
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A free standing Ru–TiC nanowire array/carbon textile cathode with enhanced stability for Li–O2 batteries†
Chenjuan
Liu
a,
Zhen
Qiu
b,
Willian R.
Brant
a,
Reza
Younesi
 a,
Yue
Ma
c,
Kristina
Edström
a,
Torbjörn
Gustafsson
a and
Jiefang
Zhu
*a
a,
Yue
Ma
c,
Kristina
Edström
a,
Torbjörn
Gustafsson
a and
Jiefang
Zhu
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 538, SE-751 21 Uppsala, Sweden. E-mail: jiefang.zhu@kemi.uu.se; Fax: +46 18 51 3548; Tel: +46 18 471 3722
bDepartment of Engineering Sciences, Solid State Physics, Uppsala University, Box 534, 75121 Uppsala, Sweden
cState Key Laboratory of Solidification Processing, Center for Nano Energy Materials, School of Materials Science and Engineering, Northwestern Polytechnical University, 710072 Xi'an, China
First published on 24th October 2018
Abstract
The instability of carbon cathode materials is one of the key problems that hinder the development of lithium–air/lithium–oxygen (Li–O2) batteries. In this contribution, a type of TiC-based cathode is developed as a suitable alternative to carbon based cathodes, and its stability with respect to its surface properties is investigated. Here, a free-standing TiC nanowire array cathode was in situ grown on a carbon textile, covering its exposed surface. The TiC nanowire array, via deposition with Ru nanoparticles, showed enhanced oxygen reduction/evolution activity and cyclability, compared to the one without Ru modification. The battery performance of the Li–O2 cells with Ru–TiC was investigated by using in operando synchrotron radiation powder X-ray diffraction (SR-PXD) during a full cycle. With the aid of surface analysis, the role of the cathode substrate and surface modification is demonstrated. The presented results are a further step toward a wise design of stable cathodes for Li–O2 batteries.
Introduction
Non-aqueous Li–air (Li–O2) batteries have been considered as a promising candidate for energy storage due to their significantly higher theoretical energy density, compared to conventional Li-ion batteries.1,2 The functioning of a Li–O2 battery relies on the reversible deposition and decomposition of the discharge product, Li2O2, on the surface of the cathode during cycling. Although the working mechanism of Li–O2 batteries appears straightforward, some challenges, such as low energy efficiency, high over-potential, and poor cyclability, hinder the commercialization of Li–O2 batteries. Today, it is well accepted that these problems mostly originate from the nature of the discharge products. These partly reduced oxygen species are highly reactive, can attack the electrolyte and the cathode, and induce irreversible side reactions.3 Up to now, some progress has been achieved in improving the electrochemical performance of Li–O2 batteries during both discharge and charge.4,5 However, fundamental questions remain, concerning the chemical and electrochemical stability of all functional components during cycling.6–10In this study, with the aid of the SR-PXD technique, we focus on improving cathode stability. Among all cathode materials, carbon is mostly used in non-aqueous Li–O2 batteries due to its abundance, light weight, high conductivity and tuneable porous structure.11–13 However, many groups have identified the instability of the carbon materials that have been used in the Li–O2 system.14,15 Consequently, a few strategies have been tried, aimed at increasing the cathode substrate stability.16–19 Owing to their outstanding anti-corrosion ability and good conductivity, carbides as a cathode material have recently aroused increasing interest.18,20,21 TiC was reported as a promising cathode material when coupled with a DMSO electrolyte and a LiFePO4 anode, showing >98% capacity retention after 100 cycles.18 This excellent performance was attributed to the good electronic conductivity of TiC and the stability of a TiO2-rich layer formed on the TiC surface. Later, Adams et al. showed that the oxygen evolution reaction (OER) upon charging was strongly dependent on the TiC cathode surface properties. The thickness of the TiO2 layer on the TiC surface was a key parameter determining the function of the TiC cathode.21 Kozmenkova et al. found that a clean TiC surface could be easily oxidized by Li2O2, and an oxidation treatment using molecular oxygen could improve TiC stability. However, they also claimed that such a complicated protective coating procedure is not practically useful for a real battery system.22 In addition, there are some shortcomings in the commonly used cathode design. Conventional cathodes with a powder and binder mixture introduce instability problems due to binder decomposition.9,23 To facilitate electron transfer to and from insulating Li2O2 during cycling, a rational design of electronic and ionic pathways in cathodes is essential. Compared to powder particles, one-dimensional (1D) nanostructures on the cathode surface can offer directional charge transportation along the 1D structure, diminishing boundary resistance.24,25 1D nanostructures, e.g. nanowires, can provide facile charge transfer, exhibit improved surface chemistry and catalytic properties, and enhance the kinetics of electrochemical reactions.26 Besides, a metallic current collector is normally used in a Li–O2 battery, which increases the weight of the battery, thus decreasing the total gravimetric capacity. It would be very meaningful to replace metallic current collectors with light conductive materials.
The formation and decomposition of Li2O2 have been studied by ex situ X-ray diffraction (XRD), Raman spectroscopy, transmission electron microscopy (TEM), nuclear magnetic resonance spectroscopy, FT-IR and X-ray absorption near edge structure spectroscopy.12,13,16,27–29 However, artifacts from sample preparation are difficult to avoid and very limited information on the operation mechanism and reaction kinetics of Li2O2 formation and oxidation could be obtained. In operando techniques including spectroscopy, microscopy and diffraction with their feature of collecting data during electrochemical cycling without unpacking the cell, have provided valuable information on the reversibility of Li2O2.30–35 However, it is still difficult to obtain the quantified efficiency of the cell by these techniques. Although differential electrochemical mass spectroscopy (DEMS) can provide quantification information in real-time, the targets are limited to gas species which are only evolved from the charge process.36 To date, only a few studies have successfully performed in situ or in operando measurement by in-house XRD to investigate the evolution of Li2O2.37 However, due to the limited X-ray intensity of in-house X-ray sources and the poor scattering ability of Li2O2, a long irradiation time is required and the kinetic information obtained during the process is lost. As summarized in Table S1,† with a higher X-ray intensity and better time resolution, in operando synchrotron radiation powder X-ray diffraction (SR-PXD), which was recently developed by us,38 can provide an efficient, accurate and reliable evaluation of the efficiency of Li–O2 electrochemical reactions during both discharge and charge.
Based on the aforementioned considerations, we aim to design and fabricate an electrochemically stable and efficient cathode for Li–O2 batteries. One of our co-authors developed a biotemplating method for TiC nanowire synthesis.50 Inspired by that work, here a TiC nanowire array (TiC NA) with a TiO2 surface layer was in situ grown on a carbon textile (CT). By this method, a binder-free cathode was obtained. Since the performance of Li–O2 batteries largely hinges on the surface chemistry of the cathode, the surface of TiC NAs/CT was modified by loading Ru nanoparticles. Ru- or RuO2 based electrocatalysts have been reported as bifunctional catalysts with a much lower cost compared to Pt, Pd and Au,19,39,40 and do not promote electrolyte decomposition.41 Galvanostatic tests and X-ray photoelectron spectroscopy (XPS) analysis confirmed the crucial effect of these Ru NPs. Unlike previous reports on TiC cathodes, this study features unique aspects: (1) the usage of polymer binders is avoided by the direct growth of TiC on the CT; (2) the flexible CT functions as a conductive backbone with its surface coated by TiC NAs, which act as a physical barrier to prevent the exposure of the carbon cloth to the electrolyte; (3) the surface of the TiC nanowires is decorated with Ru NPs, which enhance the electrochemical performance of the Li–O2 battery and improve the stability of TiC during battery operation; (4) SR-PXD and in-house XRD quantitatively track the phase evolution inside the Li–O2 battery.
