
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterconnected metal oxide CNT fibre hybrid networks for current collector-free asymmetric capacitive deionization†
Cleis
Santos
 ab,
Julio J.
Lado
*b,
Enrique
García-Quismondo
b,
Inés V.
Rodríguez
b,
Daniel
Hospital-Benito
b,
Jesús
Palma
b,
Marc A.
Anderson
bc and
Juan J.
Vilatela
*a
ab,
Julio J.
Lado
*b,
Enrique
García-Quismondo
b,
Inés V.
Rodríguez
b,
Daniel
Hospital-Benito
b,
Jesús
Palma
b,
Marc A.
Anderson
bc and
Juan J.
Vilatela
*a
aIMDEA Materials Institute, Tecnogetafe. Eric Kandel, 2, 28906, Getafe, Madrid, Spain. E-mail: juanjose.vilatela@imdea.org
bIMDEA Energy Institute, Parque Tecnológico de Móstoles, Avda. Ramón de la Sagra, 3, 28933, Móstoles, Madrid, Spain. E-mail: julio.lado@imdea.org
cDepartment of Civil and Environmental Engineering, University of Wisconsin–Madison, 53706, WI, USA
First published on 29th May 2018
Abstract
Capacitive deionization (CDI), a water desalination technology based on the electro-deposition of salt ions in porous electrodes, is considered a simple, non-energy intensive method to produce clean water. This work introduces a new current collector-free CDI architecture based on electrodes consisting of a porous metal oxide (MOx) network interpenetrated into porous fibres of carbon nanotubes (CNTf). The full CDI device, comprising a stack of γ-Al2O3/CNTf and SiO2/CNTf anodes and cathodes, respectively, has a large salt adsorption capacity of 6.5 mg g−1 from brackish water (2.0 gNaCl L−1) and very high efficiency of 86%, which translates into a low energy consumption per gram of salt removed (∼0.26 W h g−1). This is an 80% improvement compared with reference devices based on activated carbon electrodes and titanium foil current collectors. The remarkable efficiency obtained is due to the morphology of the electrodes, in which the CNT fibres act simultaneously as a current collector, active material and support for the metal oxide. Such architecture leads to high capacitance while minimizing internal resistance, as confirmed by cyclic voltammetry and electrochemical impedance spectroscopy. The fact that full electrodes can be made continuously, as demonstrated on 4 km of CNTf, makes the fabrication process more attractive.
Introduction
Capacitive deionization (CDI) is an emerging desalination technology capable of reducing the salt concentration of a water stream by electrostatic adsorption of ions in porous electrodes under a potential, equivalent to the charging step in a generic electric double-layer capacitor (EDLC).1–3 Once the electrodes are saturated, ion desorption is forced by short-circuiting the cell (capacitor discharge step). In this fashion, CDI technology is able to remove salt and ionic contaminants from water while also storing energy in the electrical double-layer (EDL).4 The energy stored can be subsequently retrieved in the electrode discharge step using the same working principle as in a capacitor (see the ESI†).5–10 Amongst its envisaged advantages over other desalination methods are: lower energy consumption to treat medium salt concentration streams (2–10 g L−1),6,8,11–14 low fouling and scaling risk15–17 and higher water recovery rates, thus achieving substantial reductions in the volume of brine.12,18,19The design and choice of materials for CDI and capacitive dye removal follows similar principles to standard EDLC, hence most cells consist of two porous carbon electrodes supported on a current collector;20 in the case of CDI a corrosion-resistant metal, typically Ti, or a non-metallic substrate such as expanded graphite is used. Nanocarbons such as carbon nanotubes (CNTs), carbon nanofibres (CNFs) and graphene are particularly attractive as active materials for this application because of their combination of high specific surface area (SSA) and electrical conductivity.21–27 Their main advantage over carbon aerogels or activated carbons (ACs) is the possibility to integrate them as electrodes with a mesoporous interconnected network,28,29 leading to a continuous charge distribution in the electrode surface, lower equivalent series resistance, faster ion diffusion and thus higher power density.30–33
Graphene-based electrodes for CDI have been studied since 2009![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 and recently reviewed by Liu et al.35 El-Deen et al.,36 for example, did extensive work on asymmetric CDI cells based on carboxymethyl cellulose and quaternary ammonium cellulose coatings on flexible graphene. They studied the effects of graphene-based materials and flow-rate conditions on salt adsorption capacity (SAC) as well as on charge efficiency. The result showed ∼23 mg g−1 and 85% of charge efficiency, although under salt concentration conditions (300 ppm NaCl) as low as in drinking water, and under a potential (1.4 V) high enough to produce hydrolysis and pH changes.
34 and recently reviewed by Liu et al.35 El-Deen et al.,36 for example, did extensive work on asymmetric CDI cells based on carboxymethyl cellulose and quaternary ammonium cellulose coatings on flexible graphene. They studied the effects of graphene-based materials and flow-rate conditions on salt adsorption capacity (SAC) as well as on charge efficiency. The result showed ∼23 mg g−1 and 85% of charge efficiency, although under salt concentration conditions (300 ppm NaCl) as low as in drinking water, and under a potential (1.4 V) high enough to produce hydrolysis and pH changes.
There has also been recent work using CNT-based CDI electrodes. A common embodiment consists in dispersing CNTs together with a binder and then depositing them onto a Ti current collector, following established methods to fabricate activated carbon electrodes. Using this technique, Wang et al.37 reported, for example, a SAC of ∼5 mg g−1 at 1.2 V in 2 g L−1 NaCl electrolyte, although requiring extremely long charging times (200 min).
Other studies on half-cells have highlighted the potential of CNTs-containing electrodes when combined with activated carbon,38 mesoporous carbon or carbon nanofibres.22 Particularly interesting are examples of the use of flexible conducting CNT arrays as current collectors in CDI electrodes, with the view of avoiding the use of metallic current collectors to reduce costs and enable the fabrication of devices with complex non-planar shapes. In this context, CNT fibres are ideal electrode materials because they combine exceptional electrochemical stability, surface area around 250 m2 g−1, mechanical toughness above that of carbon fibre and electrical conductivity approaching that of copper,39 and can already be produced on a semi-industrial scale.40 Liu et al.38 analysed commercial CNT yarns from Teijin and obtained a capacitance of around 5 F g−1 in 5–500 mM salt concentration, which could be increased to 30 F g−1 after coating them with AC. They also coated the yarns with an ion exchange membrane and demonstrated their use for electrical power generation from water of different salinity contents (Capmix-experiments). Benson et al.41 fabricated electrodes for supercapacitors and CDI by electrodeposition of polyaniline (PANI) onto nonwoven CNT fibre fabric. They obtained a capacitance as high as 200 F g−1 in 1 M NaCl. However, the electrochemical characterization was performed on samples supported on gold foil as the current collector.
