
Two-dimensional nanostructures for sodium-ion battery anodes
Jianfeng
Mao
a,
Tengfei
Zhou
 a,
Yang
Zheng
a,
Hong
Gao
a,
Hua kun
Liu
a and
Zaiping
Guo
*ab
a,
Yang
Zheng
a,
Hong
Gao
a,
Hua kun
Liu
a and
Zaiping
Guo
*ab
aInstitute for Superconducting and Electronic Materials, University of Wollongong, North Wollongong, New South Wales 2500, Australia. E-mail: zguo@uow.edu.au
bSchool of Mechanical, Materials, Mechatronics and Biomedical Engineering, University of Wollongong, Wollongong, New South Wales 2522, Australia
First published on 24th January 2018
Abstract
Sodium-ion batteries (SIBs) have attracted great attention recently due to the abundance of sodium resources, particularly for large-scale electric energy storage applications for renewable energy and smart grids. More and more nanostructured anode materials have been developed with the aims of high energy density, high cycling stability, and excellent rate capability, in which two-dimensional (2D) nanostructures are showing promise due to their shortened paths for sodium ion transportation and larger surface areas for sodium ion absorption. Moreover, 2D materials (e.g. graphene) have been proved to be excellent supporting and conducting agents in SIB anodes due to their high electrical conductivity and structural stability, in which synergetic effects between the graphene and the active materials are generally observed. This review is devoted to the recent progress in the use of 2D active materials and in composites consisting of both 2D supports and active materials as anodes for SIBs. Based on the manner of sodium storage, their electrochemical performance for sodium storage is discussed in terms of four classifications, including carbonaceous materials (graphene and carbon nanosheets), alloy based materials (Sn, Sb, and P), conversion materials (phosphides/oxides/sulfides/selenides), and intercalation materials (Ti-based compounds). Finally, the main challenges for and perspectives on 2D nanostructures for sodium storage are discussed.
 Jianfeng Mao | Jianfeng Mao received his Ph.D. in materials science from the University of Wollongong, Australia, in 2012. After working at the Max-Planck-Institute für Kohlenforschung, University of Glasgow and University of Maryland, he moved back to the University of Wollongong in 2015. His research interests are in inorganic materials for rechargeable batteries, catalysis, and hydrogen storage. |
 Tengfei Zhou | Tengfei Zhou is currently an associate research fellow at the Institute for Superconducting and Electronic Materials (ISEM), School of Mechanical, Materials, Mechatronic and Biomedical Engineering, University of Wollongong (UOW), Australia. He received his Ph.D. degree from the University of Wollongong in 2016, under the supervision of Professor Zaiping Guo. His research interests include the design and fabrication of two-dimensional functional nanomaterials and their applications in new-generation energy storage and conversion, such as Li/Na/K-ion batteries, supercapacitors, catalysts, etc. |
 Yang Zheng | Yang Zheng is currently a PhD candidate at the Institute for Superconducting and Electronic Materials (ISEM), University of Wollongong, Australia under the supervision of Prof. Zaiping Guo. His research focuses on the design and fabrication of novel nanomaterials for the applications in energy storage and conversion, such as Li/Na ion batteries and catalysis. |
 Hong Gao | Hong Gao is a PHD student at Institute for Superconducting and Electronic Materials (ISEM), University of Wollongong (UOW), Australia. Her current research interest is to optimize the sodium/lithium-storage properties of promising anode materials. |
 Hua Kun Liu | Hua Kun Liu received an award of Doctor of Science at University of Wollongong (20/07/2017). She is a distinguished Professor at Institute for Superconducting and Electronic Materials, UOW, Australia, and a Fellow of Australian Academy of Technological Science and Engineering. She received the UOW Vice-Chancellor's Award for Research Excellence Senior Researcher in 2013, and won the most competitive ARC Australian Professorial Fellowships in 1994–1998, 1999–2003, 2003–2005 and 2006–2010. She has supervised 71 PhD and 40 postdoctoral and visiting fellows. She was selected as a highly cited researcher by Thompson Reuters in 2016. Her publications have attracted citations 40000, H-index: 85. |
 Zaiping Guo | Zaiping Guo received her PhD in Materials Engineering from the University of Wollongong in December 2003. After APD fellowship in the Institute for Superconducting & Electronic Materials, she joined Faculty of Engineering and Information Sciences, University of Wollongong as a Lecturer in 2008, and was promoted to Professor in 2012, and then Senior Professor in 2013. Her current research interests focus on the design and application of nanomaterials for energy storage and conversion, including rechargeable batteries, hydrogen storage, and fuel cells. |
1 Introduction
Batteries are continually being developed to power an increasingly diverse range of applications, from microchips to vehicles and stationary energy storage devices.1 For application in large scale stationary storage, high energy density will become less critical, instead, low cost, high safety and long cycle life are crucial.2,3 Thus, Li-ion batteries (LIBs) are only expected to contribute in part to large-scale energy storage, which is needed to support the widespread use of renewable energy, due to their low abundance and high cost.2,3 These requirements call for the next generation of energy storage alternatives to have low cost and good sustainability. Sodium-ion batteries (SIBs) have again attracted increasing attention for large-scale energy storage of renewable energy and smart grid applications in the past few years, because of the high abundance and low cost of sodium resources, and their decent energy densities.4–8 More importantly, sodium has similar chemical properties to lithium, which makes the knowledge acquired from developing LIBs valuable for the design of SIB electrodes. Challenges still remain, however, since the ionic radius of Na+ is larger than that of Li+ (0.98 Å vs. 0.69 Å). The larger Na+ ions will lead to slower ion transport, more sluggish reaction kinetics, and larger volume changes upon Na+/Na insertion/extraction, which usually result in limited rate capability and Na storage reversibility. For example, graphite, as the most commonly used anode material in LIBs, can only provide very small capacity in SIBs under normal conditions. Therefore, the development of high-capacity and long-cycle-life anode materials is critical for the success of SIBs. In the past several years, research on SIBs has been accelerated, and significant improvements have been made in developing SIB anode materials, with the aim of increasing the cycling stability, rate capability, and energy density.6,7 In addition to the materials themselves, the structural properties would mostly affect the electrochemical performance, since different structures exhibit different surface and structural characteristics. For example, zero-dimensional (0D) structures such as nanoparticles possess short diffusion lengths and the minimum surface area, but there tends to be agglomeration during cycling. One-dimensional (1D) structures such as nanotubes, nanowires, and nanorods have fast electron transport along the 1D direction and short ion diffusion lengths along the radial direction, but the static structure and fixed size limit the nonadjustable specific surface area and porosity properties. In contrast, two-dimensional (2D) structures such as nanosheets show unique advantages for SIB applications. With atomic or molecular thickness and infinite planar lengths, 2D nanomaterials have different atomic structures from their bulk counterparts, including differences in atomic arrangement, chemical valence, coordination number, and bond length. Moreover, their more exposed interior atoms would inevitably induce the formation of various defects, which would have non-negligible effects on their chemical and physical properties. Actually, 2D nanomaterials have already displayed fascinating properties in the energy storage field, including good mechanical flexibility, short ion diffusion lengths, and a large exposed surface for electrochemical processes.9–14 There are many 2D nanomaterials, such as graphene and its derivatives, elemental nanosheets such as phosporene and antimonene, transition metal oxide nanosheets, transition metal dichalcogenide nanosheets, and MXene nanosheets, that have been reported for high performance sodium ion battery anodes. In addition, 2D nanomaterials, such as graphene, are well-known for acting as functional substrates for incorporating active materials into SIBs, which renders them electrochemically more active. Several advantages could usually be expected when using graphene-based composites as electrode materials for SIBs. For example, graphene acts as a support for electroactive nanomaterials, and prevents them from re-stacking by lowering the van der Waals interactions among the layers. Moreover, the elastic and highly conductive nature of graphene improves the electrical conductivity of composites and buffers the volume expansion of electrode materials during cycling. Therefore, graphene-containing nanocomposites have the ability and variety for applications as the advanded electrode materials in SIBs. The diversity of structures reported for graphene-containing nanocomposites can be classified into six different models, as summarized by Raccichini et al. (Fig. 1).15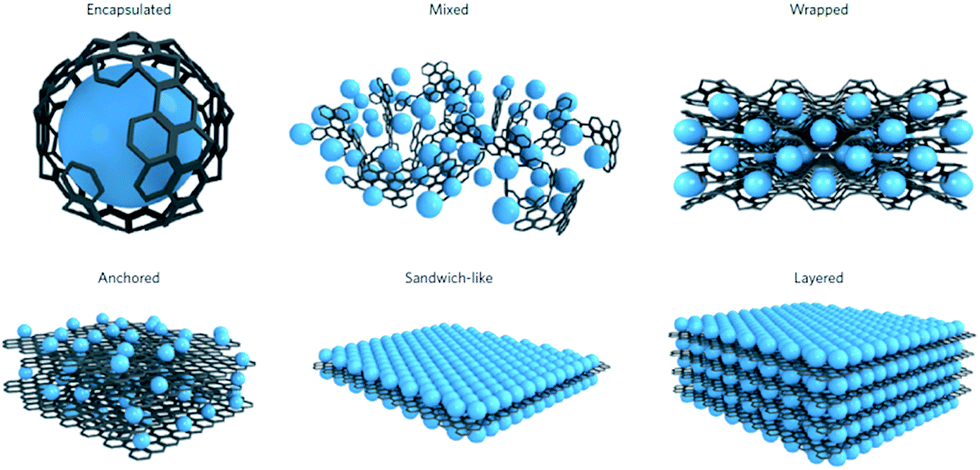 | ||