Results and discussion
Characterization of Ru–TiC nanoarrays
The morphology of the as-prepared TiC electrode was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The pristine CT is composed of carbon fibres with a diameter about 8 μm (the inset of Fig. 1a). Fig. 1a–d show that the TiC NAs were grown homogeneously and perpendicular to the surface of the CT skeleton without using any binder. Here the CT served as both the current collector and the structural support of the TiC NAs. As shown in Fig. 1a–c, the whole piece of the CT is covered with TiC NAs with a length of 30–50 μm and a width of 200–500 nm. The cross section (the inset of Fig. 1b) of one TiC NAs/CT shows the core–shell configuration, which can not only protect the CT from attack by partly reduced oxygen species, but also facilitate electron transfer throughout the cathode. The two strong peaks at 1348 and 1590 cm−1 (Fig. 1g) correspond to the D and G bands of carbon cloth, respectively. The Raman spectra shown in Fig. 1g and h reveal that the D- and G-band from the pristine CT almost vanished in the TiC NAs/CT sample. The Raman peak at 144 cm−1 can be attributed to a small amount of rutile TiO2 (as shown in Fig. S1†), while the peaks at 257, 424, and 604 cm−1 (Fig. 1h) are assigned to the vibration signals of TiC.42 This indicates the effective coverage of the CT surface by the TiC NAs. The XRD (Fig. 1i) pattern shows that the as-prepared electrode consists of TiC and CTs. The pattern peaks at 36.0°, 41.8°, 60.7°, 72.6°, and 76.5° are indexed as the (111), (200), (220), (311), and (222) reflections of the cubic TiC (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, ICSD # 1546), respectively. The HRTEM image taken from an individual TiC nanowire (Fig. 1d) shows that the d-spacing value of the adjacent lattice fringes is 0.246 nm, corresponding to the (111) lattice plane of the cubic TiC. Prior to Ru loading (Fig. 1c and d), the nanowires have a smooth surface. After loading Ru nanoparticles, the TiC nanowires still maintain the same shape (Fig. 1e and f), but the surface roughens due to the deposition of Ru nanoparticles. Fig. 1f shows the HRTEM image of Ru–TiC NAs/CT with a Ru particle size of about 2–5 nm. The lattice spacing values in Fig. 1f are 0.247 nm and 0.214 nm, corresponding to the (111) lattice plane of cubic TiC and (002) lattice plane of hexagonal-structured Ru (P63/mmc, ICSD # 40354), respectively. The EDS mapping (Fig. S2†) also shows the distribution of C, Ti, O, Ru, and Ni at the top part of the TiC NAs. The Ni NPs located at the tip of the TiC NAs (the inset of Fig. 1c) acted as the catalyst for the growth of the TiC NAs.43,44
m, ICSD # 1546), respectively. The HRTEM image taken from an individual TiC nanowire (Fig. 1d) shows that the d-spacing value of the adjacent lattice fringes is 0.246 nm, corresponding to the (111) lattice plane of the cubic TiC. Prior to Ru loading (Fig. 1c and d), the nanowires have a smooth surface. After loading Ru nanoparticles, the TiC nanowires still maintain the same shape (Fig. 1e and f), but the surface roughens due to the deposition of Ru nanoparticles. Fig. 1f shows the HRTEM image of Ru–TiC NAs/CT with a Ru particle size of about 2–5 nm. The lattice spacing values in Fig. 1f are 0.247 nm and 0.214 nm, corresponding to the (111) lattice plane of cubic TiC and (002) lattice plane of hexagonal-structured Ru (P63/mmc, ICSD # 40354), respectively. The EDS mapping (Fig. S2†) also shows the distribution of C, Ti, O, Ru, and Ni at the top part of the TiC NAs. The Ni NPs located at the tip of the TiC NAs (the inset of Fig. 1c) acted as the catalyst for the growth of the TiC NAs.43,44
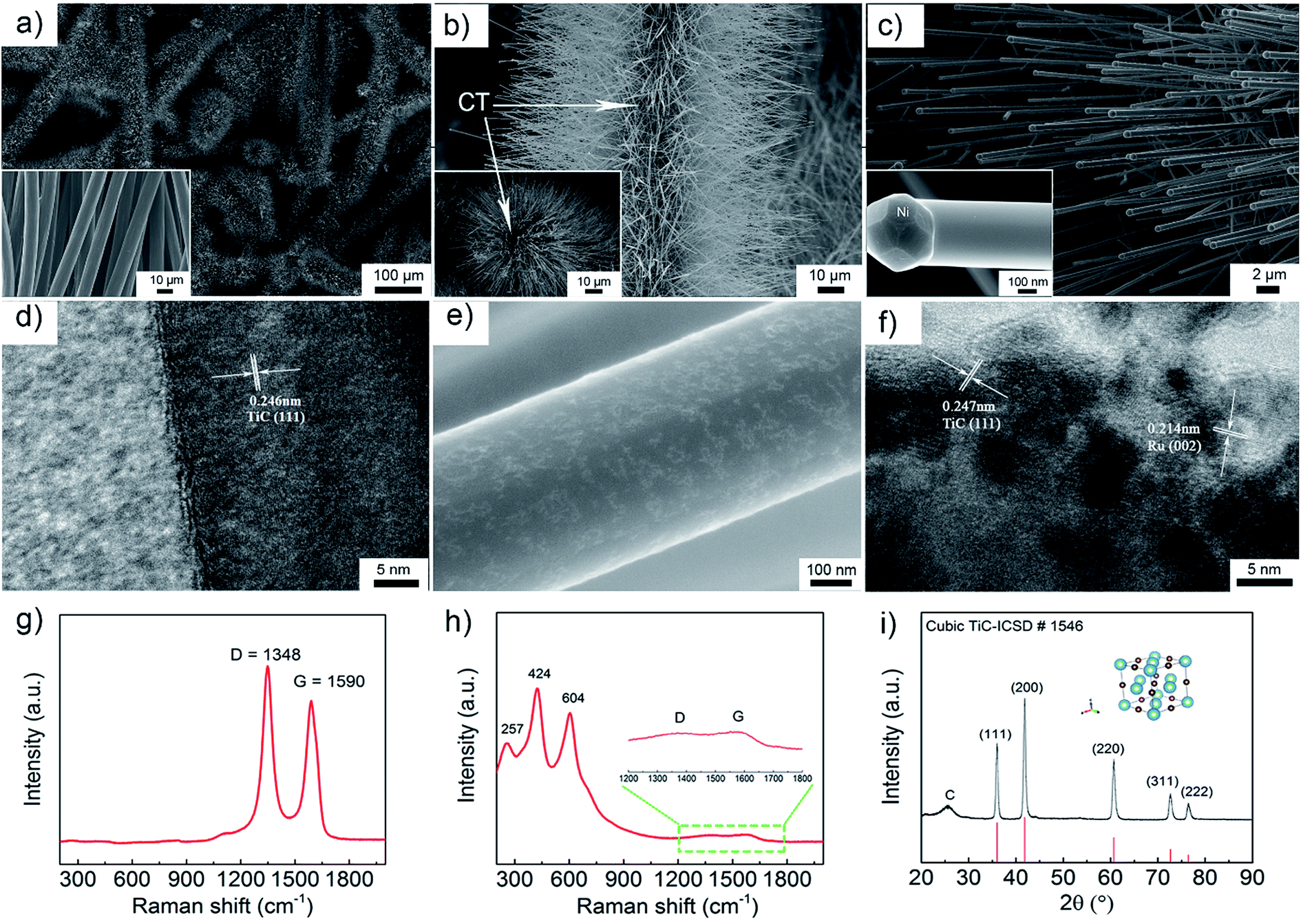 | ||
| Fig. 1 (a–c) SEM images of TiC arrays on the carbon cloth textile with different magnifications. The inset of (a) shows the pristine CT, the inset of (b) shows a cross section of TiC NAs/CT and the inset of (c) shows the tip of a TiC nanowire. (d) High magnification TEM image of a TiC nanowire. High magnification (e) SEM and (f) TEM images of Ru/TiC. Raman spectra of (g) the pristine CT and (h) TiC NAs/CT. (i) XRD pattern (λ = 1.5406 Å) of the as-prepared TiC NAs/CT. | ||
XPS was performed to investigate the elemental oxidation states and the surface composition of the as-prepared samples.