While the potential of nanocarbons is clear, there is consensus that there is ample room for further development in terms of: increasing salt adsorption capacity, increasing efficiency and thus reducing energy consumption and exploiting the extraordinary properties (mechanical, electrical) of nanocarbons in innovative CDI designs. CNT fibres would seem ideally suited to contribute to these three challenges, as they have indeed led to EDLC devices with high efficiency and augmented mechanical properties such as flexibility in bending,42,43 large stretchability41 and semi-structural properties.40,44
In parallel, there is vast body of work showing large increases in the capacitance of carbon electrodes when combined with metal oxides.45 In particular, coating with γ-Al2O3 and SiO2 has been demonstrated to be an effective technique to improve the electrochemical properties of a wide range of pre-formed carbon-based substrates such as carbon cloths,46 flat graphite,46 carbon xerogel,47 carbon sheets,48–50 activated carbon powders,51 carbon foam and nanofoams.52 Transition metal oxides53 or conducting polymers41 can provide larger increases in capacitance due to the contribution of pseudocapacitive reactions. But their integration is more cumbersome and their benefits for CDI processes, where avoiding co-ion adsorption is of paramount importance, are debatable.
Motivated by these encouraging results, here we present a new method to produce large area electrodes for CDI, by growing metal oxide (MOx) networks into macroscopic fibres of CNTs in situ as they are spun from a chemical vapour deposition (CVD) reactor in a continuous process. The resulting structure consists of interpenetrating networks that thus maximise surface area and therefore specific capacitance, while reducing internal resistance. We use electrochemically stable alumina (γ-Al2O3) and silica (SiO2) as the anode and cathode, respectively, and exploit their differences in surface potential at drinking water pH (7–8.5) to enhance ionic selectivity.45,49,50,54 In addition to a thorough structural and electrochemical characterisation of electrodes, we present results obtained with full flow-cells, showing very good salt adsorption capacity of 6.5 mg g−1 from brackish water (2.0 g L−1 NaCl) and an exceptionally high current efficiency (ξCharge) of 86%, which translates into a low energy consumption per gram of salt removed (∼0.26 W h g−1). Overall, this work demonstrates innovative highly efficient metal-free CDI devices produced using a simple fully scalable process.
Results and discussion
Fabrication and structure of electrodes
The fabrication of electrodes is based on the continuous impregnation of CNT fibres with metal oxide precursors in-line as they are spun from the chemical vapour deposition reaction, followed by calcination (see Experimental methods and the ESI†). Collecting the fibre material onto a bobbin produces the overlap of fibres into a non-woven fabric. Through capillary forces arising from solvent contact and subsequent evaporation the fibres are densified and consolidated into a robust and tough fabric material. The incorporation of the metal oxide into the CNTf electrode is done by continuously spraying a colloidal dispersion of metal oxide sol particles to the ∼4 km of fibre that makes up an individual electrode (Fig. 1a). By carrying out this process in-line and using highly optimized stable dispersions (zeta potential measurement of the suspensions is available in the ESI†) we ensure that the entire electrode is uniformly loaded with metal oxide.1,45,55,56 This overcomes the challenge of uniformly depositing nanoparticles onto thick porous electrodes, which are prone to inhomogeneous deposition due to inherent particle diffusion limitations. The method used here is also attractive to make electrodes with arbitrary shapes, although in this work we study planar electrodes with the aim to compare with established technologies. The impregnation of CNT electrodes has the purpose of increasing their capacitance by three mechanisms: promoting close association of counter-ions on the surface of the metal oxide by the protonation and de-protonation of the amphoteric surface sites;45,50,57 increasing specific surface area and enabling wetting by aqueous electrolytes of the otherwise hydrophobic graphitic CNT electrode. With these objectives in mind we chose SiO2 and γ-Al2O3, which have the added benefit of not undergoing oxidation/reduction reactions but instead, developing an electrostatic EDL surface potential in the pH range of seawater and drinking water. The potential difference between nanoporous SiO2 sol (∼−25 mV) and γ-Al2O3 (∼+30 mV) at this given pH range greatly assists in the selective electro-adsorption of ions and improved efficiency in CDI processes (the electrophoretic mobility curves as a function of the pH of MOx sol particles in aqueous solutions of NaNO3 are included in the ESI†).45,49,50,54 | ||
| Fig. 1 CDI electrodes based on CNTf and metal oxides. (a) Photograph showing in-line integration of MOx sol particles into the CNTf fabric (4 km of CNT fibre). (b) SEM images of pristine CNTf showing its mesoporous structure, (c) SEM images of samples with 21 wt% SiO2 and (d) SEM images of samples with 43 wt% γ-Al2O3. | ||
The content of metal oxide can be varied by adjusting the concentration of sol in the dispersion. Table 1 presents the range of concentrations used and the final MOx content determined. The resulting microstructure after hybridisation of the CNTf material with MOx can be observed in Fig. 1. A reference sample without MOx (Fig. 1b) shows that the CNTf consists of a network structure of interconnected CNTs. The long length of the CNTs (≈1 mm) enables the coexistence of a large porosity with long crystalline domains, leading to a porous electrode with efficient charge and stress transfer and thus high electrical conductivity and toughness. The electron micrographs (Fig. 1c and d) of hybrid electrodes with 21 wt% MOx (after calcination) confirm that the samples hybridised in-line also have a high porosity and present a uniform distribution of metal oxide on the supporting CNTf network. In the case of the sample with SiO2 for example, the SEM micrograph clearly shows the presence of an interpenetrating SiO2 network throughout the hybrid electrode sample (Fig. 1c). A comparison of electron micrographs in secondary and back-scattered mode further assists in the identification of the different phases and confirms the uniformity of the porous structure observed (see the ESI†). Such a comparison also shows the changes in microstructure when increasing MOx fraction, ultimately leading to blockage of the CNTf network.
| γ-Al2O3/CNTf electrodes | wt% MOx | SSA [m2 g−1] | Cc [mF cm−2] | η [%] | SiO2/CNTf electrodes | wt% MOx | SSA [m2 g−1] | Cc[mF cm−2] | η [%] |
|---|---|---|---|---|---|---|---|---|---|
| 6 wt% γ-Al2O3–CNTf | 6 ± 2 | 222 | 25.9 | 85 | 21 wt% SiO2–CNTf | 21 ± 5 | 156 | 15.7 | 99 |
| 21 wt% γ-Al2O3–CNTf | 21 ± 3 | 244 | 11.6 | 99 | 36 wt% SiO2–CNTf | 36 ± 9 | 16 | 7.5 | 98 |
| 38 wt% γ-Al2O3–CNTf | 38 ± 5 | 272 | 11.4 | 99 | 60 wt% SiO2–CNTf | 60 ± 8 | 561 | 5.4 | 98 |
| 43 wt% γ-Al2O3–CNTf | 43 ± 3 | 274 | 9.2 | 99 |
Closer inspection by transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy elemental mapping of the structure of the hybrid electrodes with 21 wt% MOx is presented in Fig. 2. It shows that the metal oxide consists of interconnected nanoparticles distributed throughout the CNT fibre. The hydrophobic nature of the CNTs prevents the formation of a continuous conformal coating of MOx over the individual CNTs, which would in fact rapidly reduce SSA. Instead, a key feature of the structures in Fig. 2 is that the carbon and inorganic phases form interpenetrating nanostructured networks. As such, they preserve a high specific surface area while minimising the distance between the MOx surface and the MOx/CNTf interface, hence reducing electrical resistance. The hybrid can be visualised as having the current collector built into the porous metal oxide.