| Fig. 1 Structural models refer to composites in which graphene and the active material are synthesized through one-pot processes. Encapsulated: single active-material particles are encapsulated by graphene. Mixed: graphene and active materials are synthesized separately and mixed mechanically during the electrode preparation. Wrapped: the active-material particles are wrapped by multiple graphene sheets. Anchored: electroactive nanoparticles are anchored to the graphene surface. Sandwich-like model: graphene is used as a template to generate active material/graphene sandwich structures. Layered model: active-material nanoparticles are alternated with graphene sheets to form a composite layered structure. Reproduced with permission from ref. 15. Copyright 2014, Springer Nature. | ||
There have been several recent reviews on sodium-ion battery anodes; a critical review that is focused on 2D nanostructured anode materials for application in SIBs is lacking, however. The notion of 2D favored structures will be discussed in terms of both 2D active materials and 2D nanocomposites. Depending on the distinct sodium storage mechanisms, four different types of nanomaterials are presented, including carbonaceous materials, sodium alloys/compounds, phosphides/oxides/sulfides/selenides, and titanium based materials. The unprecedented functionalities and promising applications of these materials are described first, and then the difficulties and challenges for these materials are discussed. Subsequently, we summarize the latest efforts toward addressing these limitations in order to reveal the structural features of the 2D nanoarchitectures. Finally, we also outline future prospects for the major challenges and opportunities facing the 2D nanostructures. The advances discussed here will lead to a broader understanding of SIB anode materials and rational structural design, which also can illuminate the material innovations in other energy conversion and storage realms.
2 Two-dimensional carbonaceous anodes
Carbonaceous anodes are extensively applied in batteries due to their wide availability. Graphite, the bulk form of graphene, is the dominant commercial anode material in LIBs, but it only provides a very small capacity in SIBs under normal conditions.16–19 A reasonable capacity was only obtained in modified graphite via enlarging the interlayer distance or through co-intercalation with ether-based electrolytes.20–22 Thus, interest has been attracted to other carbonaceous materials with low graphitization, where the Na storage is realized by combining Na intercalation and Na filling of pores or defects. Hard carbon is widely recognized as a potential anode material for the sodium ion battery due to the large interlayer d-spacing and rather high capacity (∼300 mA h g−1).23–28 Nevertheless, these materials are limited by their poor rate capability and cyclability because of their intrinsically low graphitization. In addition, their low charging potential may induce electrode instability and safety issues. For carbonaceous anodes, it is widely accepted that the sodium insertion mechanism strongly depends on the particle size, the degree of graphitization, the structural or textural disorder, and the porosity.29 Therefore, controlling the morphology is an effective strategy to improve the dynamic performance of carbonaceous materials. Two-dimensional carbonaceous materials, especially those having structures with large surface area, have excellent electronic conductivity and chemical stability, because this kind of structure can bring the materials into adequate contact with the electrolyte, promote the transport of ions, and facilitate strain relaxation during the charge–discharge process.15,30Graphene, a unique 2D carbon material, with large surface area, excellent chemical stability, and outstanding electronic conductivity, exhibits extraordinary advantages in energy storage systems.15,31 In addition to its use as a functional supporting matrix, graphene itself can serve as an active material in the electrochemical reaction.31–33 The use of reduced graphene oxide (rGO) as an anode material in SIBs has been found to deliver a high capacity of 141 mA h g−1 at 0.2C (40 mA g−1), with a long cycle life of over 1000 cycles and a reversible capacity of 95.6 mA h g−1 at the high current rate of 5C (1000 mA g−1).32 Such performance is attributed to the unique structure of rGO, in which the interconnected graphene nanosheets are able to greatly reduce the diffusion length of sodium ions. Moreover, Datta et al.34 discovered that Na could be absorbed into defective graphene, which is not the case for pristine graphene. The capacities of Na+ batteries reached 1450 mA h g−1 when the divacancy defect density reached its limit. In addition, the introduction of aliovalent elements such as boron, sulfur, and phosphorus into graphene helps to generate abundant defect sites to absorb the sodium ions and improves the surface hydrophilicity of the graphene, which facilitates electrode–electrolyte interactions and hence leads to higher Na-storage capacity.35–40 For example, Wang et al. utilized polydopamine, a conformal and uniform coating material, as the nitrogen source to modify graphene oxide by a pyrolysis method to synthesize N-doped graphene sheets, which showed quite stable cycling performance, with no capacity decay after 600 cycles.36 Sulfur-doped graphene, with phenyl disulfide as the sulfur source, was also synthesized as an anode material for sodium storage.40 The sulfur atoms covalently bonded to the graphene led to changes in the electronic structure of the graphene sheets. The obtained material exhibited long cycling stability and superior rate capability. In addition to graphene, heteroatom doped carbon nanosheets also showed excellent performance, Yang et al. synthesized S-doped, N-rich carbon (S–N/C) nanosheets by simply treating N-rich carbon nanosheets (N/C) with H2S/Ar mixed gas.41 It was found that pyrrolic-N substituted for S with retention of pyridinic-N and the formation of strong –C–S–C– bonds, which expanded the interlayer distance and enlarged the surface area, hence providing more active sites to create highly reversible sodium storage reactions. As a result, the N,S-codoped porous carbon nanosheets displayed high capacity and excellent rate performance as SIB anodes.
On the other hand, the construction of hierarchically porous structure is effective for improving the dynamic performance of the carbon because the electrolyte can enter the holes and the diffusion distance of the Na-ions can be decreased.42–44 Therefore, the combination of a porous structure and 2D morphology should be an excellent strategy to improve the electrochemical performance of materials. Xu et al. prepared well-defined three-dimensional (3D) mesoporous nitrogen-doped graphene foams, which not only possessed a superior 3D mesoporous graphene structure, but also induced defects by N-doping (Fig. 2a).45 All these factors were combined to make contributions to the significant improvement of the electrochemical performance. As a result, the material showed 852.6 mA h g−1 initial reversible capacity (at the current density of 500 mA g−1) and 594 mA h g−1 after 150 cycles, corresponding to capacity retention of 69.7% (Fig. 2b).
