interphase. The SEI layer formed at a lower cut-off voltage level possibly contains a higher amount of Li2O, Li2CO3, and/or carbonate materials. On the other hand, the SEI layer grown at the higher voltage levels includes a higher amount of transition metal species or by-products (e.g. MnF2, MnF3, NiF3, NiO, and MnO). This observation can be discussed by using the energy vs. density of states diagram, which can be seen in a later section (Fig. 7). A higher cut-off voltage corresponds to a lower Fermi level of the cathode, which makes the higher oxidation state of the transition metals accessible. In addition, the loss of Li+ through electrolyte decomposition and SEI trapping results in the decline of the cathode Fermi level, leading to a higher degree of the transition metal (3d)–O(2p) hybridization. Hence, during the subsequent Li+ deintercalation, electrons will be extracted from both the transition metal and the oxygen ion 2p orbital. This will be discussed in detail in a later section. As displayed in Fig. 2a, the accumulation of the transition metals was distinct when the cells had been cycled at 2.0–4.5 V and 2.0–4.8 V. However, the properties of the transition metals as SEI species are likely to be different at the cut-off voltages. It appears that the precipitation properties of the transition metals at 2.0–4.5 V are similar to those of the pristine state. However, the transition metal species precipitated at 2.0–4.8 V was notably different from the pristine electrode (NMC5 REF). The dynamic redox reactions between the electrode and the electrolyte (LiPF6 in EC/DC/DMC) at 2.0–4.8 V is likely to be the major cause of the dramatic changes in the local atomic environment of the transition metals. It is likely that the by-products of the decomposed active material accumulate onto the electrode surface, when it was cycled in the higher voltage range. The magnetic measurement also indicates the accumulation of some phase transformed cathode particles on the electrode surface, revealing higher concentration of low-spin Mn3+ and Ni3+ in the 2.0–4.8 V cycle (Fig. S1†). On the other hand, the trend in the spectral shifts for Ni 2p XPS was slightly different as compared with that of Mn and Co. For the pristine sample (REF), the Ni (2p3/2) and Ni (2p1/2) peaks were found at 854.4 eV and 872.6 eV, respectively, followed by shake-up peaks at 861.0 eV and 879.3 eV, as shown in Fig. 4b. This observation indicates that Ni mainly exists as the oxidation state of +2 at REF. Also, two minor peaks were detected at 856.4 eV and 874.2 eV, and these peaks can be assigned to Ni3+. The coexistence of Ni2+ and Ni3+ in the pristine composite would also be explained by the electron transfer between Mn4+ and Ni2+. When the cathode was cycled to 4.2 V, the Ni 2p peaks vanished. This trend is similar to that of Mn and Co. However, in 2.0–4.5 V cycling, the Ni 2p XPS spectra underwent a significant change, showing a sharp peak at approximately 857.9 eV, and this peak shifted towards 857.3 eV, when the cell was cycled in the voltage range of 2.0–4.8 V (Fig. 4b). The appearance of the sharp peak could be attributed to the surface reconstruction of the active material particles with NiO.57 The surface reconstruction from a layered phase into a spinel/rock-salt phase is a commonly observed process in layered Ni-rich cathodes, during a charge–discharge at high voltages.19,34,58 However, there is a slight difference in terms of the surface reconstruction process by NiO at the cut-off voltage levels. At a lower voltage, the surface of the active material particles is partially reconstructed by small NiO clusters, but they become fully covered with NiO in a higher voltage cycling.34 This intense peak at ∼858 eV can be also attributed to the F(KLL) transition, which is likely to be originated from decomposed products (e.g. NiF2: 857.8 eV) with the electrolyte species.43,59 On the other hand, there has been a small peak at a lower energy level (∼850 eV) of Ni 2p, which only occurs for the electrodes cycled at the higher cut-off voltages. This peak can be ascribed to the Auger peak from fluorine (F), and it is in accordance with the major peaks of F 1s for NMC5 4.5 V and NMC5 4.8 V.60 The C 1s spectrum of the pristine cathode (REF) mainly consists of the conductive carbon (284.3 eV) and the PVDF binder components (Fig. 4d). The voltage dependent spectral changes of C 1s were also similar to that of Mn and Co. For O 1s, the lattice oxygen and the metal carbonate and/or the Li2CO3 peaks were identified at 529.1 eV and 531.2 eV, respectively, for the pristine cathode (REF). There has been a marked increase in the Li2CO3 peak, when the cell was cycled between 2.0 V and 4.2 V, as can be seen from Fig. 4e. Li2CO3 is known as a major SEI species formed near the electrode substrate, if a carbonate based electrolyte was