The C 1s core level XPS spectra of the pristine CT, TiC NAs/CT and Ru–TiC NAs/CT are compared in Fig. 2. For pristine CT, the carbon peak was deconvoluted into five peaks at 284.5 eV, 285.0 eV, 286.6 eV, 287.6 eV and 290.3 eV, assigned to C–C, C–H, C–O, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –CO3, respectively.45 For the TiC NAs/CT composites, the C–Ti bond in the carbide phase and the C–C bond in the carbon matrix phase were clearly found at 281.6 eV and 284.5 eV, respectively. In addition, a contribution from TiOxCy at ∼282.4 eV was observed in accordance with previous studies.22 In Ru–TiC NAs/CT, the peaks at ∼280.2 eV and ∼284.4 eV can be attributed to the two spin orbit coupling peaks of Ru.
O and –CO3, respectively.45 For the TiC NAs/CT composites, the C–Ti bond in the carbide phase and the C–C bond in the carbon matrix phase were clearly found at 281.6 eV and 284.5 eV, respectively. In addition, a contribution from TiOxCy at ∼282.4 eV was observed in accordance with previous studies.22 In Ru–TiC NAs/CT, the peaks at ∼280.2 eV and ∼284.4 eV can be attributed to the two spin orbit coupling peaks of Ru.
 | ||
| Fig. 2 C 1s and Ru 3d core-level XPS spectra of pristine CT, TiC NAs/CT and Ru–TiC NAs/CT fitted with multiple envelopes. | ||
Efficiency of Li2O2 formation and decomposition
In operando SR-PXD (λ = 0.994 Å) was applied to track the evolution and quantify the amount of Li2O2 during the cycling of the Li–O2 cell using a Ru–TiC NAs/CT cathode and 1 M LiClO4 DMSO-based electrolyte. The cell was run for a complete discharge and charge cycle at a current density of 0.1 mA cm−2. A series of diffraction patterns containing a Si peak and two Li2O2 peaks are shown in Fig. 3a. A known amount of Si was added as a reference to quantify the amount of Li2O2 during the cycling. When the Li–O2 cell is discharged/charged with a constant current, a linear formation/decomposition of Li2O2 with time is expected in the absence of side reactions. According to our previous results, the amount of Li2O2 was fitted into two linear regions during discharge (Fig. 3b).38 During the first discharge stage, the rate (the slope in Fig. 3b) of Li2O2 formation is kf1 = 0.068 mg h−1, which is slightly lower than the theoretical value of ktheo = 0.086 mg h−1. After about 10 hours of discharge, a lower formation rate was observed (kf2 = 0.042 mg h−1). The decreasing formation rate is probably due to side reactions occurring during the later stage of discharge, such as the decomposition of the electrolyte. This two-stage model agrees with the two-step Li2O2 growth during discharge.38 Due to an uncontrollable beam loss, we could not collect diffraction patterns from 14 h to 24 h, which covered the end of discharge and the beginning of charge. As charging proceeded, a linear decrease of the electrochemically formed Li2O2 can be observed during the later stage of the charge with kd = −0.073 mg h−1, which is close to the ideal decomposition rate of Li2O2, ktheo = −0.086 mg h−1. This indicates an oxidation of bulk Li2O2 with fewer parasitic reactions. We also notice that the (101) diffraction peak of electrochemically formed Li2O2, at around 22° in Fig. 3a, is broad and overlaps with the TiC (111) peak. This peak broadening does not affect the quantification, since the quantification refinement was determined by the integrated peak area.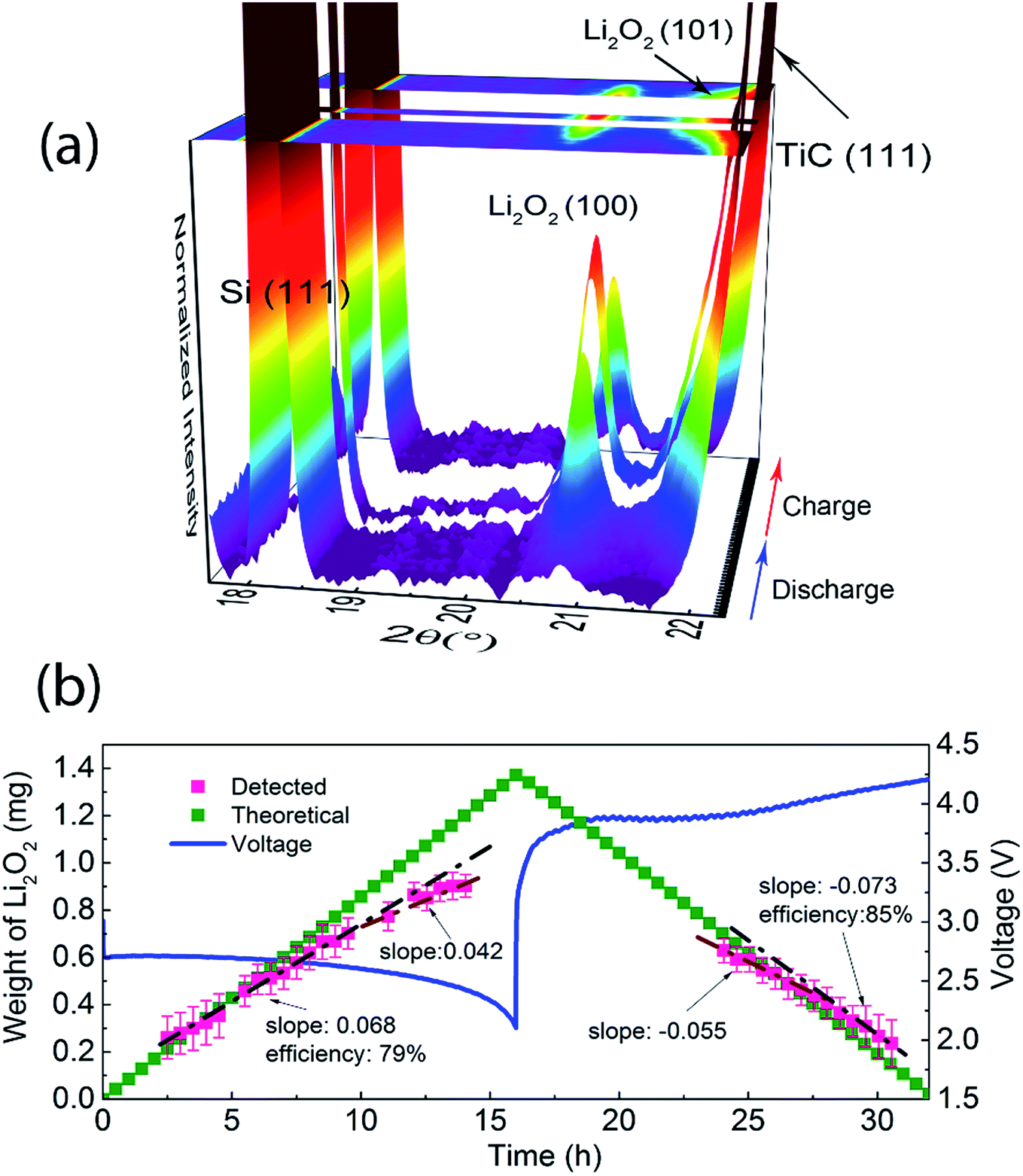 | ||
| Fig. 3 (a) In operando SR-PXD pattern (λ = 0.994 Å) of a Ru–TiC NAs/CT based Li–O2 battery, collected every 30 min during the first cycle at a constant current of 0.1 mA cm−2, (b) galvanostatic discharge–charge voltage of the Li–O2 cell and the Li2O2 mass evolution during the first cycle. The gap in the 3D plot and in the Li2O2 mass evolution corresponds to an unexpected beam loss. | ||
To investigate the OER process while eliminating the influence of electrolyte decomposition on discharge, a cell with a Ru–TiC NAs/CT cathode loaded with crystalline commercial Li2O2 powder was studied from the beginning of the charging process. As the commercial Li2O2 has better crystallinity than the electrochemically formed one, sharper diffraction peaks were observed (Fig. 4a). This results in a better resolution between the Li2O2 (101) and the TiC (111) diffraction peaks. The fading of the diffraction peaks at 32.7° and 34.8° (X-ray wavelength λ = 1.5406 Å) in Fig. 4a, corresponding to the (100) and (101) peak of Li2O2 (ICSD # 25530 P63/mmc space group), proves the electrochemical decomposition of Li2O2 during the charge process. Following the evolution of Li2O2 during charge (Fig. 4b), the oxidation of Li2O2 took place in two stages. The residual ratio of Li2O2 (calculation details are described in the ESI†) was static at the short initial step, and then declined linearly during the next stage. At the beginning of charging, the detected amount of Li2O2 was almost constant, probably due to the preferential decomposition of the surface impurities on Li2O2. The major stage shows a decomposition rate of −1.28 h−1, and the theoretical value (−1.38 h−1) is also given as a reference in Fig. 4b. This indicates that Ru–TiC NAs/CT is an efficient cathode, providing an oxidation efficiency around 93%. As demonstrated in Fig. 4b, the charge potential of the commercial Li2O2-loaded cell was higher than that with electrochemically formed Li2O2 shown in Fig. 3b, probably due to the larger particle size of commercial Li2O2 and its poorer contact with the electrode. Different from a carbon based electrode, we did not observe a sharp voltage rise and drop at the initial stage of charge here, which is due to the reaction between carbon and Li2O2 at their interface.32 This further confirms the good protection of the carbon surface by the TiC NAs in the prepared samples.