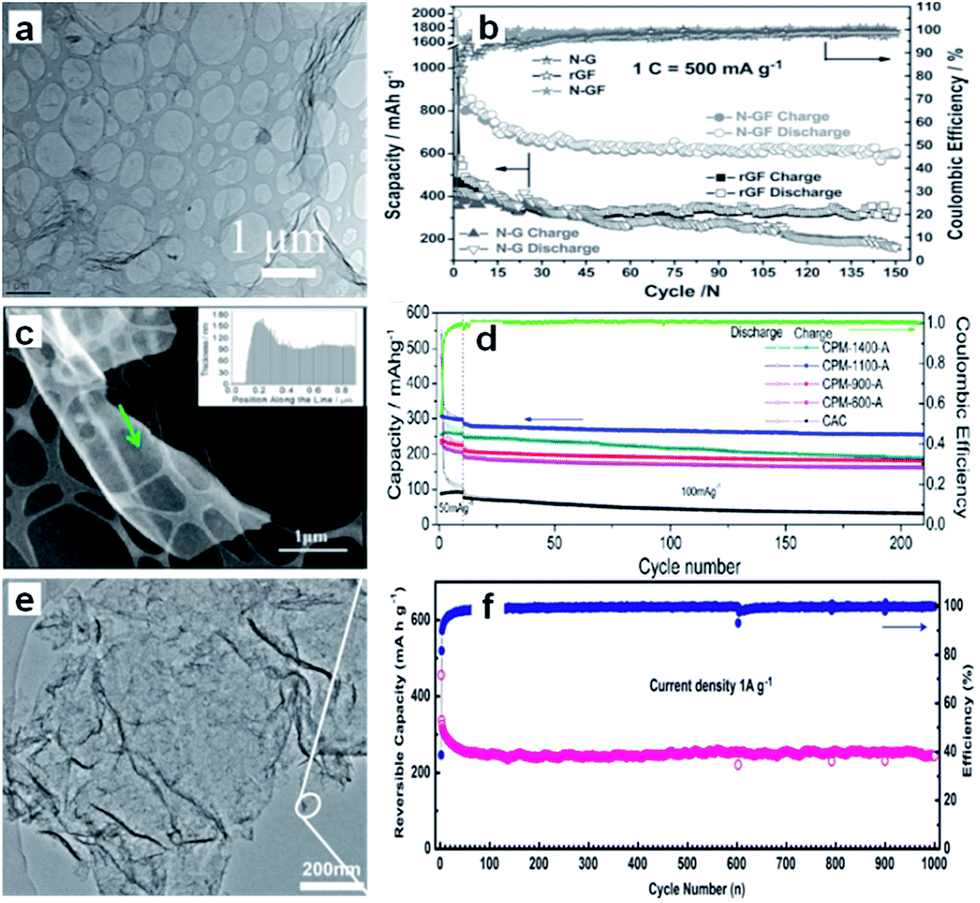 | ||
| Fig. 2 (a) Scanning transmission electron microscope (STEM) images of the nitrogen–graphene foam (N–GF), (b) cycling performance of the nitrogen doped graphene (N–G), reduced graphene foam (rGF), and N–GF at 1C in the voltage range of 0.02–3.0 V (assuming 1C = 500 mA g−1; Scapacity = specific capacity). Reprinted with permission from ref. 45. Copyright 2015, WILEY-VCH Verlag GmbH & Co. (c) High-angle annular dark field-transmission electron microscope (HAADF TEM) micrograph and electron energy loss spectroscopy (EELS) thickness profile (inset) of the carbon strand marked by the arrow in CPM-1100-A, (d) extended cycling performance of the CPM-A and CAC electrodes, with the coulombic efficiency of the CPM-1100-A electrode. Reprinted with permission from ref. 46. Copyright 2013, American Chemical Society. (e) TEM image of the graphene–hierarchically porous carbon (G@HPC) composite; and (f) cycling performance under 1 A g−1. Reprinted with permission from ref. 47. Copyright 2014, WILEY-VCH Verlag GmbH & Co. | ||
Ding et al. synthesized three-dimensional macroporous interconnected networks of carbon nanosheets from peat moss by carbonization and activation (Fig. 2c).46 The carbon nanosheets form highly ordered pseudographitic arrays with a much larger intergraphene spacing (0.388 nm) than graphite (c/2 = 0.3354 nm). Due to the 3D macroporous interconnected networks and the dilated graphene interlayer spacing, the carbon nanosheets allowed for fast sodium intercalation and extraction. The demonstrated specific capacity was up to 300 mA h g−1 at the current density of 50 mA g−1 and 100 mA h g−1 at 1 A g−1 (Fig. 2d). Furthermore, Yan et al. designed a sandwich-like hierarchically porous carbon/graphene nanocomposite by a facile ionothermal process, which overcomes the low graphitization problem of the hard carbon materials. The graphene, sandwiched by hierarchically porous carbon on both sides, facilitates electron transport (Fig. 2e).47 The nanocomposite exhibited capacity of 250 mA h g−1 at the current density of 1 A g−1, even after 1000 cycles (Fig. 2f), in which the hierarchical porous carbon facilitates Na+ insertion and the graphene guarantees high electronic conductivity for the supply of electrons.
3 Alloy based anodes
Alloys are anticipated to be promising anode materials in SIBs due to their high volumetric and gravimetric capacities. A number of elements, such as Sn, Sb, Bi, Pb, Ge, and P, can form binary alloys with Na. They can principally be used as anodes for sodium ion batteries.48–60 Only Sn, Sb, and P have been intensively investigated, however, owing to their relatively higher specific capacity and excellent electrochemical performance. Hence, this review of the alloy anodes is mainly focused on these three elements and their related compounds with 2D nanoarchitectures.3.1 Tin and antimony based alloy anodes
Tin (Sn) can deliver a theoretical capacity of 847 mA h g−1 according to the formation of Na15Sn4.48,52,53 Electrochemical sodiation/desodiation for the Na–Sn system occurs in a series of steps: Sn, NaSn5, NaSn, Na9Sn4, and Na15Sn4.49,50,52,53,59 In contrast, antimony (Sb) can deliver a theoretical capacity of 660 mA h g−1 according to the formation of Na3Sb.54,55,60 During sodiation, crystalline Sb is first electrochemically reduced to an amorphous phase, NaxSb. Then, the amorphous NaxSb is reduced to crystalline Na3Sb with a hexagonal lattice through the formation of cubic Na3Sb as a metastable intermediate phase. Upon desodiation, the crystalline Na3Sb is oxidized into amorphous Sb.54 It is believed that the formation of the intermediate amorphous phase may act as a buffer to relieve strain and hence improve the cycling performance. Thus, unlike the bulk Sn, the bulk Sb particles showed good electrochemical performance, although the nanosized Sb (10–20 nm) particles exhibited enhanced rate capability and higher cycling stability.60 The volume expansion of pure Sn upon alloying to Na15Sn4 and upon the alloying of Sb to Na3Sb is 420 and 293%, respectively, which is extremely high. This enormous volume expansion will lead to continuous and cumulative pulverization, leading to the loss of electrical contact and resulting in capacity decay during cycling. One strategy to deal with the volume expansion is to introduce a secondary matrix. Such a matrix can act as a buffer to suppress the volume changes and particle aggregation of the metal during the sodiation/desodiation cycling, and hence, allow a much higher number of cycles. 2D carbon materials such as graphene and carbon nanosheets are excellent matrix materials due to their high surface area and highly electrically conductive properties.61–75 For example, a porous scaffold composed of both reduced graphene oxide (GO) and graphene can be produced via camera flash reduction as the host for Sn (Fig. 3a).68 The porosity and mechanical properties were controlled by the mixing ratio between the reduced GO (RGO) and graphene (G); the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of (2RGO + G) was the best composition for high porosity and mechanical strength. The RGO–graphene scaffold enables uniform Sn deposition on a large surface area and offers sufficient free space for accommodating the Sn volume changes (Fig. 3b–d). Therefore, the Sn-coated (2RGO + G) electrode in particular showed a high reversible capacity of 615 mA h g−1, stable capacity retention (>84% after 50 cycles), and excellent rate capability (Fig. 3e). Liu et al. designed a sandwich-like structure graphene-supported Sb nanocomposite (denoted as Sb@RGO@Sb) through a facile electrostatic and chemical reduction process (Fig. 3g), in which ultrafine Sb nanoparticles are uniformly anchored on an RGO surface (Fig. 3h and i).75 When this designed Sb@RGO@Sb composite was used as the anode material for sodium-ion batteries, it exhibited superior sodium storage properties. It delivered a high mass-specific capacity of approximately 430 mA h g−1 at a current density of 100 mA g−1, even after 200 cycles, and an excellent rate performance of 330 mA h g−1 at 5 A g−1 (Fig. 3f). Ideally, the Sn or Sb nanoparticles are embedded or partially encapsulated in the matrix, thus better accommodating the stress and strain without being pulverized, shortening the ionic and electronic transport pathways, and preventing loss of electrical contact.