used.61–63 This peak shifted towards 531.8 eV, which can be assigned to oxygen ions with low coordination, after 2.0–4.5 V cycling.64 The low coordination oxygen ion is possibly associated with NiO resulting from the surface reconstruction. The peak tends to become broader, when the cell was cycled in the voltage range of 2.0–4.8 V, indicating a higher amount of metal carbonate in the SEI layer.65,66 For F 1s, the spectrum for the pristine material (REF) was fitted with a peak that can be assigned to the PVDF binder at approximately 687.0 eV (Fig. 4f).40,45,67 This peak is likely to overlap with that of the LixPOyFz composite, which is the salt reduction product from the electrolyte.68 After the electrochemical cycles, a LiF peak occurred at around 685.0 eV and it dramatically rose when the cell was cycled between 2.0 V and 4.5 V. This peak became broad in a higher voltage cycling up to 4.8 V. The appearance of the LiF peak in 2.0–4.2 V cycling and its dramatic increment at 2.0–4.5 V can be related to the decomposition of the electrolyte as follows:
| | | PF5 + H2O → POF3 + 2HF | (5) |
| | | Li2CO3 + 2HF → 2LiF + H2O + CO2 | (6) |
The broadening of the F 1s XPS peak at the higher cut-off voltage (2.0–4.8 V) can be attributed to the formation of the metal fluorides, i.e., NiF2, CoF2 and MnF2.43,69 The formation of HF is the dominant factor that leads to the dissolution of the transition metals during charge and discharge.45,68 The XPS spectra of P 2p also revealed a voltage dependent spectrum change, as presented in Fig. 4g. While the P2O5 (∼135.5 eV) and LixPFy (∼136.2 eV) peaks were dominant in 2.0–4.5 V and 2.0–4.8 V electrochemical cycles, the peak of LixPOyFz was intense when the cell was charged in the voltage range of 2.0–4.5 V (Fig. 4g). A number of studies have confirmed that LiF, LixPFy, and LixPOyF are the major lithiated species decomposed from the electrolyte, during charge–discharge.40,68 The present findings suggest the different nature of the surface environment, which leads to the dissimilar formation of SEI layers at cut-off voltage levels. To correctly understand the involvement of the SEI components in the capacity fading mechanism, the chemical properties of the SEI layer were examined by mass spectroscopy with sputtering (Fig. 5). There has been a gradual increase of the peak intensity at approximately 47 amu, which can be assigned to the PO from the electrolyte decomposition (Fig. 5a). The peak at around 44 amu is likely to be assigned to the C2F component, which is related to the PVDF binder and/or a SEI species. One interesting observation is the occurrence of the LiF peak at approximately 27 amu, and the peak intensity was remarkable when the cell was cycled at 2.0–4.5 V. The marked increase in the peak intensity at around 44 amu seems to be related to the LiF compound. The superior cell performance of NMC5 4.5 V during 50 cycles (Fig. 1a) could be attributed to the higher concentration of LiF in the SEI layer, as can be seen in Fig. 5b. It has been studied that LiF can lower the interfacial resistance and extend the cell life.70 In addition, a previous study on the density functional theory (DFT) of the energy barrier for Li+ diffusion revealed that LiF can enhance the ionic diffusivity.71 Those findings are in a good agreement with our SIMS visualizations (Fig. 2a and b) and EIS results (Fig. 2d). The precipitation process of the transition metal species was also identified for all cycled samples at around 55–61 amu, as shown in Fig. 5c. The peak intensity associated with the transition metals was considerable for NMC5 4.5 V and NMC5 4.8 V, while the intensity was weak for the NMC5 4.2 V sample. There has also been a dissimilarity in the trend of the transition metal spectra between the NMC5 4.5 V and NMC5 4.8 V electrodes. While the NMC5 4.5 V cathode showed a consistency in the concentration with the depth after the initial sputtering, the transition metal concentration gradually increased with the depth for the NMC5 4.8 V cathode. Another crucial finding is the appearance of the OF2 peak at around 52 amu, which is originated from the electrolyte decomposition. Along with the transition metal peak, the OF2 peak intensity was also considerable for NMC5 4.5 V and NMC5 4.8 V cells. Fig. 5d presents the formation of 58NiO (74 amu) as a part of the active material surface reconstruction, which is known as an inherent problem for Ni-rich transition metal oxide cathodes. The distinct change of the Ni state at the high cut-off voltage was shown by the Ni 2p XPS measurement, and this is likely to be connected to the surface reconstruction with NiO, as evidenced by the mass spectroscopy analyses. It appears that the cell cycled at 2.0–4.8 V underwent a higher degree of the NiO surface reconstruction during charge–discharge, as compared to the NMC5 4.5 V cell.