 | ||
| Fig. 2 High-resolution TEM micrographs and associated EDX elemental maps of samples with (a) 21 wt% SiO2 and (b) 21 wt% γ-Al2O3 showing the network structure of both the CNT fibre and the metal oxides. | ||
Electrochemical tests and electrode optimisation
The content of metal oxide can be varied by adjusting the concentration of sol in the dispersion. Table 1 presents the range of concentrations used and the final MOx content determined by thermogravimetric analysis (TGA). Specific surface areas measured by gas adsorption are also included for reference, although as we discuss later in the paper, BET surface area is not a good indicator of capacitance. (Examples of thermograms and detailed textural properties are included in the ESI.†)To investigate the performance of the different hybrid materials produced as CDI electrodes, various electrochemical measurements were first performed in small-scale 0.785 cm2 Swagelok® cells. Cyclic voltammetry (CV) was carried out to investigate charge-storage capability as a function of metal oxide content, and to analyse specific electrochemical features of the two different oxides (γ-Al2O3 and SiO2). A comparison of voltammograms for all the samples produced is included in the ESI† and the results summarised in Table 1. The data indicate that the optimum MOx mass fraction is in the range 6–21 wt%. Higher mass fractions led to a lower storage capacity as a consequence of MOx build-up and CNT blockage, thus reducing the effective surface of the hybrid material. Similarly, samples with a lower MOx mass fraction exhibited a low coulombic efficiency (η), mainly associated with oxygen evolution and redox reactions (ca. 0.6 V). Indeed, the contribution of faradaic processes in similar MOx–graphite systems is known to be very sensitive to mass fraction.58 With these results in mind, further analysis is presented only for samples combining high capacity and efficiency, as these two parameters are key to obtain a low energy consumption per gram of salt removed in full flow cells (vide infra).
Fig. 3 presents CV curves of pristine CNTf and samples with 21 wt% γ-Al2O3 (21 wt% γ-Al2O3–CNTf) and a 21 wt% SiO2 (21 wt% SiO2–CNTf). The different MOx/CNTf composites and pristine CNTf have a quasi-rectangular shape, typical of a highly conducting EDLC, slightly distorted due to the reminiscent chemical (quantum) capacitance of the CNTf.39 The voltammograms demonstrate that geometric capacitance (Cc), expressed in mF cm−2, is significantly higher for the hybrid electrodes than that of the uncoated pristine CNTf. At 100 mV s−1 the values are 12 mF cm−2 and 16 mF cm−2 for 21 wt% γ-Al2O3–CNTf and 21 wt% SiO2–CNTf hybrids, respectively, compared with <5 mF cm−2 for the bare CNTf. The corresponding gravimetric capacitance of these electrodes is 50–66 F g−1, which is in a similar range to that of electrodes based on commercial Picactif high SSA (2410 m2 g−1) activated carbon deposited on a Ti current collector (85 F g−1), albeit with a much higher coulombic efficiency, η ∼ 99%.
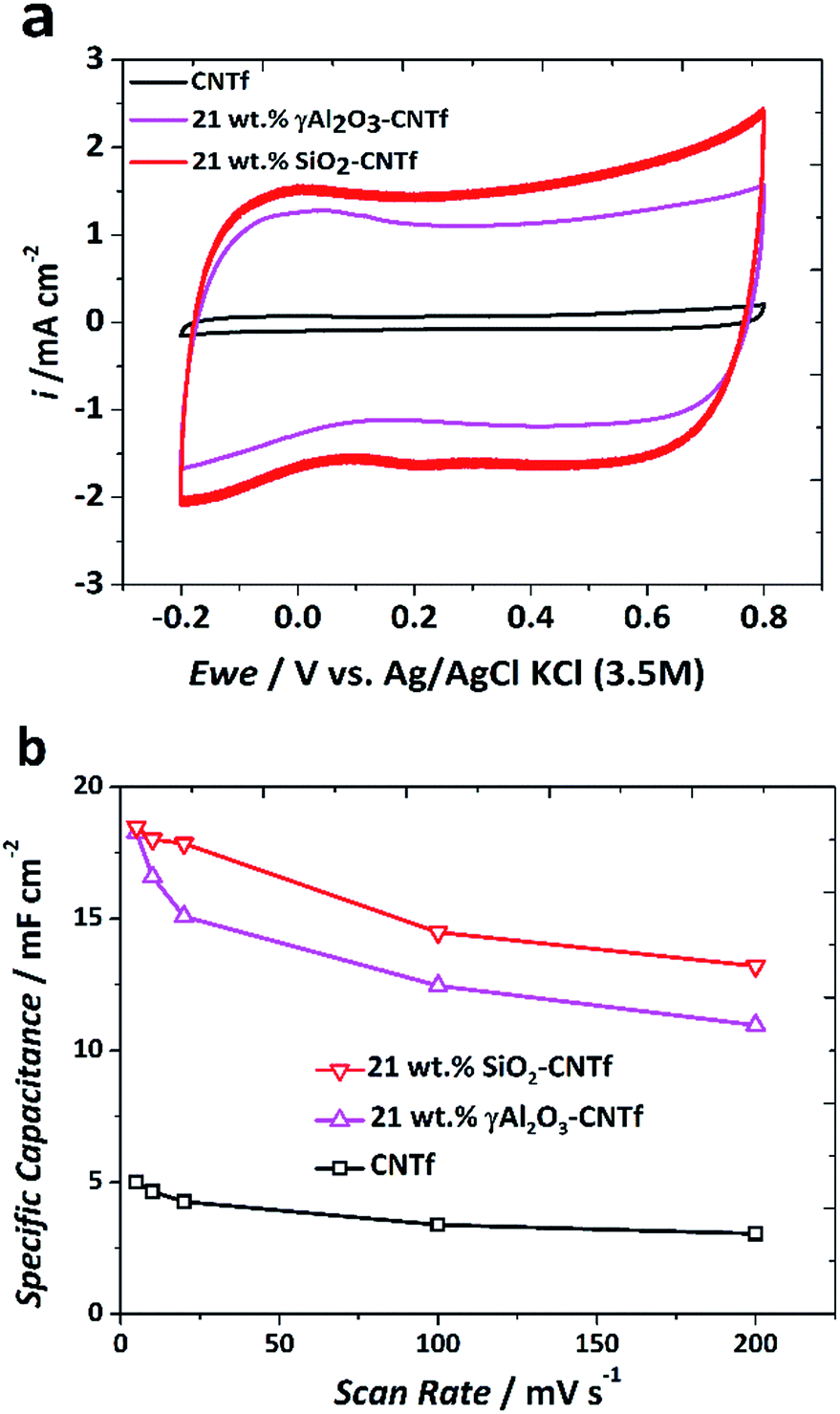 | ||
| Fig. 3 Cyclic voltammetry tests in neutral aqueous media (K2SO4 0.5 M). (a) Cyclic voltammograms at 100 mV s−1 scan rate j (mA cm−2). (b) Specific capacitances (mF cm−2) vs. scan rate (mV s−1) of the CNTf and MOx/CNTf composites. cm2 refers to the electrode geometrical area. | ||
The comparison of samples with different MOx also shows that electrodes with SiO2 have a slightly higher BET-SSA-normalised capacity than those with γ-Al2O3. The data in Fig. 3, for example, translate into values of 5.7 μF cmBET−2 and 8.2 μF cmBET−2, for 21 wt% γ-Al2O3–CNTf and 21 wt% SiO2–CNTf hybrids, respectively. These results suggest that the effective surface area of the hybrid material taking part in the EDL formation is sensitive to the morphology of the MOx network structure supported on the CNTs and confirm the view that BET surface area measurements have limited use as predictors of capacitance in these hybrids.