:1 ratio of (2RGO + G) was the best composition for high porosity and mechanical strength. The RGO–graphene scaffold enables uniform Sn deposition on a large surface area and offers sufficient free space for accommodating the Sn volume changes (Fig. 3b–d). Therefore, the Sn-coated (2RGO + G) electrode in particular showed a high reversible capacity of 615 mA h g−1, stable capacity retention (>84% after 50 cycles), and excellent rate capability (Fig. 3e). Liu et al. designed a sandwich-like structure graphene-supported Sb nanocomposite (denoted as Sb@RGO@Sb) through a facile electrostatic and chemical reduction process (Fig. 3g), in which ultrafine Sb nanoparticles are uniformly anchored on an RGO surface (Fig. 3h and i).75 When this designed Sb@RGO@Sb composite was used as the anode material for sodium-ion batteries, it exhibited superior sodium storage properties. It delivered a high mass-specific capacity of approximately 430 mA h g−1 at a current density of 100 mA g−1, even after 200 cycles, and an excellent rate performance of 330 mA h g−1 at 5 A g−1 (Fig. 3f). Ideally, the Sn or Sb nanoparticles are embedded or partially encapsulated in the matrix, thus better accommodating the stress and strain without being pulverized, shortening the ionic and electronic transport pathways, and preventing loss of electrical contact.
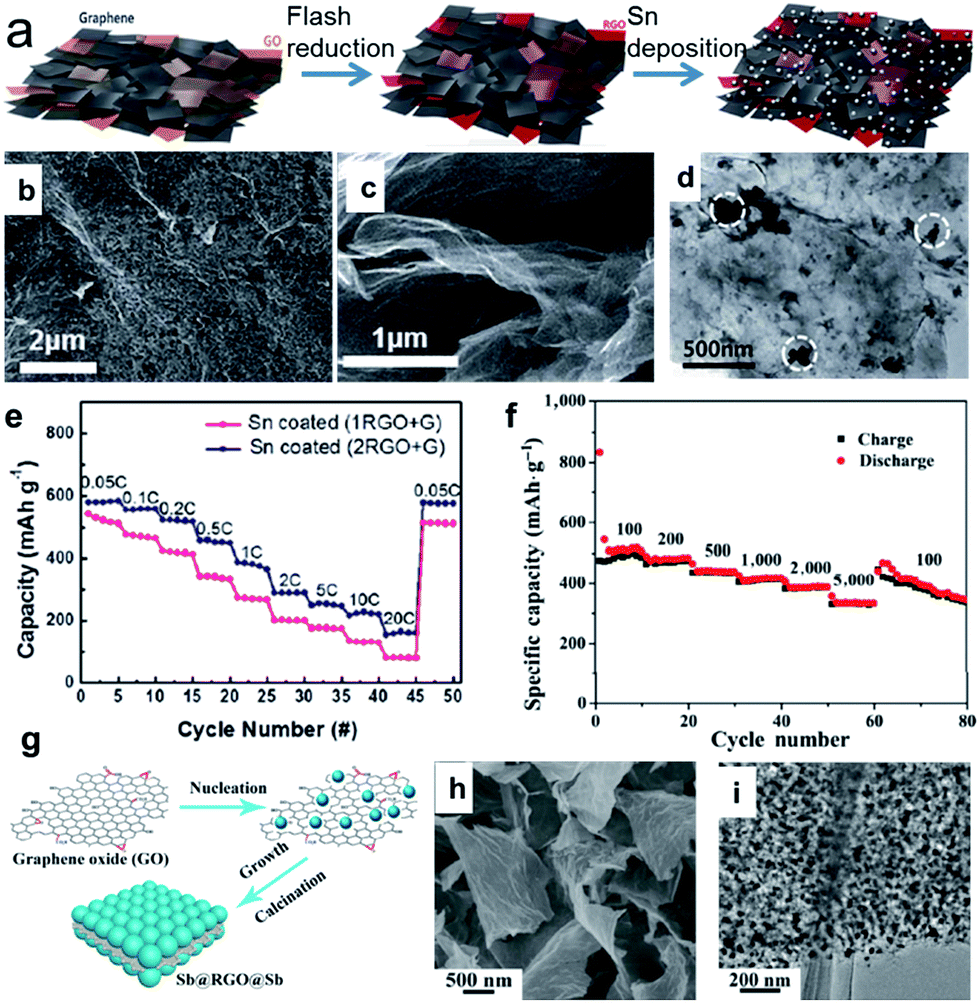 | ||
| Fig. 3 (a) Schematic illustration of the electrode fabrication process, which involves flash reduction and Sn electrophoretic deposition; (b) top view and (c) cross-sectional view SEM images of the Sn coated (2RGO + G) composite. (d) TEM image of Sn coated (2RGO + G) composite; (e) rate capabilities of Sn coated composite at various C rates from 0.05C to 20C. Reprinted with permission from ref. 68. Copyright 2016, Royal Society of Chemistry. (g) Schematic illustration of the formation of sandwich-like Sb@RGO@Sb nanosheets; (h) and (i) SEM and TEM images of the Sb@RGO@Sb nanosheets, respectively; (f) rate capabilities of the Sb@RGO@Sb nanosheets at current densities from 0.1 to 5 A g−1. Reprinted with permission from ref. 75. Copyright 2017, Springer. | ||
Interestingly, metallic antimony (Sb) in its gray allotrope is also a graphite-like layered material, though it has rarely been considered from the viewpoint of a 2D layered system, in which the Sb layers consist of fused, ruffled, six-membered rings. The weak bonding between the layers enables it to be a potential candidate for the top-down fabrication of Sb nanosheets. Recently, Gu et al. proposed an efficient strategy to fabricate free-standing metallic Sb nanosheets via liquid-phase exfoliation of gray Sb powder in an isopropyl alcohol (IPA) solution with a constant concentration of sodium hydroxide (Fig. 4a).76 Remarkably, the resultant metallic Sb nanosheets have ultrathin (∼4 nm), foldable features and large aspect ratios. Moreover, several hybrid films composed of metallic Sb nanosheets and graphene with tunable densities were achieved, in which the notorious volume changes of metallic Sb can be efficiently alleviated with the aid of the good flexible graphene, and the whole density of electrode films can be significantly improved by harnessing the high density of Sb nanosheets. As a consequence, the optimized metallic Sb nanosheet-graphene (SbNS-G) film displayed a high volumetric capacity of 1226 mA h cm−3, high-rate capability, and good cycling performance for sodium storage (Fig. 4b and c).76