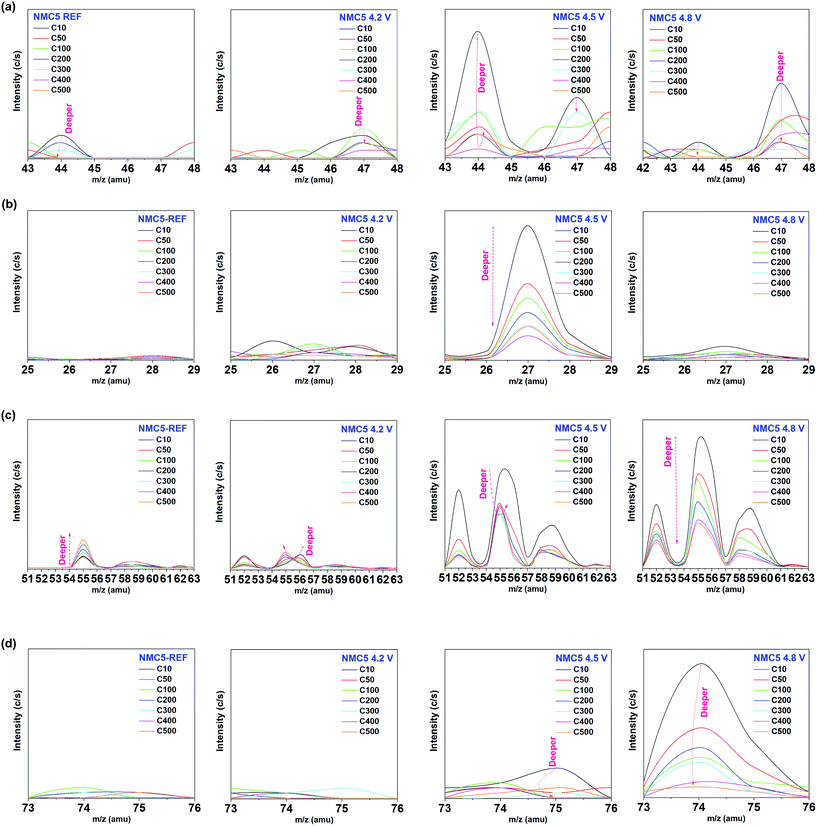 |
| | Fig. 5 Mass spectroscopy with Ar-sputtering. C10, C50, C100, C200, C300, C400, and C500 indicate sputtering numbers of 10, 50, 100, 200, 300, 400 and 500, respectively. The species at the atomic mass unit of (a) 43.00–48.00 amu, (b) 25.00–29.00 amu, (c) 51.00–63.00 amu, and (d) 73.00–76.00 amu have been examined for each NMC5 REF, NMC5 4.2 V, NMC5 4.5 V, and NMC5 4.8 V electrode. The cells have been cycled 100 times at 2.0–4.2 V (NMC5 4.2 V), 2.0–4.5 V (NMC5 4.5 V), and 2.0–4.8 V (NMC5 4.8 V) with a current rate of 0.4C. | |
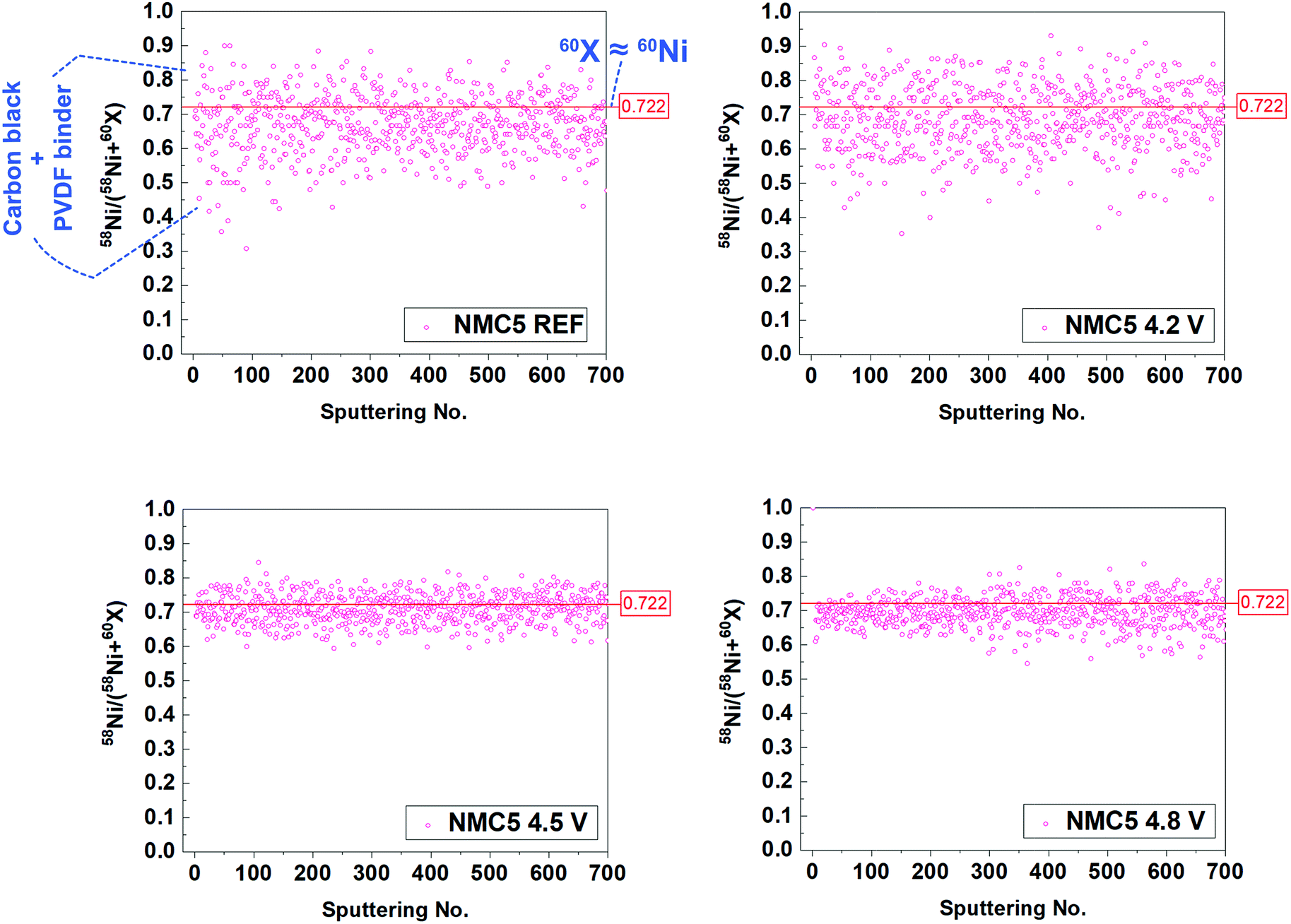
The SIMS visualization (Fig. 2a) and the XPS measurement (Fig. 4d) provide insights into the carbonic system of the electrode composite. As shown in Fig. 2a, we can observe the notable precipitation of the carbon (12C) when the cell was cycled at 2.0–4.2 V and 2.0–4.8 V, suggesting a higher amount of organic SEI species (e.g. Li2CO3 and ROCO2Li) on the surface. By contrast, the band of the carbon (12C) from NMC5 4.5 V was notably weak among the cycled cathodes. Another major effort has been made to understand the cell fading mechanism associated with the carbon additive. In particular, the C5OF bond between the carbon black and PVDF binder is likely to be associated with the electrode stability and electron conduction, thus determining the cell performance. A problem arises, however, when mass spectroscopy is chosen for the investigation into the carbon stability, since the total mass of 12C5 (60 amu) overlapped with that of 60Ni (60 amu). In order to distinguish the influence on the fading process by 12C5 from 60Ni at different cut-voltage levels, an approach with isotope concentration has been adopted, as presented in Fig. 6. The most abundant isotopes of nickel (Ni) under natural conditions are 58Ni and 60Ni.72 On the basis of the two nickel isotopes, the isotope concentration of 58Ni should correspond to approximately 0.722. Therefore, the data spots deviated from this ratio can be assigned to 12C5 from C5OF species (Fig. 6). There has been little impact on the material stability of the carbon additive and the PVDF binder, when the cell was cycled between 2.0 V and 4.2 V. With the increase of the cut-off voltages, however, 12C5 appears to be degraded with the formation of NiO during the electrochemical cycles.
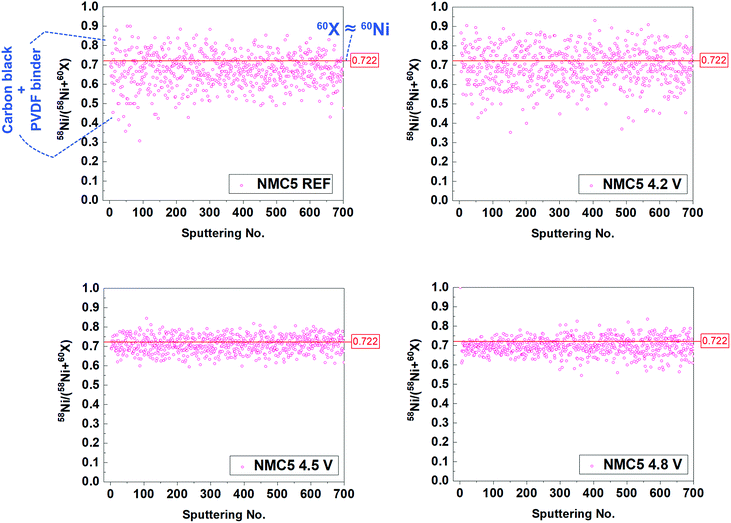 |
| | Fig. 6 Isotope concentration between 58 amu and 60 amu species. Total mass of 12C5 (60 amu) overlapped with 60Ni (60 amu), but only 58Ni/(58Ni + 60Ni) can correspond to 0.722 in nature. The data plot that is not in line with the ratio of 0.722 can be assigned to the 12C5 species. The cells have been cycled 100 times at 2.0–4.2 V (NMC5 4.2 V), 2.0–4.5 V (NMC5 4.5 V), and 2.0–4.8 V (NMC5 4.8 V) with a current rate of 0.4C. | |