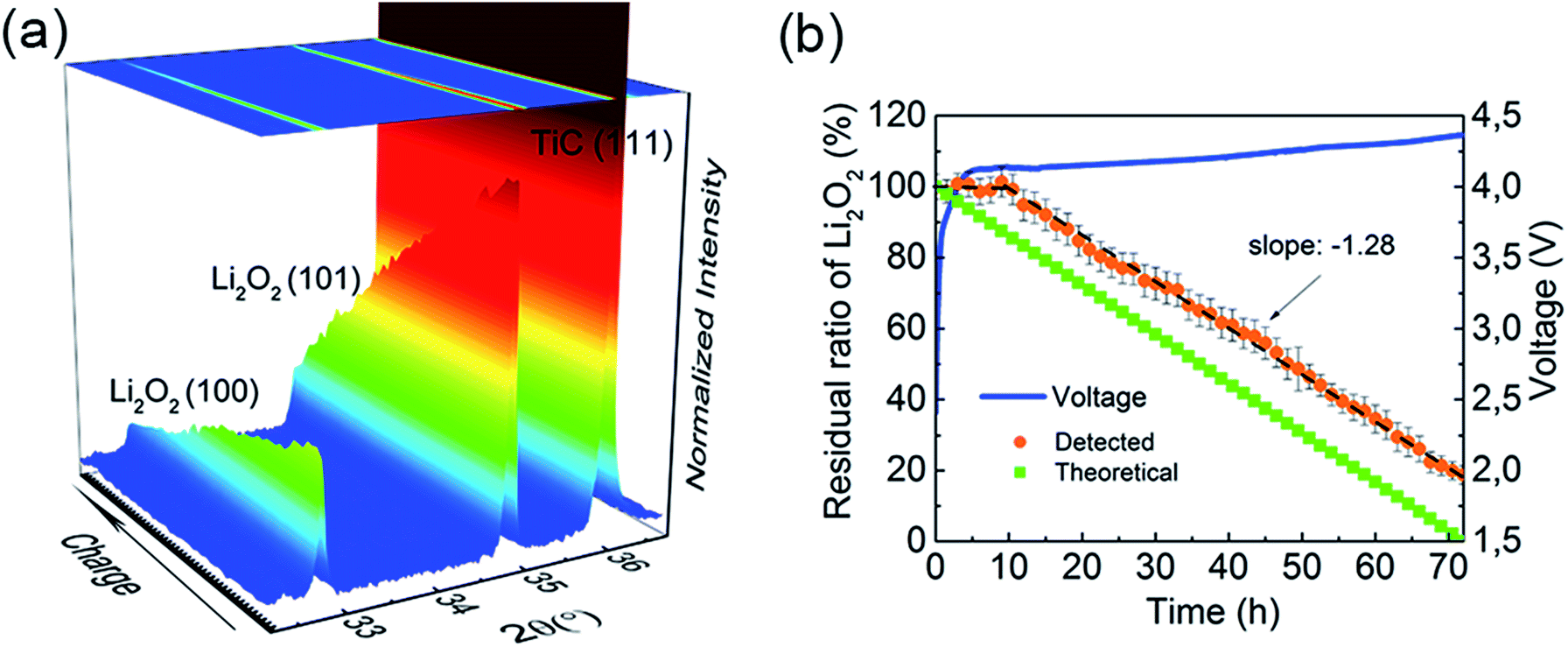 | ||
| Fig. 4 Galvanostatic charging of commercial Li2O2. (a) 3D plots of the diffraction patterns (λ = 1.5406 Å) presented as a function of time during charge with a constant current of 0.2 mA cm−2. (b) The charging profile, integrated curve and linear fitting of Li2O2 decomposition during the in operando XRD measurement. | ||
Morphology evolution
Since Li2O2 is very sensitive to the electron beam of the TEM, which could lead to Li2O2 degradation,46,47 SEM was employed to elucidate the morphological evolution of the discharge product after different stages of cycling. As shown in Fig. 5a and b, after the first discharge process, small particles were uniformly distributed on the Ru–TiC nanowire surface with an average size around 200 nm. These particles have been suggested to be Li2O2.48,49 After the first charge process, the particles disappeared and the smooth Ru–TiC surface reappeared (Fig. 5c and d). During the second discharge process, as shown in Fig. 5e and f, Li2O2 appeared again. Overall, the SEM images confirmed that Li2O2 was reversibly formed and decomposed on the surface of Ru–TiC, at least for the first few cycles. | ||
| Fig. 5 SEM images with different magnifications of the Ru/TiC cathode after the (a and b) 1st discharge, (c and d) 1st charge, and (e and f) 2nd discharge at 0.05 mA cm−2 current density with a limited capacity of 0.2 mA h cm−2. | ||
Battery performance
To highlight the merits of the as-prepared electrode, bare CT and TiC NAs/CT were used as the cathodes for comparison. Fig. 6a displays the complete discharge curves (with a cut off potential of 2.3 V) of the cells with bare CT, as-prepared TiC NAs/CT and Ru–TiC NAs/CT cathodes at a current density of 0.1 mA cm−2. The Li–O2 cell with pristine CT exhibits a capacity of about 0.6 mA h cm−2, while the cells with TiC NAs/CT and Ru–TiC NAs/CT show a capacity of about 1.2 mA h cm−2 and 1.6 mA h cm−2, respectively. According to the mass of TiC (determined from the thermal gravimetric analysis (TGA) in Fig. S3† and the inductively coupled plasma (ICP) analysis in Table S3†), the TiC NAs/CT and Ru–TiC NAs/CT cathodes showed a capacity of about 352 mA h gTiC−1 and 468 mA h gTiC−1, respectively (corresponding to 155 mA h g−1 and 206 mA h g−1 based on the total weight of the cathode, respectively; ESI, Fig. S4†). The current density used here was about 29.3 mA gTiC−1 (corresponding to 12.9 mA h g−1 based on the total weight of the cathode). The discharge voltage of Ru–TiC NAs/CT is slightly higher than those of pristine CT and TiC NAs/CT (Fig. 6b). This result was further supported by the electrochemical impedance spectroscopy (EIS) results in Fig. S5.† As shown in Fig. 6b, compared with using pristine CT, the first discharge–charge overpotential can be significantly decreased by using TiC NAs/CT, and can be further reduced by using Ru–TiC NAs/CT. Based on the first discharge–charge curves in Fig. 6b, the overpotentials of the cells can be estimated to be ∼1.65 V, ∼1.12 V and ∼0.91 V for the CT, TiC NAs/CT and Ru–TiC NAs/CT cathodes, respectively. The reduced overpotential of Ru–TiC NAs/CT can be ascribed to the intrinsic contact between the TiC NAs and the CT as well as the catalytic activity of Ru nanoparticle-modified TiC. In addition, the cyclic voltammetry (CV) measurements (Fig. S6†) also reveal that with Ru modification, the Ru–TiC NAs/CT cathode exhibits a higher catalytic activity, compared to the TiC NAs/CT cathode.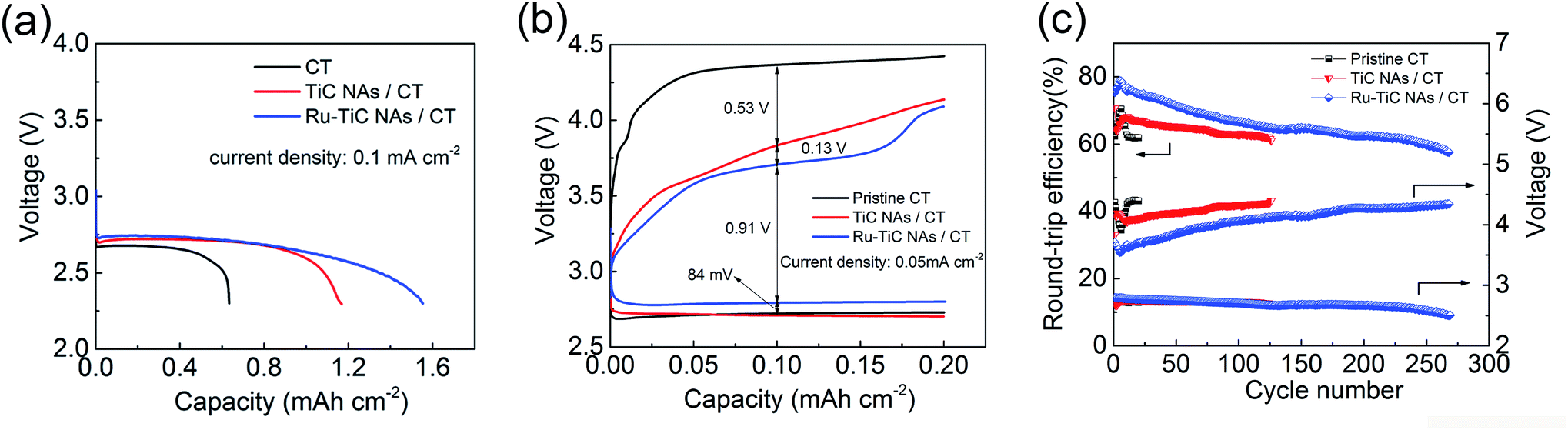 | ||
| Fig. 6 (a) Full discharge of the Li–O2 cells with bare CT, as-prepared TiC NAs/CT and Ru–TiC NAs/CT cathodes with a cut-off voltage at 2.3 V and a current density of 0.1 mA cm−2. (b) First discharge–charge curves of the Li–O2 cells with bare CT, as-prepared TiC NAs/CT and Ru–TiC NAs/CT cathodes at a current density of 0.05 mA cm−2. (c) Round-trip efficiencies and half-capacity voltages for each cycle of the Li–O2 batteries with pristine CT, TiC NAs/CT and Ru–TiC NAs/CT cathodes at a current density of 0.05 mA cm−2 with a limited capacity of 0.2 mA h cm−2. | ||