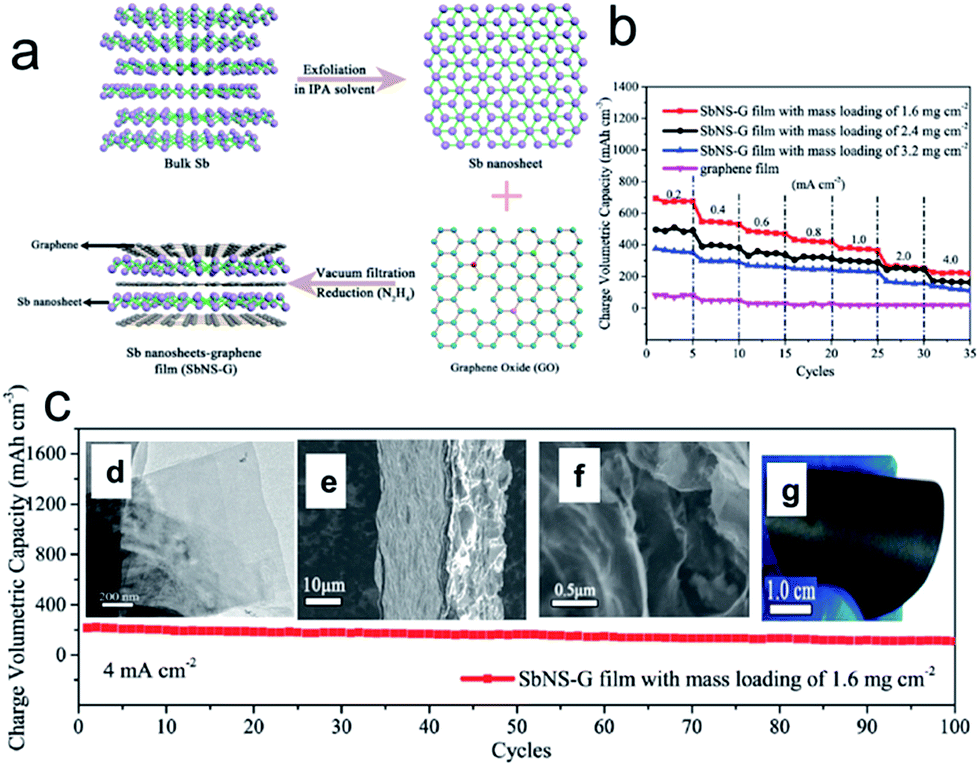 | ||
| Fig. 4 (a) Schematic illustration of the fabrication process for SbNS-G film. Bulk Sb powders were first exfoliated in isopropyl alcohol (IPA) solvent to obtain ultrathin Sb nanosheets; subsequently, the as-prepared Sb nanosheets were mixed with graphene (Sb:GO = 4:1) and then collected by vacuum filtration and chemically reduced to a uniform SbNS-G film; (b) rate performances of SbNS-G and graphene films at various current densities from 0.2 to 4.0 mA cm−2; (c) cycling performance of SbNS-G film at a current density of 4.0 mA cm−2. Insets of (c) shows TEM images of Sb nanosheets (d), SEM images of SbNS-G film from cross-sectional views (e, f), and a digital photograph of SbNS-G film (g), which shows its flexibility. Reprinted with permission from ref. 76. Copyright 2017, WILEY-VCH Verlag GmbH & Co. | ||
3.2 Phosphorus based alloy anodes
Phosphorus (P) has the highest theoretical capacity of 2596 mA h g−1 according to the formation of Na3P.57,58 Meanwhile, its redox potential of about 0.4 V vs. Na+/Na is appropriate for an anode in SIBs. The volume change between P and Na3P is huge (490%), however. P has three main allotropes: white, red, and black, but only red P (RP) and black P (BP) are suitable as electrode materials for battery application. RP is amorphous and commercially available, although it has low electronic conductivity (∼1 × 10−14 S cm−1). Thus, a secondary matrix with high conductivity is necessary to achieve high performance and stable cyclability.77–83 Graphene nanosheets78–83 were utilized as the conducting matrix to improve the electrical conductivity of red phosphorus and alleviate the volume changes during the sodiation/desodiation process. For example, Liu et al. developed a method to deposit red phosphorus nanodots densely and uniformly onto reduced graphene oxide sheets (P@RGO) by the physical vapor deposition (PVD) method, which allows the growth of red phosphorus particles with diameters varying from several hundreds to tens of nanometers on RGO (Fig. 5a).82 The resultant P@RGO flexible anode achieved 1165.4, 510.6, and 135.3 mA h g−1 specific charge capacity at 159.4, 31878.9, and 47818.3 mA g−1 charge/discharge current density in rate capability testing, respectively, and a 914 mA h g−1 capacity after 300 deep cycles in a cycling stability test at 1593.9 mA g−1 current density (Fig. 5b), which marks a significant performance improvement for red phosphorus anodes for sodium-ion chemistry. Gao et al.83 developed a new advanced synthesis protocol to incorporate red P nanoparticles (NPs) into a porous carbon on graphene aerogel (C@GA) matrix with a uniform distribution through a localized in situ vapor-redistribution strategy (Fig. 5c).83 The red P NPs 10–20 nm in size were uniformly distributed and firmly sealed in a well defined porous C@GA framework, which not only provides a 3D conductive network for electron transfer, but also effectively accommodates/buffers the large volume changes of red P during the charge–discharge process, helping to maintain the integrity of the electrode and enhance the electrochemical performance (Fig. 5d and e). As a result, the as-prepared C@P/GA electrode delivers a high capacity of 1867 mA h g−1 after 100 cycles at 0.1C and demonstrates a significant capacity of 1095.5 mA h g−1, even at 1C after 200 cycles (Fig. 5f).