Measurements at different scan rates for these two hybrid electrode configurations show a weak dependence of capacitance on scan rate (Fig. 3b), indicating that the increase in charge storage relative to the pristine CNTf occurs without changing the dominant EDL mechanism.3 But we note that a stronger dependence on scan rate was observed in hybrids with a higher MOx mass fraction due to a higher resistance and the predominance of smaller diffusion-limiting pores, confirming that mass fraction optimisation is key for device operation. The weak dependence of capacitance on scan rate indicates relatively agile ion movement into the pore structure of the electrode even under high electric demand. This feature is particularly relevant for the implementation of electrodes in real CDI systems, in which devices operate continuously under variable charging conditions, often unpredictable and with peak power pulses, for example when integrated with renewable energy systems.
A further point of interest is to understand the origin of the large increase in capacitance obtained in the hybrids relative to the pristine CNT fibre. We attribute such improvement to two effects: the MOx enabling the electrolyte to wet the porous CNT fibre electrode, and the contribution from the differential capacitance of the non-transition metal oxides used. In the first case, the MOx acts as an agent that enables the ingress of aqueous electrolyte into the porous hydrophobic CNT network, somewhat similar to other graphite–inorganic systems,48,49,54 including recent observations using MOx particles as binders.33 Using identical CNT fibre electrodes and similar aqueous electrolytes, we have previously demonstrated large increases in capacitance through wetting by adding MnO253 or by gas-phase functionalisation with ozone.59 A rough estimate of the critical mass fraction of SiO2 and γ-Al2O3 required for the electrolyte to infiltrate the electrode can be calculated using a semi-empirical approximation widely used to assess liquid/polymer wetting.60 This value comes out at around <10%, which indicates that for the range of mass fractions produced in this work the electrodes are expected to be fully impregnated with the electrolyte (see the ESI†).
The contribution of differential capacitance from γ-Al2O3 and SiO2 is enabled by the nanoscopic size of the particles and close proximity to the built-in current collector, which reduces the potential drop across the dielectric particles. Under those conditions, an unusual electric double layer is formed at the interface between charged MOx and electrolyte interface, which is sensitive to the inherent surface potential of the MOx and to the electrolyte pH.45 It is associated with a surface complexation mechanism that leads to the reduction of the separation between the ions and the oxide surface, hence increasing capacitance. It is typically described by triple-layer models with: two constant capacitance planes (surface/electrolyte interface layer) containing the inner Helmholtz plane (protons and hydroxyl group adsorption) and the outer Helmholtz plane (other electrolyte ion adsorption), and a diffuse layer.61–63
Electrodes were further studied in symmetric devices subjected to galvanostatic charge–discharge (CD) experiments at 50 mA g−1 from 0 V to 1 V, during 10 cycles. The results confirm CV measurements. As shown in Fig. 4a, the CD voltage profiles for the hybrid and pristine electrodes exhibit the triangular line shape of an electrical double layer capacitor with high coulombic efficiency (>95%). Both gravimetric energy and capacitance are substantially higher in the hybrids than in the reference CNT fibre sample. Energy reaches 9 mW h g−1, 7 mW h g−1 and 2 mW h g−1, and capacitance 66 F g−1, 52 F g−1 and 12 F g−1, for 21 wt% Al2O3–CNTf, 21 wt% SiO2–CNTf and pristine CNTf, respectively. These properties are also higher than those obtained in CNTf made hydrophilic by chemical functionalisation (1 mW h g−1 and 30 F g−1) and which have complete wetting by a similar aqueous electrolyte,59 confirming the genuinely synergistic effect of combining high surface area non-transition MOx with CNTf built-in current collectors.
 | ||
| Fig. 4 Galvanostatic charge–discharge measurements: (a) typical shape of the charge–discharge profiles of γAl2O3–CNTf and SiO2–CNTf. (b) Impedance spectroscopy experiments pristine CNTf, 21 wt% γ-Al2O3–CNTf and 21 wt% SiO2–CNTf. Tests performed in neutral aqueous media (K2SO4 0.5 M). | ||
The low electrical resistance of the system is evident in the very small ohmic drop in CD curves. From impedance spectroscopy measurements on the hybrids (Nyquist plot in Fig. 4b), we extract a low equivalent series resistance (ESR) in the range of 15–55 Ω, which is comparable to that of dielectric-free supercapacitors.64,65 Furthermore, separation of resistance contributions gives a negligible setup resistance (Rs). As expected, the area-normalized equivalent series resistance (ANESR) increases with the addition of metal oxide (11.7 Ω cm2 to 43 Ω cm2), but is still amongst the lowest in the CDI literature based on traditional current collector-based electrodes.66
Flow cells: proof-of-concept
Aiming to put to test the electrodes studied above under relevant conditions for CDI, an asymmetric CDI (aCDI)1,21,46,67,68 full flow cell was assembled and used for brackish water (2.0 g L−1 NaCl) desalination. Considering that 2.0 g L−1 NaCl is one of the highest salt concentrations studied in nanocarbon-based electrodes for CDI,69 and that the maximum salt adsorption capacity (mg g−1) reaches a plateau followed by a decrease at high concentrations,20 higher NaCl concentrations were not considered for further tests. The aCDI flow cell contained 16 MOx/CNTf electrodes stacked in a filter-press assembly, with an exposed geometric area of 10 cm2, in which eight SiO2/CNTf electrodes were stacked and used as the cathode and another eight γ-Al2O3/CNTf electrodes as the anode, both without current collectors. A schematic of individual electrodes and cells including the recirculation hydraulic system is shown in Fig. 5. The fabrication of such relatively large amounts of electrode material entirely from precursors was possible due to the relative simplicity of the CVD and sol–gel methods and continuous infiltration process introduced here, which lend themselves to scale up to a prototype level and possibly beyond. To the best of our knowledge, this is the first report of a current collector-free electrode tested in a CDI flow-cell. | ||
| Fig. 5 (a) MOx/CNTf layer of fibres without current collectors. (b) CDI laboratory set-up. Insets: opened CDI cell with a stack of MOx CNTf fibres, zoom of the direct electrical connection to the MOx/CNTf electrode. | ||
Once the cell was assembled and the CDI system was set up, capacitive deionization experiments were performed in constant current mode (CDI-CC) using 34 mM NaCl (∼2.0 g L−1) as the electrolyte. Current densities of 1.5 A m−2 and −0.5 A m−2 were applied during charging and discharging steps, respectively. An operational potential window was established between 0 V and 1.2 V. The CDI experiment consisted of 3 charge–discharge cycles. After the experiment a short-circuit step (constant cell voltage at 0 V) was performed followed by a 10 min open circuit voltage. Finally, another 3 charge–discharge cycle experiment was conducted. Results were compared with a reference sample consisting of a filter-press CDI cell system with activated carbon (Picactif BP10) paste electrodes supported on highly conductive titanium current collectors.