Considering the rate performance of the Ru–TiC NAs/CT cells as shown in Fig. S7,† a current density of 0.05 mA cm2 was chosen to cycle all the batteries. The selected voltage profiles of the pristine CT, TiC NAs/CT and Ru–TiC NAs/CT cathodes with a limited capacity of 0.2 mA h cm−2 are presented in Fig. S8.† An increase in the discharge and charge potentials in the 260th cycle is observed with the Ru–TiC NAs/CT cathode, compared to the 10th cycle, which could be attributed to the electrolyte decomposition and the accumulation of side products during cycling. The capacity limiting cycling method applied here does not present the whole image of the cyclability of the Li–O2 cell. A large number of cycles do not guarantee good reversibility, since they could still originate from side reactions. However, comparing the cycling performance for different cathode materials at the same cycling conditions can, to some extent, reflect the activity and the stability of the cathode materials toward the Li2O2 formation and decomposition process. As summarized in Fig. 6c, with the same current density and capacity limitation, the cell with Ru–TiC NAs/CT can operate for around 270 cycles, which is much longer than for the cells with TiC NAs/CT (around 125 cycles) and pristine CT electrodes (about 15 cycles). The charge potentials of all the cells decreased during the first few cycles and then increased continuously upon cycling. As shown in Fig. 6c, the average charge potential of the cell with Ru–TiC NAs/CT is lower than those of the cells with TiC NAs/CT and pristine CT. To calculate the round-trip efficiencies, the average discharge potential was divided by the average charge potential, and the results are shown in Fig. 6c. The round-trip efficiency of the cell with Ru–TiC NAs/CT is consistently higher than those of the cells with TiC NAs/CT and pristine CT. In addition, the instability of the cell with the CT electrode was also revealed by ex situ XRD results. As shown in Fig. S9,† LiOH was formed during discharge, which indicates the instability of the DMSO electrolyte in contact with the carbon cathode.
Stability information from the XPS study
The stability of the TiC cathode in Li–O2 batteries was reported to be related to the surface chemistry, where a layer of TiO2 was formed during cycling.18,22 However, the thickness of the oxidized layer could be the factor that determines the electron transfer kinetics through it. In this study, due to the surface modification using Ru NPs, we observed better cyclability of the cell with Ru–TiC NAs/CT than that with TiC NAs/CT. To investigate the function of Ru decoration on the TiC surface, XPS measurements were conducted. As shown in Fig. 7, the XPS results indicate that the chemical states of Ti on the two pristine electrodes (TiC NAs/CT and Ru–TiC NAs/CT) were identical (except for the presence of Ru), but their surface chemistry differed after cycling. As calculated from the peak areas of the fitted Ti 2p XPS spectra, the surface ratios of TiC/TiOxCy/TiO2 are 0.64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.17:0.19 and 0.62:0.17:0.21 for pristine TiC NAs/CT and pristine Ru–TiC NAs/CT, respectively. However, after the first discharge, the surface ratios of TiC/TiOxCy/TiO2 changed to 0.46:0.22:0.32 and 0.60:0.12:0.29 for TiC NAs/CT and Ru–TiC NAs/CT, respectively. Without the surface modification using Ru NPs, the proportion of TiO2 on the TiC NAs/CT cathode increased more quickly and the proportion of TiC decreased faster than on the Ru–TiC NAs/CT cathode during cycling. Combined with the Raman spectrum in Fig. 1h showing a small peak of rutile TiO2 and the XRD data in Fig. 3a showing only TiC diffraction peaks, we conclude that the surface layer of TiO2 on the as-prepared TiC is either very thin or amorphous. However, the increased amount of TiO2 on the surface of TiC NAs/CT during extended cycling hinders electron transfer.21 After 125 cycles, the TiC NAs/CT cathode surface was dominated by TiO2 and unknown species, indicated by binding energies around 460 eV and 466 eV, which did not show up in the Ru–TiC NAs/CT sample. We conclude that with Ru modification, the stability of the TiO2-rich TiC surface was largely improved.