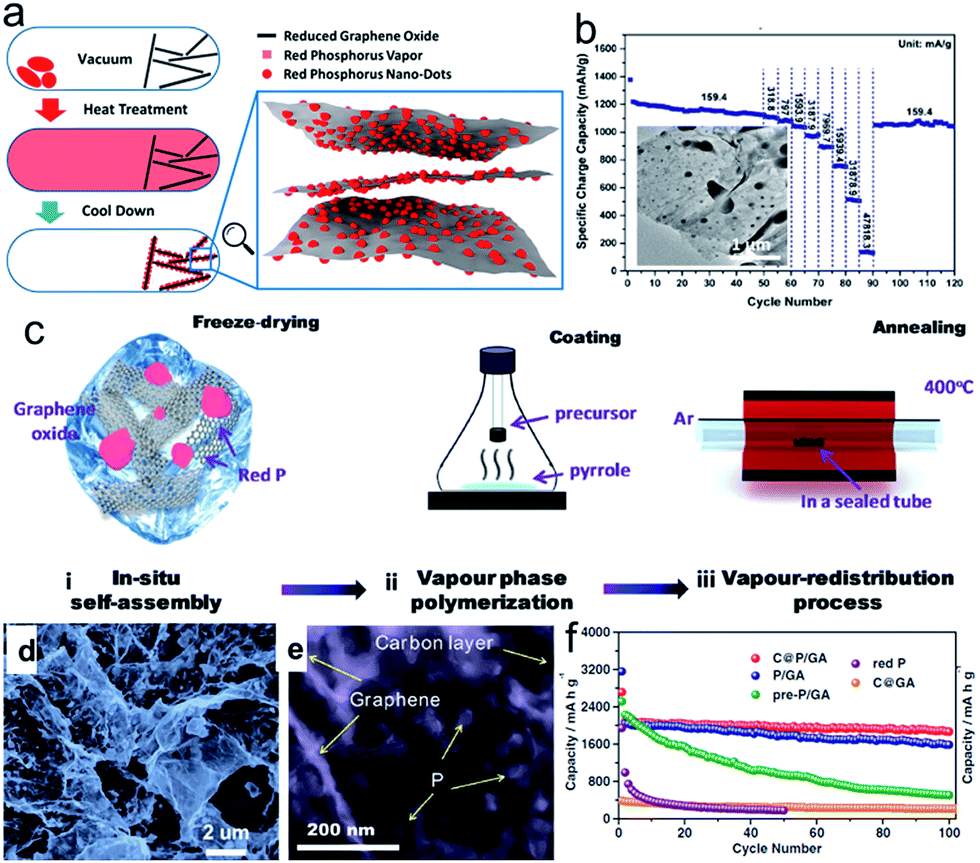 | ||
| Fig. 5 (a) Schematic description of P@RGO synthesis; (b) rate performance of the P@RGO anode; inset of (b) shows a TEM image of the P@RGO composite. Reprinted with permission from ref. 82. Copyright 2017, American Chemical Society. (c) Schematic illustration of the synthesis process for C@P/GA composite in three steps; (d, e) SEM and TEM images of pre-P/GA composite; (f) cycling performance of pre-P/GA, P/GA, C@P/GA, and C@GA composites at 0.1C, with red P shown for comparison. Reprinted with permission from ref. 83. Copyright 2016, WILEY-VCH Verlag GmbH & Co. | ||
Orthorhombic black phosphorous (BP, layered crystal structure in Fig. 6a) is thermodynamically the most stable allotrope, and it is nonflammable and insoluble in most solvents. With its layered structure of puckered sheets and good electrical conductivity, BP is very similar to graphite in terms of appearance, structure, and properties. Indeed, single layer BP, called “phosphorene”, can be mechanically exfoliated just like graphene from graphite.84 A BP/C nanocomposite synthesized by a particular ball-milling unit has been investigated as SIB anode.85 It was found that the composite can display impressive initial capacity, at the level of approximately 1300 mA h g−1 (per total weight of the composite). Nevertheless, the capacity gradually deteriorates when the cycling is performed within the potential window of 2.0–0.01 V vs. Na/Na+. In contrast, an attractive stable cycling performance at the level of 350–400 mA h g−1 can be observed if the potential window is restricted to 2.0–0.33 V vs. Na/Na+. The post-cycling SEM studies revealed that electrodes gradually disintegrate and delaminate from their current collectors when the electrochemical testing is conducted within wider potential windows. Such an effect is absent for the restricted potential windows, leading to stable cycling performance in these potential windows.
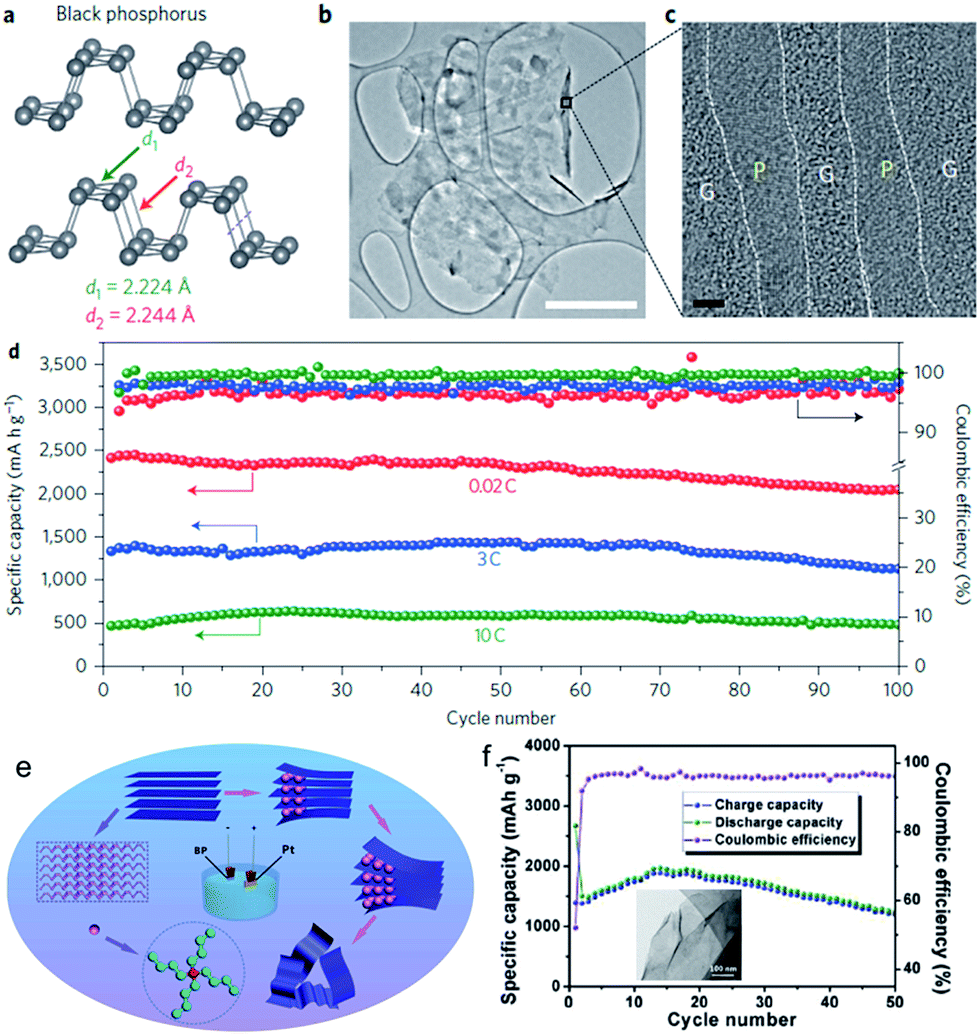 | ||
| Fig. 6 (a) Structure of black phosphorus. (b) TEM image of the phosphorene–graphene hybrid. Scale bar, 2 μm. (c) Cross-sectional HRTEM image of the phosphorene–graphene hybrid (the turned-up right edge in (b)). Scale bar, 2 nm. (d) Reversible desodiation capacity and coulombic efficiency for the first 100 galvanostatic cycles of the phosphorene/graphene (48.3 wt% P) anode tested at different current densities. Reproduced with permission from ref. 86, Copyright 2015, Springer Nature. (e) Schematic illustration of the electrochemical exfoliation process for phosphorene; (f) cycling performance and coulombic efficiency of FL-P-5 (phosphorene obtained at −5 V) at 100 mA g−1; inset of (f) shows a TEM image of FL-P-5. Reprinted with permission from ref. 87. Copyright 2017, WILEY-VCH Verlag GmbH & Co. | ||
Recently, Cui et al. demonstrated a BP/graphene nanostructured composite fabricated as a sandwich-structure comprising a few phosphorene layers alternating with several graphene layers (Fig. 6b and c).86 This phosphorene/graphene structure delivered an extremely high capacity of 2440 mA h g−1 at 50 mA g−1 and featured 83% capacity retention for 100 cycles (Fig. 6d). They discovered that the graphene layers not only function as an electrical conductor but also serve as an elastic buffer for accommodating the volume expansion during cycling. More recently, electrochemical cationic intercalation has been introduced to prepare phosphorene, through which large-area few-layer phosphorene (FL-P) without surface functional groups can be efficiently obtained (less than 1 h) (Fig. 6e).87 More importantly, its layer number (from 2 to 11 layers) can be manipulated by changing the applied potential. When directly utilized as an anode material in a SIB, phosphorene delivered a capacity of 1968 mA h g−1 at a current density of 100 mA g−1 (Fig. 6f).