Fig. 6 displays the charge–discharge graphs for the MOx/CNTf CDI system operated in constant current mode.5,7 Their profile shows the EDL formation expected in an efficient CDI device, confirming that the electrodes can indeed be used without the need for a current collector. Moreover, no electrolyte leakage was observed during operation, proving the perfect sealing of the current collector-free CDI cell. Charging and discharging resistances are small, at 0.19 ± 0.03 kΩ cm2 and 0.21 ± 0.02 kΩ cm2, respectively, both lower than the standard reference cell with the current collector and conventional active material (see Table 2). But note that the resistance at the interface between conventional planar current collectors and porous electrodes in traditional CDI devices can amount to as much as 94% of the total ESR.66
 | ||
| Fig. 6 (a) Ecell and pH vs. time profile evolution observed during the CDI-CC experiment in 2 g L−1 NaCl. (b) Main results for the current collector-free cell associated with cycle n = 3. | ||
| Electrode material | R c [kΩ cm2] | R d [kΩ cm2] | SAC [mg gAM−1] | ASAR [mg gAM−1 min−1] | ξ c charge [%] | ε [W h g−1] |
|---|---|---|---|---|---|---|
| a Current collector is required. b N.A. not available. c Cell voltage, 1.6 V. d Low NaCl concentration, <1 g L−1. e Asymmetric CDI Gcm, graphene chemically modified. f Low NaCl concentration, ∼0.3 g L−1. g Cell voltage, 1.4 V low NaCl concentration, ∼0.3 g L−1. | ||||||
| MOx/CNTf | 0.19 | 0.21 | 6.5 | 0.21 | 86 | 0.26 |
| Picactif BP10a | 0.23 | 0.35 | 4.4 | 0.20 | 55 | 1.94 |
| ZnO/AC-clotha79 | N.A. | N.A. | 7c | N.A | 78 | 0.02d |
| Carbon aerogela,e80 | N.A. | N.A. | 5e | 0.06 | 53 | 0.03f |
| 3D-Gcma,e36 | N.A | N.A | 14g | N.A | 84 | N.A. |
The low internal resistance of electrodes and contribution from MOx surface potential lead to an impressive current charge efficiency (ξcharge) of 86 ± 6% in a 2 g L−1 electrolyte, which is much higher than that of the reference sample with AC electrodes and Ti current collectors (55 ± 2%). It is also above the few reports on full cells available in the literature (43–84%) and which correspond to more favourable testing conditions in terms of initial salt concentration (250 mg L−1 to 1 g L−1).36,70,71
In addition to electrochemical measurements, pH was continuously measured and electrolyte ion content determined by ionic chromatography (IC) after each cycle. The analysis of the pH evolution (see Fig. 6a) indicates a slight acidification of the electrolyte solution during the first cycle, most likely attributed to faradaic reactions associated with the reduction of dissolved oxygen72,73 and thus not present in subsequent cycles. Accordingly, due to these pH fluctuations conductivity measurements could be inaccurate and masking capacitive performance. Therefore, IC analysis was selected as the most reliable technique to measure ion removal.
From IC measurements we extracted a value of SAC of 6.5 mg g−1 and an Average Salt Adsorption Rate (ASAR) of 0.21 mg g−1 min−1 for the MOx/CNTf cell. This is above the reference AC CDI cell (SAC: 4.4 mg g−1, ASAR: 0.20 mg g−1 min−1). Moreover, they are substantially higher than reported values for systems based on CNTs (SAC: 0.7–5.2 mg g−1, ASAR: 0.009–0.026 mg g−1 min−1), carbon nanofibres (SAC: 3.2–4.6 mg g−1, ASAR: ∼0.0017 mg g−1 min−1) and on CNT-CNF composites (SAC: 3.3 mg g−1, ASAR: 0.11 mg g−1 min−1).22
The reported SAC value is also on par with recent reports on electrodes comprising MnO2 deposited on multi-walled CNTs (SAC: 6.6 mg g−1, ASAR: 0.12 mg g−1 min−1).74 However, the literature work corresponds to electrodes produced from a slurry of active material deposited on a stainless steel mesh, and a CDI system was operated at 1.8 V, with the associated risk of faradaic reactions, electrode corrosion and long-term reduction of electrosorption capacity,75,76 combined with a higher energy consumption. Very importantly, the SAC for the hybrid aCDI system (6.5 mg g−1) was achieved consuming only 0.38 W h g−1 and with no degradation observed in the profile of the electrodes (see Raman spectra before and after cycles in the ESI†). This value represents an 80% reduction of the energy demand compared with the reference AC CDI cell (1.94 W h g−1). Such energy consumption is also much lower than conventional brackish water treatments by energy intensive methods like reverse osmosis (ca. 1.5 W h g−1).77,78
Finally, we note that the value of energy consumption per gram of salt removed of 0.38 W h g−1 is conservative, since it does not take into account the energy recovered during discharge in the second step of the CDI cycle, a unique feature of CDI relative to other desalination technologies. To take this effect into account, we first calculate the round-trip efficiency, a key figure of merit for the operation of CDI systems that are based on the current efficiency of the charging step and the energy released during discharge.7 This comes out as 34%, with large room for improvement using new operational modes.9 The resulting net energy consumption, at 0.26 W h g−1, is exceptionally low (Table 2), nearly an order of magnitude lower than optimised systems based on AC tested under similar salt concentrations.