:0.17:0.19 and 0.62:0.17:0.21 for pristine TiC NAs/CT and pristine Ru–TiC NAs/CT, respectively. However, after the first discharge, the surface ratios of TiC/TiOxCy/TiO2 changed to 0.46:0.22:0.32 and 0.60:0.12:0.29 for TiC NAs/CT and Ru–TiC NAs/CT, respectively. Without the surface modification using Ru NPs, the proportion of TiO2 on the TiC NAs/CT cathode increased more quickly and the proportion of TiC decreased faster than on the Ru–TiC NAs/CT cathode during cycling. Combined with the Raman spectrum in Fig. 1h showing a small peak of rutile TiO2 and the XRD data in Fig. 3a showing only TiC diffraction peaks, we conclude that the surface layer of TiO2 on the as-prepared TiC is either very thin or amorphous. However, the increased amount of TiO2 on the surface of TiC NAs/CT during extended cycling hinders electron transfer.21 After 125 cycles, the TiC NAs/CT cathode surface was dominated by TiO2 and unknown species, indicated by binding energies around 460 eV and 466 eV, which did not show up in the Ru–TiC NAs/CT sample. We conclude that with Ru modification, the stability of the TiO2-rich TiC surface was largely improved.
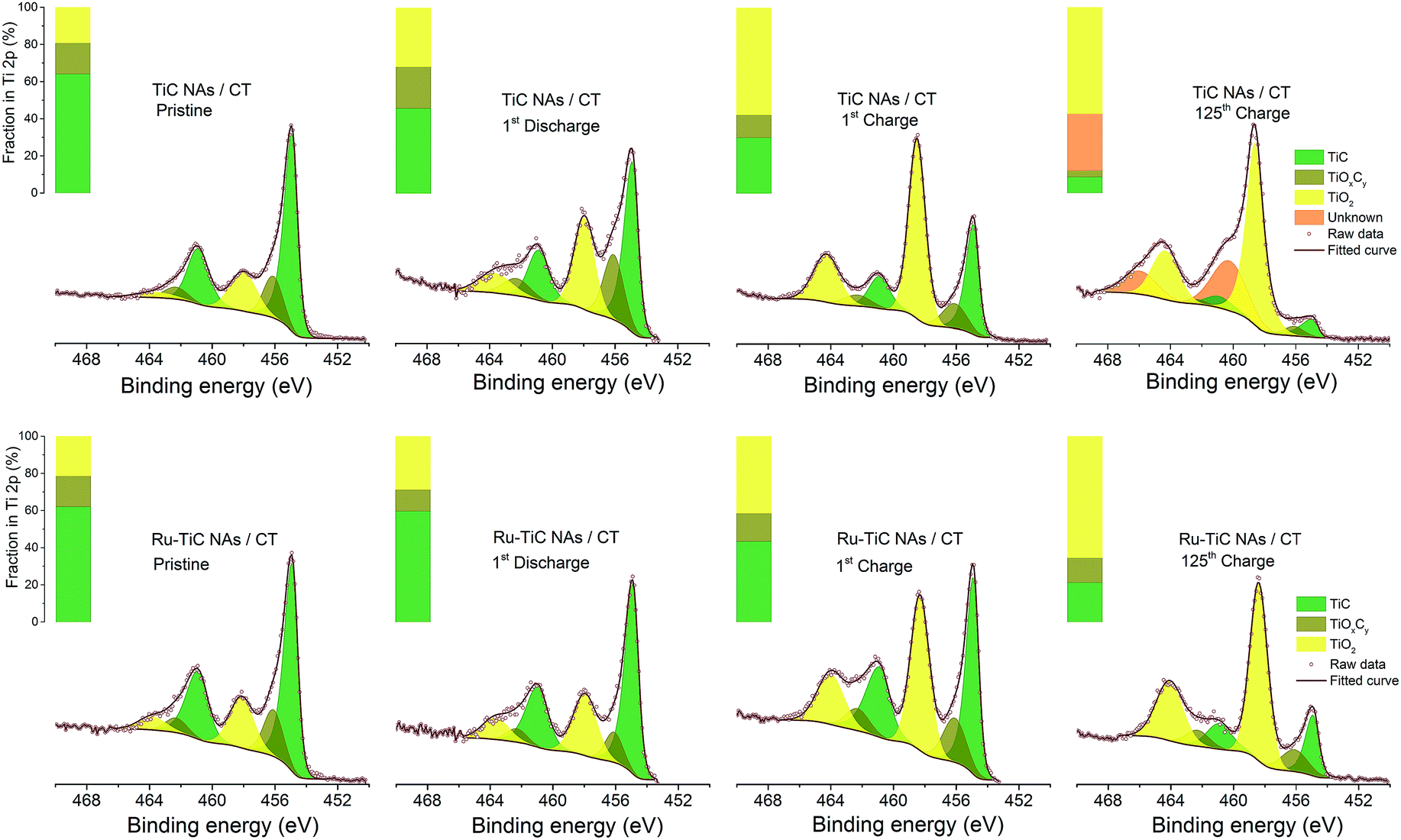 | ||
| Fig. 7 Ti 2p XPS spectra of TiC NAs/CT and Ru–TiC NAs/CT at different cycle stages. | ||
Conclusions
Having a stable cathode material that can promote highly reversible formation and decomposition of Li2O2 is one of the key requirements to successfully operate a rechargeable aprotic Li–O2 cell. As a step towards this goal, we have designed a highly efficient and stable cathode of Ru NP decorated TiC for Li–O2 batteries, as a free standing electrode. The oriented TiC NAs provide not only a large surface for the deposition of discharge products, but also a pathway for electrical and ionic conduction. In addition, this cathode configuration prevents the direct exposure of carbon to the electrolyte, and ensures reliable electron transfer and structural integrity of the electrode. Furthermore, it avoids unnecessary usage of polymeric binders. The surface of TiC is easily oxidized to form a TiO2 protective layer during cycling. However, progressive oxidation of the TiC surface should be avoided. Here, we introduce Ru NPs, which not only benefits the discharge and charge process, but also slows down the continuous TiC surface oxidation, increasing the stability of the cathode material. The Ru–TiC NAs/CT cathode demonstrated an improved areal capacity, lower overpotential, longer cyclability, and higher stability towards TiC surface degradation. The high kinetics and efficiency of the formation and decomposition of Li2O2 in Ru–TiC NAs/CT based Li–O2 batteries were confirmed by in operando XRD. All of these improvements arise from the integration of favourable compositional and structural properties of the Ru–TiC NAs/CT composite. This study provides a viable approach toward high-performance and stable cathode materials for Li–O2 batteries.Experiments and methods
Synthesis of TiC NAs
The TiC NA preparation was modified from a previously reported work.50 The synthetic strategy for fabricating TiC NAs/CT cathodes is illustrated in Scheme 1. Briefly, Ni(NO3)2·6H2O (30 mg) and NaCl (58 mg) were dissolved in 10 mL of ethanol. 160 mg of TiO2 powder (anatase, < 25 nm, 99.7%, Aldrich chemistry) was then dispersed into the above solution by ultrasound irradiation for 1 h to form a homogeneous Ni–Ti–Cl emulsion. Then, 300 mg of carbon cloth (pre-punched into a 13 mm diameter disc) was added into the emulsion. After magnetic stirring overnight, the carbon cloth was transferred into an oven, and dried at 60 °C for 2 h to remove ethanol. With nickel, chlorine, and titanium being loaded onto the textile surface, the carbon cloth was placed between two slices of titanium foam, and placed in a graphite crucible covered with a lid. The sample was put into a tube furnace, heated to 1250 °C with a ramp of 5 °C min−1, kept at this temperature for 3 h in pure Ar (100 sccm), and further calcined for 3 h under a continuous flow of 5% H2/95% Ar gas mixture (5 sccm/95 sccm) to reduce the possible intermediate TiOx into TiC.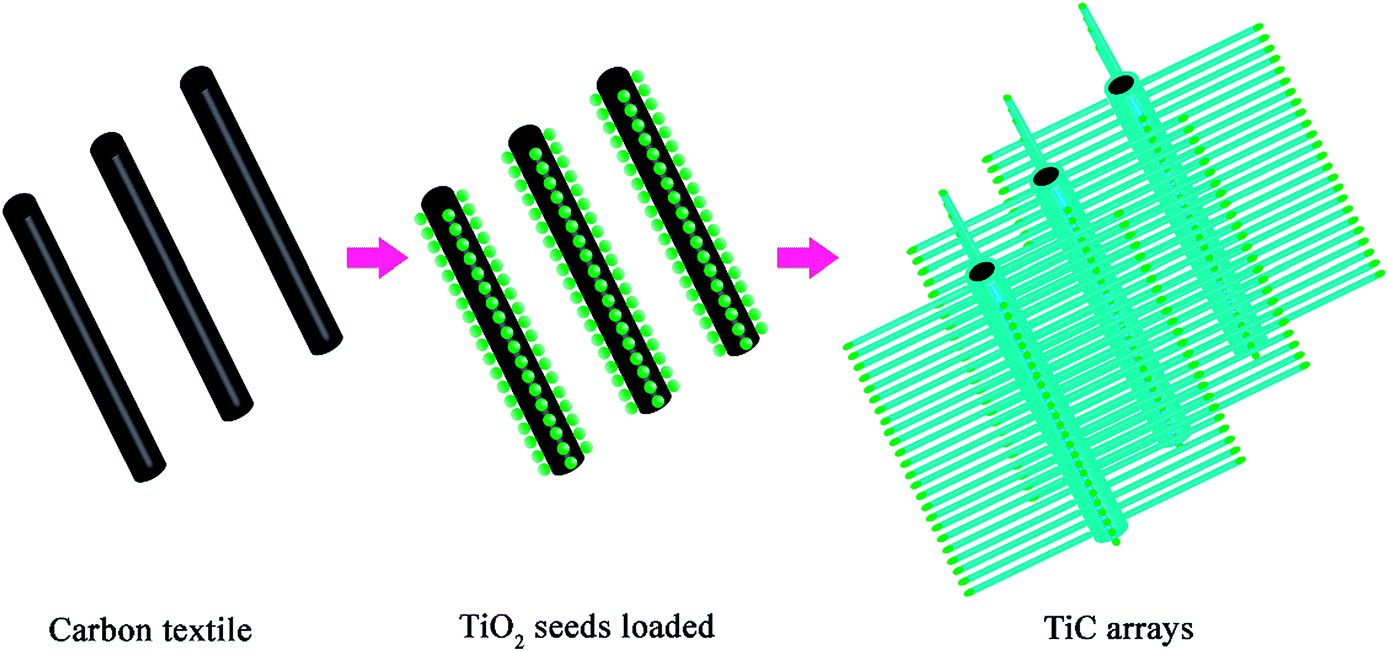 | ||
| Scheme 1 Schematic illustration of the preparation of TiC NAs. | ||
Preparation of the Ru–TiC NAs/CT cathode