Taken together, the fading mechanism associated with the SEI formation at different cut-off voltages can be summarised in Fig. 7. The present study reveals the discrepancy in the properties of the SEI at different cut-off voltages in a Ni-rich transition metal oxide cathode. The illustration in Fig. 7 shows the different surface environments with the information of the transition metal spin-states, crystal structures, visualized mass spectroscopy profiles, and the schematic of the energy vs. density of states diagrams. The work function (Wf) of the electrodes was measured by UPS (Fig. S1†) to approximate the trend of the Wf energy shift under different Li+ deintercalation conditions. A higher work function (Wf) indicates a lower Fermi energy (EF) relative to the vacuum level (Evac) of the transition metal oxide cathode. The schematics of the energy vs. density of states for the cycled electrodes are based on the charge state with each cut-off voltage. At the reference state (NMC5 REF), the electrode consists of the carbon black, PVDF binder, and active material with the trigonal crystal phase, which signifies a good Li-layered structure, showing the highest lithium diffusion coefficient (DLi). As for the spin-state, a small amount of low-spin (LS) Mn3+ and high-spin (HS) Ni3+ was identified, due to the charge transfer between Mn4+ and Ni2+ in the pristine cathode. When the cell was cycled in the voltage range of 2.0–4.2 V, Li+ trapping was observed in the SEI layer, which is connected to the shift of the energy diagram of the transition metals with higher oxidation states (or lower d-orbital) in the octahedron. The Li+ loss during the deintercalation process into the cathode tends to lower the energy band of the transition metals, which in turn results in the overlapping of the low d-orbital (e.g. t2g) of the transition metals (Ni, Mn, and Co) with the O2− (2p) band.8,33 At a fully discharged state, after multiple electrochemical cycles, the Fermi energy level (Ef) would be reduced as compared with that of the reference state. This reduced Ef is the new energy level of the initial state for the next subsequent cycles. During the charge, Ef would decline due to the extractions of the electrons from the cathode composite. At a lower cut-off voltage, such as 4.2 V, the electron extraction is likely to be associated with the lower oxidation state of the transition metals or high d-orbital (e.g. eg), and thus has no impact on the O2− (2p) band, although the transition metal bands were overlapped with the O2− (2p) band. The Ni3+ existed as a low-spin at the reference state, but it changed into a high-spin Ni3+ after the electrochemical cycles at 2.0–4.2 V. This result suggested a notable local-atomic environment change in Ni, as it actively contributes to the charge compensation mechanism. Also, a new crystal phase occurred, namely, the spinel phase (cubic: Fdm), after the cycling, but the amount was very small. On the other hand, if the cell was cycled in the voltage range between 2.0 V and 4.5 V, there has been little Li+ trapping in the SEI layer, indicating little energy shift of the transition metal with a higher oxidation state (or a lower d-orbital) to a lower energy. This