Experimental
Electrode fabrication
CNT fibres were synthesized by the direct spinning chemical vapor deposition method81 under reaction conditions leading to highly graphitised CNTs with few layers (3–5), average diameter < 7 nm and length in the millimeter range. The CNTs associate in bundles with diameter in the range of 10–30 nm. Electrodes are formed by winding multiple CNTfs onto a bobbin with a paper support. The overlay of incoming CNT fibres on top of each other as a fabric makes them consolidate into a continuous CNT bundle network structure, with electrochemical properties as reported before.39 The hybrid electrodes were produced by spraying the CNTf at its exit from the vertical CVD reaction and before it was deposited onto the paper support. The spray solution contained metal oxide sol nanoparticles (SiO2 or γ-AlOOH) suspended in a volatile solvent. Nanoparticles of acidic SiO2 were synthesized from tetraethylorthosilicate (TEOS 98% Sigma Aldrich) following ref. 82 and used as an acidic SiO2 sol at a concentration of 35 g L−1 in ethanol at pH 3. To obtain γ-Al2O3 nanoparticles, γ-AlOOH sols were prepared using the reported methods.54,56 The final step to manufacture the fibre-metal oxide composite consists of annealing the samples at 350 °C for 2.5 h (temperature rate 5 °C min−1). That calcination temperature was high enough to ensure crystallisation of the metal oxides, as observed by X-ray diffraction, which also confirmed the transformation of γ-AlOOH into γ-Al2O3 according to 2γ-AlOOH → γ-Al2O3 + H2O (see thermogravimetric analysis (TGA) and X-ray diffraction (XRD) data in the ESI† for further information).52Structural characterization
Thermogravimetric analysis (TGA) of pristine CNT fibres and MOx-based composites was performed at 5 °C min−1 under air flow. Nitrogen (N2) adsorption at 77 K of the composites was measured with a Quantachrome Instrument (Quadrasorb SI, version 5.03). Samples were previously degassed at 150 °C under vacuum for 20 h. The specific surface areas and pore size distributions (PSDs) were calculated by using the BET equation and Quadrachrome ASiQwin™ software. Electron micrographs were obtained with a FIB-FEGSEM Helios NanoLab 600i (FEI) used at 15 kV and a JEOL JEM 3000F TEM used at 300 kV.Electrochemical tests
Small-scale cells. Sample size was 0.785 cm2, corresponding to a circular-shape Swagelok® cell electrode. Cyclic voltammetry, galvanostatic charge–discharge measurements and impedance spectroscopy experiments were performed by using an electrochemical workstation (Biologic VMP3 multichannel potentiostat–galvanostat coupled with EC-Lab v10.44 software).Two types of cell configurations were used for performing the electrochemical characterization: three-electrode “T-Type” connection and two-electrode connection (see the ESI†). In both set-ups, a neutral electrolyte (0.5 M K2SO4) was infused in a cellulosic separator between the working and counter electrode. Firstly, “T-type” Swagelok® cells were employed for cyclic voltammetry tests at scan rates from 5 mV s−1 to 200 mV s−1, and a voltage window from −0.2 V to 0.8 V. In this configuration, the working electrode (We) consisted of a MOx/CNTf based composite; expanded graphite was used as the counter electrode (Ce) and an Ag/AgCl reference electrode to complete the set-up. Capacitances were calculated by cyclic voltammetry tests. Electrode capacitance is directly related to the charge storage in the electrodes, thus with knowledge of capacitance during the charge process it is possible to estimate the amount of ions adsorbed in the electric double layer. From the voltammogram's area, capacitance (C) is calculated following eqn (1):
 | (1) |
A second configuration, the two-electrode Swagelok® cells, with both We and Ce made of MOx/CNTf electrodes, was used in galvanostatic charge–discharge measurements as well as in impedance spectroscopy experiments without the need for an additional current collector. In the two-electrode configuration, galvanostatic CD experiments were carried out at 0.045–0.0012 mA cm−2 from 0 V to 1 V, during 10 cycles. Data were extracted from the last cycle. The frequency range for impedance spectroscopy experiments was 200 kHz to 10 mHz at a bias voltage of 0 V.
Stacking and CDI cell assembly
Cell electrodes were prepared from stacks of layers of MOx/CNTf, cut out to match the CDI cell shape (50 cm2). The thickness of the electrode stacks was between 0.80 and 1.15 mm and the mass loading of the SiO2/CNTf and γ-Al2O3/CNTf stacks was 14.07 mg cm−2 and 13.93 mg cm−2 respectively. The self-standing MOx/CNTf electrodes were placed in the CDI cell and directly connected to conventional crocodile clips. Reference samples consisted of a filter-press CDI cell system with activated carbon (Picactif BP10) paste electrodes (geometrical area exposed: 10 cm2, electrode thickness of 800 ± 35 μm and mass loading of 26 ± 3 mg cm−2) supported on highly conductive titanium current collectors.Except for the lack of metallic current collectors, the CDI cell had the conventional widely used filter-press configuration.3,20 The flow cell used has: electrode geometrical area exposed to the electrolyte of 10 cm2, γ-Al2O3 as a working electrode (positively polarized during charging) and SiO2–CNTf counter electrode (negatively polarized during the charging), an insulating paper separator to avoid shorting and a 2 mm flow distributor. A reference electrode (Ag/AgCl) was employed to control each of the electrode potential (see the ESI†). Charge–discharge experiments were carried out at current densities of 1.5 A m−2 and -0.5 A m−2 during charging and discharging, respectively, with potential from 0 V to 1.2 V during 3 cycles.
During operation, brackish water was fed from the bottom of the CDI reactor and was collected, once processed, at the top of the opposite end. In this manner, we could flood the entire device and avoid forming preferential channels – short circuiting the device (see the ESI†). Experiments were performed using 60 mL of an aqueous NaCl brackish solution of 34 mM (∼2.0 g L−1). The pump was calibrated to provide a flow rate of 1.5 mL min−1 through the CDI reactor. To evaluate the ion's removal performance, samples were taken at three different times: at the beginning of the experiment, just once the charge step (deionization) is completed, which also corresponds to the initial point of the discharge stage (electrode regeneration) and when the discharge is complete. These samples were analyzed by ionic chromatography (930 Compact IC Flex Model). pH and ionic conductivity were also measured during the adsorption–desorption cycles.
Conclusions
This work introduces a new type of CDI electrodes based on unidirectional fabrics of CNT fibres hybridised with MOx, γ-Al2O3 and SiO2 as the anode and cathode, respectively. The CNT porous fabric plays the role of a built-in current collector and support for the nanostructured MOx. Such electrodes have a high capacitance and efficiency in aqueous electrolytes, as a result of the hybrid structure consisting of two interconnected porous networks. This architecture maximises the effective surface area for EDL formation while reducing internal resistance.The use of a relatively simple fabrication process, particularly the infiltration of fabrics with sol particles in-line during fibre spinning, enables fabrication of large electrode samples and testing of a full CDI flow cell for brackish water desalination. The results show very high SAC and unprecedented energy efficiency, particularly when considering energy recovery upon discharge. These electrodes outperform control samples based on commercial high SSA AC as well as reported materials tested under similar salt concentrations, and lead to much lower energy consumed per gram of salt removed than established water purification methods.
Further improvements in CDI performance can be expected after optimisation of electrode morphology. Of particular interest is the increase in CNT fibre specific surface area, the use of other metal oxides and development of asymmetric devices.21,67,68 It would also be beneficial to gain more insight into the role of the various interfaces in the three phase system (electrolyte, MOx, CNTs) during CDI for water purification and ion removal. From a technological point of view, the high conductivity and flexibility in bending of the CNT fibre fabrics opens the possibility to make current collector-free electrodes with complex non-planar shapes that simplify integration and improve device performance. Work in this direction is in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr V. Reguero and Dr L. Cabana for the fabrication of CNTf-based electrodes, to Mr E. Senokos for discussions on CNTf electrochemical performance and cell assembly assistance and to Dr Yang Wang for discussions on capacitive deionization performance. J. J. Vilatela acknowledges financial support from the European Union Seventh and Eighth Framework Programs under Grant Agreement No. 678565 (ERC-STEM) and from the Ministry of Economy, Industry and Competitiveness (MINECO, RyC-2014-15115, Spain). Moreover, J. J. Lado acknowledges Comunidad de Madrid for the postdoctoral fellowship as part of the Young Talent Attraction Program (2016-T2/AMB-1310). IMDEA Energy acknowledges financial support from the MINECO through the National Program for Research Aimed at the Challenges of Society Program (RTC-2015-3969-5 (DC-SOIAS project)) and the cooperation of its participants (GS Inima Environment, S.A. Imdea Energy and Proingesa) is greatly acknowledged.Notes and references
- M. A. Anderson, A. L. Cudero and J. Palma, Electrochim. Acta, 2010, 55, 3845–3856 CrossRef.