4 Conversion materials
In addition to carbon-based and alloy anode materials, conversion-type materials have also been intensively studied as candidates for SIB anodes due to their high theoretical specific capacities. The conversion materials, usually including metal phosphides, oxides and sulfides/selenides, can store Na+ ions through a conversion reaction, or by a combined conversion reaction and Na-alloy reaction. In this section, we mainly focus on the typical application of two-dimensional (2D) active nanosheets and graphene-supported active materials as anodes in sodium storage.4.1 Metal phosphides
As discussed above, phosphorus has a high theoretical capacity, but suffers from huge volume changes during cycling.88 In order to buffer the volume expansion, a number of elements, such as Ni, Co, Fe, and Sn, have been introduced to form P based binary compounds, such as Ni2P,89 FeP,90 CoP,91 and Sn–P (Sn4P3, SnP3).92–96 It was demonstrated that the full sodiation of M–P (M = Ni, Co, Fe) electrode leads to the formation of Na3P electrode embedded with inactive M nanoparticles. Fast capacity fading was observed for these three samples, although the performance can be enhanced through synthesizing graphene-based nanocomposites.89–91 This capacity fading could be attributed to the agglomeration of active elements and the continuous pulverization of alloy anodes during the repeated sodiation/desodiation cycles because of the volume changes. If these elements could reform the starting compounds after Na extraction, the pulverization could be partially repaired, and the accumulation of pulverization can be terminated. An excellent example is a layered structure material, Sn4P3, which exhibited a high reversible capacity of 718 mA h g−1, with negligible capacity fading over 100 cycles, even on the ball-milled samples.92–96 Mechanism studies showed that Sn4P3 was first converted into Na3P and Sn during sodiation through the conversion reaction, and the Sn was further sodiated to form Na15Sn4 after full sodiation through the alloy reaction. After desodiation, the Sn4P3 was recovered through the reversible conversion reaction (Sn4P3 + 9Na ↔ 3Na3P + 4Sn).92,94 The excellent electrochemical performance of Sn4P3 was attributed to synergistic sodium storage reactions of the Sn and P components, where the Sn-based phase worked to provide electronic channels to enhance the electrical conductivity of the P component, and the elemental P and Na3P acted as a shielding matrix to relieve the volume expansion during the Na insertion reaction. On the other hand, the reversible reaction is critical for enhanced performance, because it repairs the cracks, damage, and aggregation of Sn particles that occur in the alloying process, 4Sn + 15Na ↔ Na15Sn4 during cycling, and hence terminates the pulverization.94 Moreover, the electrochemical performance of Sn4P3 can be further improved by introducing graphene.97,98 For example, Sn4P3/RGO hybrids with adjustable graphene content were synthesized through an in situ low-temperature solution-based phosphorization chemical transformation route from Sn/RGO (Fig. 7a).97 The monodisperse Sn4P3 nanoparticles with a small diameter of 6 nm can be loaded on the graphene nanosheets uniformly (Fig. 7b and c). The Sn4P3/RGO-2 nanohybrid with a graphene concentration of about 10.4 wt% exhibited a higher sodium storage capacity of 656 mA h g−1 at a current density of 100 mA g−1 over 100 cycles (Fig. 7d). In addition, longer cycling performance, with a capacity of 362 mA h g−1 at a current density of 1.0 A g−1, could be obtained after 1500 cycles. More recently, a nanocomposite of Sn4P3 and phosphorus (Sn:P = 1:3) embedded in a graphene matrix has been synthesized via a novel mechanochemical transformation method (Fig. 7e). This composite exhibits ultrastable and much improved capacity retention at a high current rate (>550 mA h g−1 over 1000 cycles at 1 A g−1) and unrivalled rate capability (>815 mA h g−1 at 0.1 A g−1, ∼585 mA h g−1 at 2 A g−1, and ∼315 mA h g−1 at 10 A g−1) (Fig. 7f).98
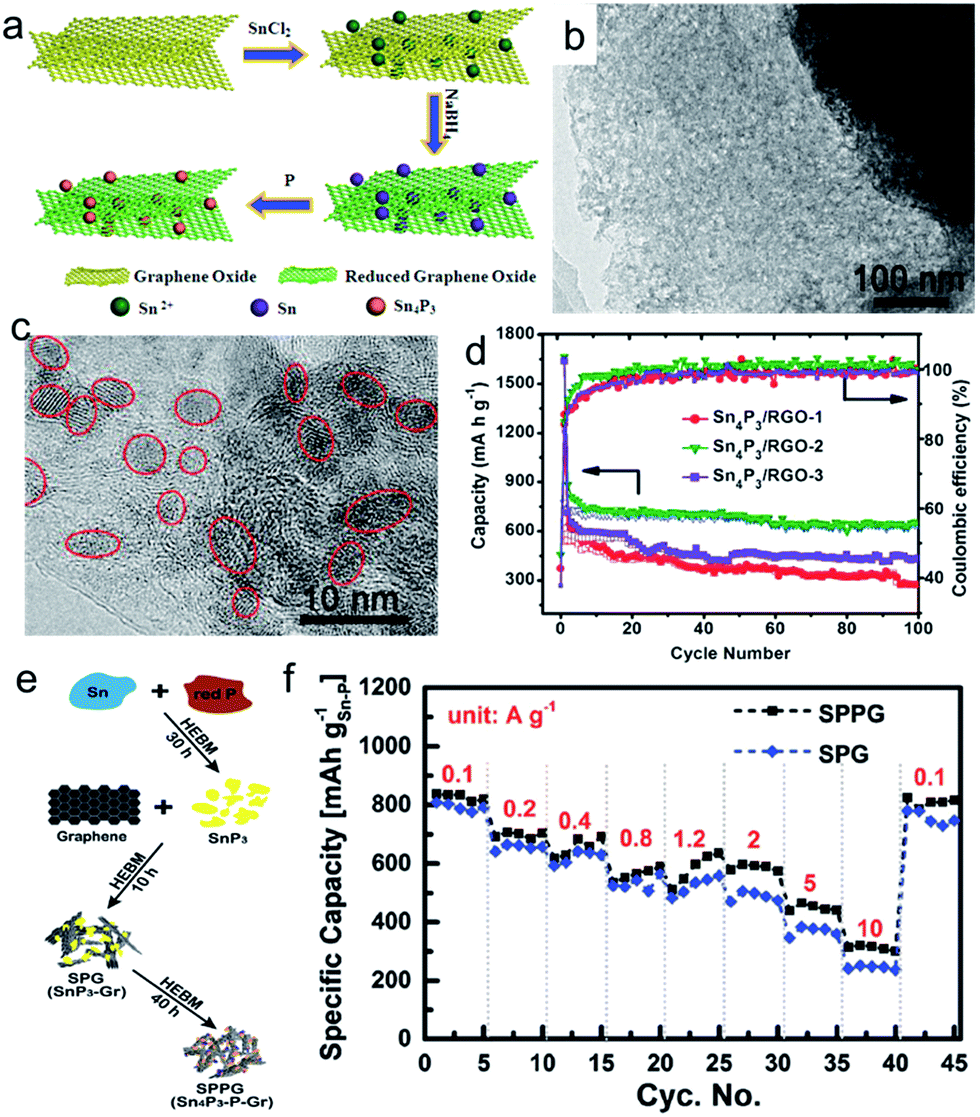 | ||
| Fig. 7 (a) Schematic illustration of the synthesis process for the Sn4P3/RGO hybrid sample; (b, c) low-magnification and high-magnification TEM images of Sn4P3/RGO sample; (d) cycling performance of Sn4P3/RGO samples at a current density of 100 mA g−1 in the voltage range of 0.01–3.00 V versus Na+/Na. Reprinted with permission from ref. 97. Copyright 2016, WILEY-VCH Verlag GmbH & Co. (e) Schematic illustration of the mechanochemical synthesis process (HEBM: high energy ball milling); (f) rate capabilities of SPPG (Sn4P3–P@graphene nanocomposite) and SPG (SnP3@graphene composite) based Na ion anodes. Reprinted with permission from ref. 98. Copyright 2017, WILEY-VCH Verlag GmbH & Co. | ||
In addition, the ball milled Sn4P3/C composite shows excellent electrochemical performance when used as a anode material for potassium-ion batteries (PIBs), which delivered a reversible capacity of 384.8 mA h g−1 at 50 mA g−1 and a good rate capability of 221.9 mA h g−1, even at 1 A g−1.99 In particular, the Sn4P3/C electrode displays a discharge potential plateau of 0.1 V in PIBs, slightly higher than for sodium-ion batteries (SIBs) (0.01 V), and well above the plating potential of metal. This diminishes the formation of dendrites during cycling, and thus Sn4P3 is relatively safe as a PIB anode.