The loading of Ru nanoparticles onto the surface of the TiC NAs was carried out via a simple impregnation method. The as-prepared TiC NAs were immersed into a 1 mg mL−1 RuCl3 (Alfa Aesar) ethanol solution overnight. The obtained electrode was dried in air and calcined at 300 °C for 3 h under a flow of 5% H2/95% Ar gas mixture at 100 sccm. This work and ref. 50 used different textile templates for TiC synthesis. The main differences between ref. 50 and this work also lie in the deposition of noble metal and the calcination of TiC in a reducing atmosphere.A cathode loaded with commercial Li2O2 was prepared in an argon filled glovebox. Commercial Li2O2 powder (90%, Sigma Aldrich) was dispersed in dimethyl carbonate by sonication. The dispersion was added dropwise to the TiC NAs and Ru–TiC NAs/CT cathodes. The loading amount of Li2O2 was obtained from the weight gain of each cathode.
Material characterization
SEM and TEM were carried out with a SEM/EDS-Zeiss 1550 microscope operated at 10 kV and a JEOL JEM-2100F microscope operated at 200 kV, respectively. The TEM samples were prepared in an Ar-filled glove box and loaded into a JEOL vacuum transfer holder. TEM images were recorded using a Gatan Ultrascan 1000 or Orius 200D camera. Energy-filtered TEM images were acquired using a Gatan Imaging Filter (GIF Trideim 863). Raman spectra were measured using a Renishaw micro Raman system equipped with a Leica LM optical microscope, a CCD camera and a 532 nm laser source. The chemical composition of the samples was analysed with a Perkin-Elmer 7000 DV ICP-AES, following a total digestion using alkaline fusion (NaOH–Na2O2). In-house XRD patterns were obtained on a Stoe & Cie GmbH Stadi X-ray powder diffractometer equipped with a Ge monochromator (Cu Kα1) operating at 45 kV and 40 mA in transmission mode. A pouch cell was used for this measurement. With a polymer coated thin Al-foil as the pouch material, the pouch cell enabled the X-rays to penetrate (Fig. S10†). The residual ratio of Li2O2 was determined in comparison with the constant TiC via Rietveld refinement using the FullProf program (calculation details can be found in the ESI†). SR-PXD was performed at beamline I711, MAX II synchrotron with a wavelength (λ) of 0.994 Å at MAX IV Laboratory in Lund, Sweden (Fig. S11†). The in operando cell was prepared using a similar design as given in our previous work (Fig. S12†).38 The cell was mounted in transmission mode with an Oxford diffraction Titan CCD area detector to collect the diffraction patterns. LaB6 was used to calibrate the diffraction parameters of the instrument. In order to avoid beam damage, measurement with short exposures (30 s) and long waiting times (30 min) was implemented.Electrochemical measurements
Dimethyl sulfoxide (DMSO, >99.5%, Sigma-Aldrich) was distilled before use and LiClO4 (>99.9%, Sigma-Aldrich) was dried in a vacuum oven inside an Ar-filled glove box. 0.5 M LiClO4/DMSO electrolyte was prepared in an Ar-filled glove box. The water content of the as-prepared electrolyte was less than 3 ppm, determined by Karl-Fischer titration. For the electrochemical measurements, Li–O2 cells were assembled (based on a Swagelok type design) inside an Ar-filled glovebox (O2 and H2O < 1 ppm). The cells were composed of a Li metal foil anode (11 mm in diameter and 0.25 mm in thickness), a piece of glass fiber separator (13 mm in diameter), excess electrolyte (0.5 M LiClO4/DMSO), and the as-prepared cathodes. The cells were gas-tightened except for the cathode side being exposed to a pure O2 atmosphere, and rested for 5 h before testing. Galvanostatic cycling was performed on a Digatron BTS-600 system at a current density of 0.1 mA cm−2. For in-house XRD and SR-PXD measurements, the electrochemical tests were controlled by an external potentiostat (SP-240, Bio-Logic SAS).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully appreciate the support for the synchrotron measurement from the MAX-lab staff at beam-line I711. The authors acknowledge the financial support by the Swedish Research Council, Swedish Energy Agency, StandUp for Energy, China Scholarship Council and Opening Project from the Key Laboratory for Ultrafine Materials of the Ministry of Education in the East China University of Science and Technology. Dr Maria Hahlin is gratefully acknowledged for her useful discussion and advice on XPS part. Dr Ismael Saadoune and Dr Rachid Hakkou are greatly acknowledged for their help with the ICP measurement. The authors also thank Dr Jakob Spiegelberg for his help with the in operando data refinement.Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. D. McCloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshoj, J. K. Norskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- B. J. Bergner, A. Schurmann, K. Peppler, A. Garsuch and J. Janek, J. Am. Chem. Soc., 2014, 136, 15054–15064 CrossRef CAS PubMed.