- Y. Oren, Desalination, 2008, 228, 10–29 CrossRef.
- S. Porada, R. Zhao, A. Van Der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef.
- A. Subramani and J. G. Jacangelo, Water Res., 2015, 75, 164–187 CrossRef PubMed.
- E. García-Quismondo, R. Gómez, F. Vaquero, A. L. Cudero, J. Palma and M. A. Anderson, Phys. Chem. Chem. Phys., 2013, 15, 7648 RSC.
- A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Water Res., 2016, 104, 303–311 CrossRef PubMed.
- E. García-Quismondo, C. Santos, J. Lado and M. A. Anderson, Environ. Sci. Technol., 2013, 47, 11866–11872 CrossRef PubMed.
- P. Długołecki and A. Van Der Wal, Environ. Sci. Technol., 2013, 47, 4904–4910 CrossRef PubMed.
- E. García-Quismondo, C. Santos, J. Soria, J. Palma and M. A. Anderson, Environ. Sci. Technol., 2016, 50, 6053–6060 CrossRef PubMed.
- A. M. Pernia, F. J. Alvarez-Gonzalez, M. A. J. Prieto, P. J. Villegas and F. Nuno, IEEE Transactions on Power Electronics, 2014, 29, 3573–3581 CrossRef.
- J. C. Farmer, D. V. Fix, G. V. Mack, R. W. Pekala and J. F. Poco, J. Appl. Electrochem., 1996, 26, 1007–1018 CrossRef.
- R. Zhao, S. Porada, P. M. Biesheuvel and A. van der Wal, Desalination, 2013, 330, 35–41 CrossRef.
- T. J. Welgemoed, Capacitive Deionization Technology: Development and Evaluation of an industrial prototype system, University of Pretoria, 2005 Search PubMed.
- O. N. Demirer, R. M. Naylor, C. A. Rios Perez, E. Wilkes and C. Hidrovo, Desalination, 2013, 314, 130–138 CrossRef.
- C. Huyskens, J. Helsen and A. B. de Haan, Desalination, 2013, 328, 8–16 CrossRef.
- E. García-Quismondo, C. Santos, J. Palma and M. A. Anderson, Desalin. Water Treat., 2014, 984929, 1–10 CrossRef.
- M. Mossad and L. Zou, J. Hazard. Mater., 2013, 244–245, 387–393 CrossRef PubMed.
- L. Y. Lee, H. Y. Ng, S. L. Ong, G. Tao, K. Kekre, B. Viswanath, W. Lay and H. Seah, Water Res., 2009, 43, 4769–4777 CrossRef PubMed.
- F. A. AlMarzooqi, A. A. Al Ghaferi, I. Saadat and N. Hilal, Desalination, 2014, 342, 3–15 CrossRef.
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 Search PubMed.
- P. Liu, T. Yan, J. Zhang, L. Shi, D. Zhang, A. McBride, E. C. Kumbur, Y. Gogotsi, X. M. Sun, N. Noel, Z. Luo, Z. Guo and S. Wei, J. Mater. Chem. A, 2017, 5, 14748–14757 Search PubMed.
- Y. Liu, C. Nie, X. Liu, X. Xu, Z. Sun and L. Pan, RSC Adv., 2015, 5, 15205–15225 RSC.
- K. Shi and I. Zhitomirsky, Electrochim. Acta, 2015, 174, 588–595 CrossRef.
- K. Shi, M. Ren and I. Zhitomirsky, ACS Sustainable Chem. Eng., 2014, 2, 1289–1298 CrossRef.
- M.-Y. Lee, H. Kim, J.-O. Kim and S. Kang, Desalin. Water Treat., 2017, 90, 46–53 CrossRef.
- H. Duan, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Chem. Commun., 2017, 53, 7465–7468 RSC.
- H. Wang, T. Yan, L. Shi, G. Chen, J. Zhang and D. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 3329–3338 CrossRef.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 72–88 RSC.
- L. Wang, M. Wang, Z.-H. Huang, T. Cui, X. Gui, F. Kang, K. Wang and D. Wu, J. Mater. Chem., 2011, 21, 18295 RSC.
- A. Burke, J. Power Sources, 2000, 91, 37–50 CrossRef.
- Z. H. Huang, M. Wang, L. Wang and F. Kang, Langmuir, 2012, 28, 5079–5084 CrossRef PubMed.
- J. Liu, S. Wang, J. Yang, J. Liao, M. Lu, H. Pan and L. An, Desalination, 2014, 344, 446–453 CrossRef.
- T. Takahashi, M. I. Tejedor-Tejedor, J. J. Wouters, R. Perez-Roa and M. A. Anderson, J. Electrochem. Energy Convers. Storage, 2016, 7, 147–158 CrossRef.
- H. Li, T. Lu, L. Pan, Y. Zhang and Z. Sun, J. Mater. Chem., 2009, 19, 6773 RSC.
- P. Liu, T. Yan, L. Shi, H. S. Park, X. Chen, Z. Zhao and D. Zhang, J. Mater. Chem. A, 2017, 5, 13907–13943 Search PubMed.
- A. G. El-Deen, R. M. Boom, H. Y. Kim, H. Duan, M. B. Chan-Park and J. H. Choi, ACS Appl. Mater. Interfaces, 2016, 8, 25313–25325 Search PubMed.
- S. Wang, D. Wang, L. Ji, Q. Gong, Y. Zhu and J. Liang, Sep. Purif. Technol., 2007, 58, 12–16 CrossRef.
- F. Liu, R. M. Wagterveld, B. Gebben, M. J. Otto, P. M. Biesheuvel and H. V. M. Hamelers, Colloid Interface Sci. Commun., 2014, 3, 9–12 CrossRef.
- E. Senokos, V. Reguero, J. Palma, J. Vilatela and R. Marcilla, Nanoscale, 2016, 8, 3620–3628 RSC.
- J. J. Vilatela and R. Marcilla, Chem. Mater., 2015, 27, 6901–6917 CrossRef.
- J. Benson, I. Kovalenko, S. Boukhalfa, D. Lashmore, M. Sanghadasa and G. Yushin, Adv. Mater., 2013, 25, 6625–6632 CrossRef PubMed.
- B. Zhang, F. Kang, J. Tarascon and J. Kim, J. Process Mater. Sci., 2016, 76, 319–380 Search PubMed.
- L. Wen, L. Feng and H. Cheng, Adv. Mater., 2016, 28, 4306–4337 CrossRef PubMed.