condition is potentially more stable, due to the little overlapping between the transition metals and the O2− (2p) band, and this probably mitigates the formation of the spinel phase. However, there was a higher amount of transition metal precipitates on the electrode surface, as a consequence of the deeper extractions of the electrons from the transition metal bands (Fig. 2a and 5c). This is likely to be the major reason why the cell revealed a lower capacity after 50 cycles than that of the NMC5 4.2 V cell. In addition, the NiO clustering on the surface from 4.5 V could also affect the cell performance. The degradation in the linkage between the carbon additives and the PVDF binder is another major cause of the cell fading. On the other hand, the key factor that leads to the good performance could be explained by the well-organized Li+ diffusion channel, as evidenced by mass spectroscopy and the diffusion coefficient (Fig. 7b). The Li+ conduction pathway appears to be improved due to the presence of LiF, which may assist the formation of a stable SEI layer. In the voltage range of 2.0–4.8 V, we can see a sharp drop of the charge capacity during the electrochemical cycles. There has been marked Li+ trapping in the SEI layer, indicating a decrease in the Fermi energy (EF) of the cathode. Therefore the work function (Wf) would increase after the charge–discharge process. The higher the cut-off voltage is, the deeper the band energy would be extracted. Upon charge, this will lead to the evolution of O2− into oxygen gas due to the stronger hybridization between the transition metal d-orbital and the O2− (2p) band.8,19,48 The oxygen evolution exerts a strong influence on the device stability, as it can trigger a fatal reaction that leads to a battery explosion. The deeper extraction of the electrons at the cut-off voltage of 4.8 V tends to cause a higher amount of transition metal precipitates on the electrode surface (Fig. 2a and 5c), suggesting the loss of the key resource for the battery charge capacity. The distinct formation of the P–O bond stemming from the electrolyte decomposition (Fig. 4g and 5a) can increase the instability of the SEI layer, which in turn results in the increment of the SEI resistance, as shown in Fig. 7b. Another key factor that leads to the failure of the Ni-rich transition metal battery can be assigned to the formation of NiO, which is strongly associated with the surface reconstruction of the active material particles. In addition, the increase of Ni3+ at higher cut-off voltages not only indicates the loss of the charge capacity, but also suggests some changes in the local-atomic environment, crystal structure, and SEI component. It is interesting to correlate the similarities of the Ni spin states to the battery stability. The low-spin (LS) Ni3+ is possibly associated with the crystal structure transition and/or formation of some unstable SEI species, while the high-spin (HS) Ni3+ has no significant influence on the formation of the unstable interphase.