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125 RSC.
- H. Geaney and C. O'Dwyer, J. Electrochem. Soc., 2015, 163, A43–A49 CrossRef.
- R. Younesi, G. M. Veith, P. Johansson, K. Edström and T. Vegge, Energy Environ. Sci., 2015, 8, 1905–1922 RSC.
- R. Younesi, M. Hahlin, M. Treskow, J. Scheers, P. Johansson and K. Edström, J. Phys. Chem. C, 2012, 116, 18597–18604 CrossRef CAS.
- L. J. Hardwick and P. G. Bruce, Curr. Opin. Solid State Mater. Sci., 2012, 16, 178–185 CrossRef CAS.
- B. Sun, S. Q. Chen, H. Liu and G. X. Wang, Adv. Funct. Mater., 2015, 25, 4436–4444 CrossRef CAS.
- J. Yang, D. Zhai, H. H. Wang, K. C. Lau, J. A. Schlueter, P. Du, D. J. Myers, Y. K. Sun, L. A. Curtiss and K. Amine, Phys. Chem. Chem. Phys., 2013, 15, 3764–3771 RSC.
- J. Lu, L. Cheng, K. C. Lau, E. Tyo, X. Luo, J. Wen, D. Miller, R. S. Assary, H. H. Wang, P. Redfern, H. Wu, J. B. Park, Y. K. Sun, S. Vajda, K. Amine and L. A. Curtiss, Nat. Commun., 2014, 5, 4895 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- D. M. Itkis, D. A. Semenenko, E. Y. Kataev, A. I. Belova, V. S. Neudachina, A. P. Sirotina, M. Havecker, D. Teschner, A. Knop-Gericke, P. Dudin, A. Barinov, E. A. Goodilin, Y. Shao-Horn and L. V. Yashina, Nano Lett., 2013, 13, 4697–4701 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- J. Xie, X. Yao, I. P. Madden, D. E. Jiang, L. Y. Chou, C. K. Tsung and D. Wang, J. Am. Chem. Soc., 2014, 136, 8903–8906 CrossRef CAS PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef CAS PubMed.
- F. Li, D. M. Tang, Y. Chen, D. Golberg, H. Kitaura, T. Zhang, A. Yamada and H. Zhou, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed.
- D. Kundu, R. Black, B. Adams, K. Harrison, K. Zavadil and L. F. Nazar, J. Phys. Chem. Lett., 2015, 6, 2252–2258 CrossRef CAS PubMed.
- B. D. Adams, R. Black, C. Radtke, Z. Williams, B. L. Mehdi, N. D. Browning and L. F. Nazar, ACS Nano, 2014, 8, 12483–12493 CrossRef CAS PubMed.
- A. Y. Kozmenkova, E. Y. Kataev, A. I. Belova, M. Arnati, L. Gregoratti, J. Velasco-Velez, A. Knop-Gericke, B. Senkovsky, D. V. Vyalikh, D. M. Itkis, Y. Shao-Horn and L. V. Yashina, Chem. Mater., 2016, 28, 8248–8255 CrossRef CAS.
- C. V. Amanchukwu, J. R. Harding, Y. Shao-Horn and P. T. Hammond, Chem. Mater., 2015, 27, 550–561 CrossRef CAS.
- X. J. Feng, K. Zhu, A. J. Frank, C. A. Grimes and T. E. Mallouk, Angew. Chem., Int. Ed., 2012, 51, 2727–2730 CrossRef CAS PubMed.
- N. K. Shrestha and P. Schmuki, Electrochemistry, 2013, 12, 87–131 Search PubMed.
- X. Xia, J. Tu, Y. Zhang, X. Wang, C. Gu, X. B. Zhao and H. J. Fan, ACS Nano, 2012, 6, 5531–5538 CrossRef CAS PubMed.
- Y.-C. Lu, D. G. Kwabi, K. P. C. Yao, J. R. Harding, J. Zhou, L. Zuin and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2999 RSC.
- M. Leskes, N. E. Drewett, L. J. Hardwick, P. G. Bruce, G. R. Goward and C. P. Grey, Angew. Chem., Int. Ed. Engl., 2012, 51, 8560–8563 CrossRef CAS PubMed.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518 RSC.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J. M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- H. Zheng, D. Xiao, X. Li, Y. Liu, Y. Wu, J. Wang, K. Jiang, C. Chen, L. Gu, X. Wei, Y. S. Hu, Q. Chen and H. Li, Nano Lett., 2014, 14, 4245–4249 CrossRef CAS PubMed.
- S. Ganapathy, B. D. Adams, G. Stenou, M. S. Anastasaki, K. Goubitz, X. F. Miao, L. F. Nazar and M. Wagemaker, J. Am. Chem. Soc., 2014, 136, 16335–16344 CrossRef CAS PubMed.
- J. P. Vivek, N. G. Berry, J. Zou, R. J. Nichols and L. J. Hardwick, J. Phys. Chem. C, 2017, 121, 19657–19667 CrossRef CAS.
- J. L. Shui, J. S. Okasinski, P. Kenesei, H. A. Dobbs, D. Zhao, J. D. Almer and D. J. Liu, Nat. Commun., 2013, 4, 2255 CrossRef PubMed.
- L. Zhong, R. R. Mitchell, Y. Liu, B. M. Gallant, C. V. Thompson, J. Y. Huang, S. X. Mao and Y. Shao-Horn, Nano Lett., 2013, 13, 2209–2214 CrossRef CAS PubMed.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS PubMed.
- H. Lim, E. Yilmaz and H. R. Byon, J. Phys. Chem. Lett., 2012, 3, 3210–3215 CrossRef CAS PubMed.
- C. Liu, W. R. Brant, R. Younesi, Y. Dong, K. Edström, T. Gustafsson and J. Zhu, ChemSusChem, 2017, 10, 1592–1599 CrossRef CAS PubMed.
- Z. Guo, C. Li, J. Liu, X. Su, Y. Wang and Y. Xia, J. Mater. Chem. A, 2015, 3, 21123–21132 RSC.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- J. R. Harding, Y. C. Lu, Y. Tsukada and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2012, 14, 10540–10546 RSC.
- M. V. Klein, J. A. Holy and W. S. Williams, Phys. Rev. B, 1978, 17, 1546–1556 CrossRef CAS.
- X. Y. Tao, J. Du, Y. C. Yang, Y. P. Li, Y. Xia, Y. P. Gan, H. Huang, W. K. Zhang and X. D. Li, Cryst. Growth Des., 2011, 11, 4422–4426 CrossRef CAS.
- X. Y. Tao, Y. P. Li, J. Du, Y. Xia, Y. C. Yang, H. Huang, Y. P. Gan, W. K. Zhang and X. D. Li, J. Mater. Chem., 2011, 21, 9095–9102 RSC.
- R. Younesi, M. Hahlin, F. Björefors, P. Johansson and K. Edström, Chem. Mater., 2013, 25, 77–84 CrossRef CAS.
- L. Zhong, R. R. Mitchell, Y. Liu, B. M. Gallant, C. V. Thompson, J. Y. Huang, S. X. Mao and Y. Shao-Horn, Nano Lett., 2013, 13, 2209–2214 CrossRef CAS PubMed.
- S. Basak, J. Jansen, Y. Kabiri and H. W. Zandbergen, Ultramicroscopy, 2018, 188, 52–58 CrossRef CAS PubMed.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772 RSC.
- R. R. Mitchell, B. M. Gallant, Y. Shao-Horn and C. V. Thompson, J. Phys. Chem. Lett., 2013, 4, 1060–1064 CrossRef CAS PubMed.
- Z. Qiu, H. Huang, J. Du, X. Tao, Y. Xia, T. Feng, Y. Gan and W. Zhang, J. Mater. Chem. A, 2014, 2, 8003–8008 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta10930j |
| This journal is © The Royal Society of Chemistry 2018 |