- Y. Wang, H. Wei, Y. Lu, S. Wei, E. Wujcik and Z. Guo, Nanomaterials, 2015, 5, 755–777 CrossRef PubMed.
- K. C. Leonard, W. E. Suyama and M. A. Anderson, Langmuir, 2012, 28, 6476–6484 CrossRef PubMed.
- J. J. Lado, R. E. Perez-Roa, J. J. Wouters, M. Isabel Tejedor-Tejedor and M. A. Anderson, Sep. Purif. Technol., 2014, 133, 236–245 CrossRef.
- X. Gao, J. Landon, J. K. Neathery and K. Liu, J. Electrochem. Soc., 2013, 160, E106–E112 CrossRef.
- J. J. Wouters, J. J. Lado, M. I. Tejedor-Tejedor, R. Perez-Roa and M. A. Anderson, Electrochim. Acta, 2013, 112, 763–773 CrossRef.
- J. J. Lado, J. J. Wouters, M. I. Tejedor-Tejedor, M. A. Anderson and E. Garcia-Calvo, J. Electrochem. Soc., 2013, 160, E71–E78 CrossRef.
- K. C. Leonard, J. R. Genthe, J. L. Sanfilippo, W. A. Zeltner and M. A. Anderson, Electrochim. Acta, 2009, 54, 5286–5291 CrossRef.
- E. S. Im, J.-H. Choi and K. Y. Jung, J. Electrochem. Soc., 2012, 159, E198–E203 CrossRef.
- J. J. Wouters, M. I. Tejedor-Tejedor, J. J. Lado, R. Perez-Roa and M. A. Anderson, J. Electrochem. Soc., 2016, 163, A2733–A2744 CrossRef.
- A. Pendashteh, E. Senokos, J. Palma, M. A. Anderson, J. J. Vilatela and R. Marcilla, J. Power Sources, 2017, 372, 64–73 CrossRef.
- J. J. Wouters, J. J. Lado, M. I. Tejedor-Tejedor and M. A. Anderson, J. Electrochem. Soc., 2012, 159, A1374–A1382 CrossRef.
- J. J. Lado, Study of Asymmetric Capacitive Deionization Cells for Water Treatment Applications, Universidad de Alcalá, 2014 Search PubMed.
- F. S. Schultz and M. A. Anderson, J. Am. Chem. Soc., 1999, 121, 4933–4940 CrossRef.
- J. J. Lado, R. E. Perez-Roa, J. J. Wouters, M. I. Tejedor-Tejedor, C. Federspill, J. M. Ortiz and M. A. Anderson, Sep. Purif. Technol., 2017, 183, 145–152 CrossRef.
- P. Srimuk, M. Zeiger, N. Jäckel, A. Tolosa, B. Krüner, S. Fleischmann, I. Grobelsek, M. Aslan, B. Shvartsev, M. E. Suss and V. Presser, Electrochim. Acta, 2017, 224, 314–328 CrossRef.
- D. Iglesias, E. Senokos, B. Alemán, L. Cabana, C. Navío, R. Marcilla, M. Prato, J. J. Vilatela and S. Marchesan, ACS Appl. Mater. Interfaces, 2018, 10(6), 5760–5770 Search PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef.
- D. Sverjenski, Geochim. Cosmochim. Acta, 2001, 65, 3643–3655 CrossRef.
- J. Westall and H. Hohl, Adv. Colloid Interface Sci., 1980, 12, 265–294 CrossRef.
- W. B. S. De Lint, N. E. Benes, J. Lyklema, H. J. M. Bouwmeester, A. J. Van der Linde and M. Wessling, Langmuir, 2003, 19, 5861–5868 CrossRef.
- M. M. Jaramillo, A. Mendoza, S. Vaquero, M. A. Anderson, J. Palma and R. Marcilla, RSC Adv., 2012, 2, 8439 RSC.
- G. Ayalneh Tiruye, D. Muñoz-Torrero, J. Palma, M. A. Anderson and R. Marcilla, J. Power Sources, 2015, 279, 472–480 CrossRef.
- Y. Qu, T. F. Baumann, J. G. Santiago and M. Stadermann, Environ. Sci. Technol., 2015, 49, 9699–9706 CrossRef PubMed.
- W. Cai, J. Yan, T. Hussin and J. Liu, Electrochim. Acta, 2017, 225, 407–415 CrossRef.
- P. Liu, H. Wang, T. Yan, J. Zhang, L. Shi and D. Zhang, J. Mater. Chem. A, 2016, 4, 5303–5313 Search PubMed.
- B. Jia and W. Zhang, Nanoscale Res. Lett., 2016, 11, 64 CrossRef PubMed.
- Z. Li, B. Song, Z. Wu, Z. Lin, Y. Yao, K. S. Moon and C. P. Wong, Nano Energy, 2015, 11, 711–718 CrossRef.
- X. Wen, D. Zhang, T. Yan, J. Zhang and L. Shi, J. Mater. Chem. A, 2013, 1, 12334 Search PubMed.
- D. He, C. E. Wong, W. Tang, P. Kovalsky and T. D. Waite, Environ. Sci. Technol. Lett., 2016, 3, 222–226 CrossRef.
- N. Holubowitch, A. Omosebi, X. Gao, J. Landon and K. Liu, ChemElectroChem, 2017, 4(9), 2404–2413 CrossRef.
- B. Chen, Y. Wang, Z. Chang, M. Li, X. Liu, L. Zhang and X. Wang, RSC Adv., 2016, 6, 6730–6736 RSC.
- I. Cohen, E. Avraham, Y. Bouhadana, A. Soffer and D. Aurbach, Electrochim. Acta, 2015, 153, 106–114 CrossRef.
- J. J. Lado, R. E. Perez-Roa, J. J. Wouters, M. I. Tejedor-Tejedor, C. Federspill and M. A. Anderson, J. Environ. Chem. Eng., 2015, 3, 2358–2367 CrossRef.
- A. Al-Karaghouli and L. L. Kazmerski, Renewable Sustainable Energy Rev., 2013, 24, 343–356 CrossRef.
- S. S. Shenvi, A. M. Isloor and A. F. Ismail, Desalination, 2015, 368, 10–26 CrossRef.
- K. Laxman, M. T. Z. Myint, R. Khan, T. Pervez and J. Dutta, Electrochim. Acta, 2015, 166, 329–337 CrossRef.
- X. Gao, A. Omosebi, J. Landon and K. Liu, Environ. Sci. Technol., 2015, 49, 10920–10926 CrossRef PubMed.
- Y.-L. Li, I. A. Kinloch and A. H. Windle, Science, 2004, 304, 276–278 CrossRef PubMed.
- L. Chu, M. I. Tejdor-Tejedor and M. A. Anderson, Microporous Mater., 1997, 8, 207–213 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CDI technology and experimental set-up; fabrication of MOx/CNTf electrodes; electron micrographs; TG analysis; cyclic voltammetry; theoretical analysis of water wetting/wicking electrodes with different MOx mass fraction. Fig. S1–S8 and Table S1. See DOI: 10.1039/c8ta01128a |
| This journal is © The Royal Society of Chemistry 2018 |