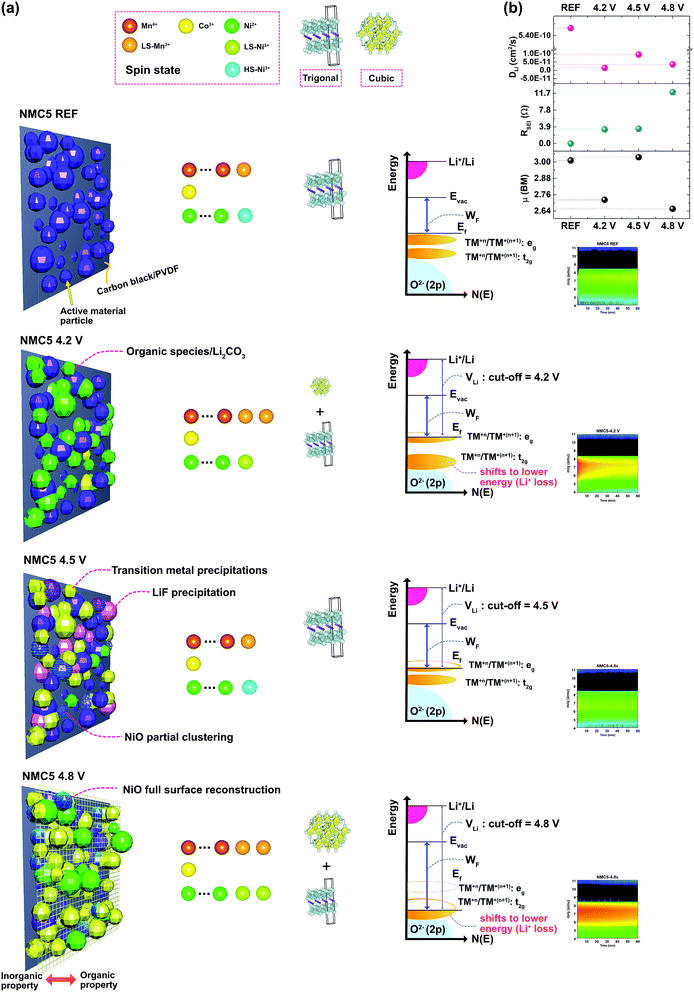 |
| | Fig. 7 (a) Proposed SEI structures/properties at different cut-off voltages with transition metal spin states, crystal structures, schematics of energy vs. density of states (N(E)) diagram, and visualizations of Li+ trapping based on the SIMS profile in the Ni-rich transition metal oxide cathode. (b) Correlation between cut-off voltages, lithium diffusivity (DLi), SEI impedance (RSEI), and magnetic moment (μB). | |
4. Conclusions
The present study identifies the dissimilar nature of the solid electrolyte interphase (SEI) at different levels of the cut-off voltage for a Ni-rich transition metal oxide cathode. Major attention has been paid to the key properties of the SEI layer underlying the battery fading mechanism. The formation of organic species at the interphase impedes the Li+ diffusion to the active material and thus results in capacity fading/voltage decay upon electrochemical cycling. In spite of the large amount of the transition metal precipitate, the existence of LiF tends to improve the Li+ diffusivity, leading to a higher capacity. However, the speciation of the transition metal elements (Ni, Mn, and Co) into diverse oxidation and spin states is the dominant process of the irreversible capacity loss. A deeper access from the lower Fermi level to the O 2p band of the Ni-rich cathode generates a high number of metallic species, i.e., NiO, MnOn, CoOn, Li2CO3, NiF2, MnF2, Li2O, LiF2, LiF, and CoF3 with some decomposed composites from the electrolyte (e.g. ROCO2Li, OF2, and PO) close to the outermost surface. Our work is expected to provide clues on tailoring the electrode material design for high-energy density Li-ion batteries with improved stability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by funding from the Energy Materials and Surface Sciences Unit of the Okinawa Institute of Science and Technology Graduate University, the OIST R&D Cluster Research Program, and the OIST Proof of Concept (POC) Program.
Notes and references
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef.
- B. V. Ratnakumar, M. C. Smart, A. Kindler, H. Frank, R. Ewell and S. Surampudi, J. Power Sources, 2003, 119–121, 906–910 CrossRef.
- J. R. Croy, A. Abouimrane and Z. Zhang, MRS Bull., 2014, 39, 407–415 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef.
- R. V. Chebiam, F. Prado and a. Manthiram, J. Solid State Chem., 2002, 163, 5–9 CrossRef.
- J. Choi and a. Manthiram, J. Electrochem. Soc., 2005, 152, A1714 CrossRef.
- C. M. Julien, A. Mauger, K. Zaghib and H. Groult, Inorganics, 2014, 2, 132–154 CrossRef.
- Z. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191 CrossRef.
- J. Reed and G. Ceder, Chem. Rev., 2004, 104, 4513–4533 CrossRef PubMed.
- J. M. Kim and H. T. Chung, Electrochim. Acta, 2004, 49, 3573–3580 CrossRef.
- Z. Li, N. a. Chernova, M. Roppolo, S. Upreti, C. Petersburg, F. M. Alamgir and M. S. Whittingham, J. Electrochem. Soc., 2011, 158, A516 CrossRef.
- Y. Koyama, I. Tanaka, H. Adachi, Y. Makimura and T. Ohzuku, J. Power Sources, 2003, 119–121, 644–648 CrossRef.
- F. Weill, N. Tran, L. Croguennec and C. Delmas, J. Power Sources, 2007, 172, 893–900 CrossRef.
- A. N. Mansour, L. Croguennec and C. Delmas, Electrochem. Solid-State Lett., 2005, 8, A544 CrossRef.
- J. R. Croy, M. Balasubramanian, D. Kim, S.-H. Kang and M. M. Thackeray, Chem. Mater., 2011, 23, 5415–5424 CrossRef.
- S. Venkatraman, J. Choi and a. Manthiram, Electrochem. Commun., 2004, 6, 832–837 CrossRef.
- W. Liu, P. Oh, X. Liu, M.-J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef PubMed.
- A. Manthiram, J. C. Knight, S. T. Myung, S. M. Oh and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef PubMed.
- Y.-K. Sun, Z. Chen, H.-J. Noh, D.-J. Lee, H.-G. Jung, Y. Ren, S. Wang, C. S. Yoon, S.-T. Myung and K. Amine, Nat. Mater., 2012, 11, 942–947 CrossRef PubMed.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef.
- S.-K. Jung, H. Gwon, J. Hong, K.-Y. Park, D.-H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- M. Dixit, M. Kosa, O. S. Lavi, B. Markovsky, D. Aurbach and D. T. Major, Phys. Chem. Chem. Phys., 2016, 18, 6799–6812 RSC.
- L. Yang, B. Ravdel and B. L. Lucht, Electrochem. Solid-State Lett., 2010, 13, A95 CrossRef.
- N.-S. Choi, J.-G. Han, S.-Y. Ha, I. Park and C.-K. Back, RSC Adv., 2015, 5, 2732–2748 RSC.
- J.-M. Lim, T. Hwang, D. Kim, M.-S. Park, K. Cho and M. Cho, Sci. Rep., 2017, 7, 39669 CrossRef PubMed.
- A. Kraytsberg, Y. Ein-Eli, A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef.
- G. Cherkashinin, D. Ensling and W. Jaegermann, J. Mater. Chem. A, 2014, 2, 3571 RSC.
- M. D. Radin, S. Hy, M. Sina, C. Fang, H. Liu, J. Vinckeviciute, M. Zhang, M. S. Whittingham, Y. S. Meng and A. Van der Ven, Adv. Energy Mater., 2017, 7(20), 1602888 CrossRef.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- C. H. Chen, J. Liu and K. Amine, J. Power Sources, 2001, 96, 321–328 CrossRef.
- G. Cherkashinin, M. Motzko, N. Schulz, T. Späth and W. Jaegermann, Chem. Mater., 2015, 27, 2875–2887 CrossRef.
- J. H. Kim, N. P. W. Pieczonka, Z. Li, Y. Wu, S. Harris and B. R. Powell, Electrochim. Acta, 2013, 90, 556–562 CrossRef.
- H. Hantsche, Scanning, 1989, 11, 257–280 CrossRef.
-
C. M. Julien and A. Mauger, Review of 5-V electrodes for Li-ion batteries: status and trends, 2013, vol. 19 Search PubMed.
- J. R. Croy, D. Kim, M. Balasubramanian, K. Gallagher, S.-H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2012, 159, A781 CrossRef.
- M. N. Ates, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A355–A363 CrossRef.
- S.-H. Kang, P. Kempgens, S. Greenbaum, a. J. Kropf, K. Amine and M. M. Thackeray, J. Mater. Chem., 2007, 17, 2069 RSC.
- T. Eriksson, A. M. Andersson, C. Gejke, T. Gustafsson and J. O. Thomas, Langmuir, 2002, 18, 3609–3619 CrossRef.
- D. Aurbach, K. Gamolsky, B. Markovsky, G. Salitra, Y. Gofer, U. Heider, R. Oesten and M. Schmidt, J. Electrochem. Soc., 2000, 147, 1322–1331 CrossRef.
- M. Noh and J. Cho, J. Electrochem. Soc., 2013, 160, A105–A111 CrossRef.
- Y. M. Lee, K. M. Nam, E. H. Hwang, Y. G. Kwon, D. H. Kang, S. S. Kim and S. W. Song, J. Phys. Chem. C, 2014, 118, 10631–10639 CrossRef.
- T. Eriksson, A. M. Andersson, A. G. Bishop, C. Gejke, T. Gustafsson and J. O. Thomas, J. Electrochem. Soc., 2002, 149, A69 CrossRef.
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358 CrossRef.
- M. Ma, N. A. Chernova, B. H. Toby, P. Y. Zavalij and M. S. Whittingham, J. Power Sources, 2007, 165, 517–534 CrossRef.
- D. Mohanty, A. S. Sefat, S. Kalnaus, J. Li, R. a. Meisner, E. A. Payzant, D. P. Abraham, D. L. Wood and C. Daniel, J. Mater. Chem. A, 2013, 1, 6249 RSC.
- N. Tran, L. Croguennec, M. Ménétrier, F. Weill, P. Biensan, C. Jordy and C. Delmas, Chem. Mater., 2008, 20, 4815–4825 CrossRef.
- K. Ben-Kamel, N. Amdouni, A. Mauger and C. M. Julien, J. Alloys Compd., 2012, 528, 91–98 CrossRef.
- Y. Wei, J. Zheng, S. Cui, X. Song, Y. Su, W. Deng, Z. Wu, X. Wang, W. Wang, M. Rao, Y. Lin, C. Wang, K. Amine and F. Pan, J. Am. Chem. Soc., 2015, 137, 8364–8367 CrossRef PubMed.
- B. J. Hwang, Y. W. Tsai, D. Carlier and G. Ceder, Chem. Mater., 2003, 15, 3676–3682 CrossRef.
- R. Prasad, R. Benedek, A. Kropf, C. Johnson, A. Robertson, P. Bruce and M. Thackeray, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 12101 CrossRef.
- Z. F. Huang, F. Du, C. Z. Wang, D. P. Wang and G. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 54411 CrossRef.
- Z.-D. Huang, X.-M. Liu, S.-W. Oh, B. Zhang, P.-C. Ma and J.-K. Kim, J. Mater. Chem., 2011, 21, 10777 RSC.
- J. Yuan, J. Wen, J. Zhang, D. Chen and D. Zhang, Electrochim. Acta, 2017, 230, 116–122 CrossRef.
- L. Li, L. Wang, X. Zhang, M. Xie, F. Wu and R. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 21939–21947 CrossRef PubMed.
- S. O. Grim, L. J. Matienzo and W. E. Swartz, J. Am. Chem. Soc., 1972, 94, 5116–5117 CrossRef.
- F. Lin, D. Nordlund, Y. Li, M. K. Quan, L. Cheng, T.-C. Weng, Y. Liu, H. L. Xin and M. M. Doeff, Nat. Energy, 2016, 1, 15004 CrossRef.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
-
C. Wagner, Handbook of X-ray photoelectron spectroscopy: a reference book of standard data for use in X-ray photoelectron spectroscopy, Physical Electronics Division Perkin-Elmer Corp., Eden Prairie Minn, 1979 Search PubMed.
- E. Peled, J. Electrochem. Soc., 1997, 144, L208 CrossRef.
- K. Kanamura, H. Tamura and Z. Takehara, J. Electroanal. Chem., 1992, 333, 127–142 CrossRef.
- D. Aurbach, Y. Gofer, M. Ben-Zion and P. Aped, J. Electroanal. Chem., 1992, 339, 451–471 CrossRef.
- J. F. Marco, J. R. Gancedo, M. Gracia, J. L. Gautier, E. Ríos and F. J. Berry, J. Solid State Chem., 2000, 153, 74–81 CrossRef.
- Y. Deng, S. Liu and X. Liang, J. Solid State Electrochem., 2012, 17, 1067–1075 CrossRef.
- S. K. Martha, J. Nanda, G. M. Veith and N. J. Dudney, J. Power Sources, 2012, 216, 179–186 CrossRef.
- M. Xu, L. Zhou, Y. Dong, Y. Chen, J. Demeaux, A. D. MacIntosh, A. Garsuch and B. L. Lucht, Energy Environ. Sci., 2016, 9, 1308–1319 RSC.
- K. Edstr, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef.
- D. T. Nguyen, J. Kang, K. M. Nam, Y. Paik and S. W. Song, J. Power Sources, 2016, 303, 150–158 CrossRef.
- Y. Lu, Z. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef PubMed.
- L. Fan, H. Zhuang, L. Gao, Y. Lu and L. Archer, J. Mater. Chem. A, 2017, 5, 3483–3492 RSC.
- J. W. Gramlich, L. A. Machlan, I. L. Barnes and P. J. Paulsen, J. Res. Natl. Inst. Stand. Technol., 1989, 94, 347–356 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta02622j |
| ‡ Present address: ARC Centre of Excellence in Exciton Science and Department of Chemical Engineering, Monash University, Clayton, VIC, 3800, Australia. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Luis K.
Ono
,
Luis K.
Ono