4.2 Metal oxides
Bare oxide anodes usually suffer from bad rate capability and poor cyclability because of their extremely large volume changes during the sodium insertion–extraction process.100 One remedy is to use 2D nanosheets, since the nanosheets could provide short paths for ion transport. For example, ultrathin NiO nanosheets (4–5 nm in thickness) were synthesized via a facile solvothermal process followed by annealing in air (Fig. 8a and b).101 For sodium storage, the NiO nanosheets deliver a high reversible specific capacity of 299 mA h g−1 at a current density of 1 A g−1, and the capacity still remains as high as 266 mA h g−1 after the 100th cycle (Fig. 8c). More recently, Yu's group reported a general two-step strategy for controlled synthesis of holey 2D transition metal oxide (TMO) nanosheets with tunable pore sizes by using graphene oxides as a sacrificial template (Fig. 8d and e).102,103 This approach has been demonstrated for the synthesis of various 2D holey TMO nanosheets, including simple oxides such as Fe2O3, Co3O4, abd Mn2O3, and mixed oxides such as ZnMn2O4 (ZMO), ZnCo2O4 (ZCO), NiCo2O4 (NCO) and CoFe2O4 (CFO). In particular, 2D holey TMO nanosheets exhibit much improved rate capability and cycling stability for both lithium and sodium ion storage, due to the increased surface areas and interfaces, facile interfacial transport, and shortened diffusion paths.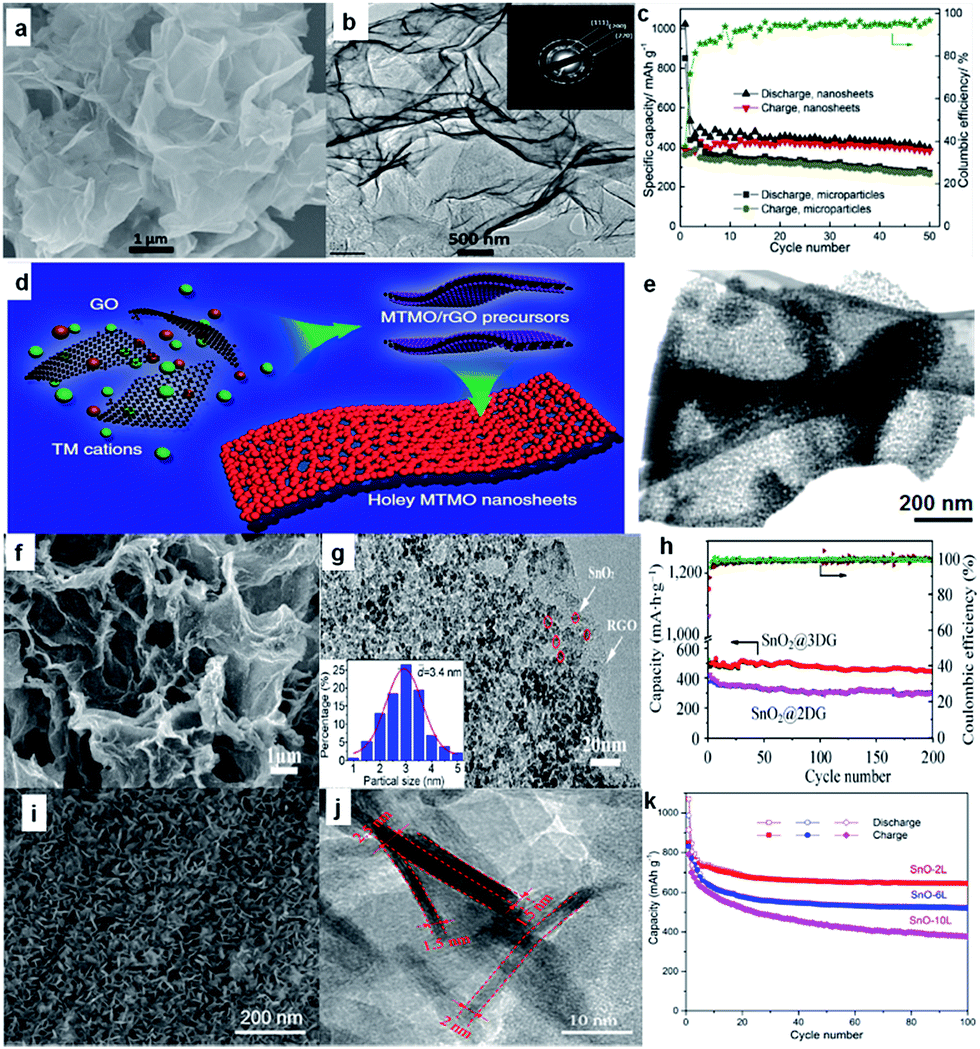 | ||
| Fig. 8 (a) SEM image of NiO nanosheets; (b) high-resolution TEM (HRTEM) image of NiO nanosheets, with the inset the selected-area electron diffraction (SAED) pattern; (c) cycling performances of SIBs with NiO nanosheet electrode at 0.1 A g−1 and NiO microparticle electrode at 0.2 A g−1. Reprinted with permission from ref. 101. Copyright 2015, Elsevier. (d) Schematic illustration showing the general strategy to synthesize 2D holey TMO nanosheets. Two transition metal (TM) cations are mixed with graphene oxide (GO) and then anchored on surfaces of reduced graphene oxide (rGO) templates during the solution-phase reaction. 2D holey mixed transition metal oxide (MTMO) nanosheets composed of interconnected MTMO nanocrystals are formed after removing the rGO templates during the post-calcination; (e) STEM image of 2D holey ZMO nanosheets shows holey nanosheets composed of interconnected ZMO nanocrystals. Reproduced with permission from ref. 102. Copyright 2017, Springer Nature. (f) SEM image of SnO2@3DG; (g) TEM image of SnO2@3DG. Inset: the corresponding particle size distribution calculated from 100 particles, (h) cycling performance of SnO2@3DG and SnO2@2DG at the current density of 100 mA g−1. Reprinted with permission from ref. 116. Copyright 2015, Elsevier. (i, j) SEM and TEM images of SnO-2L nanosheets; (k) cycling performances of bare SnO and SnO-2L electrodes at 0.1 A g−1 for 100 cycles. Reprinted with permission from ref. 117. Copyright 2017, American Chemical Society. | ||
Another strategy is to combine the oxide with a 2D support such as graphene, carbon cloth, and stainless steel mesh, which could enhance the electrical conductivity with its highly conductive network. Various oxides, including Fe2O3,104,105 Fe3O4,106 Co3O4,107,108 NiO,109 CuO,110 MoO3,111 Mn3O4,112 SnO2,112–116 and SnO117 have been investigated as SIB anodes. Among them, Sn-based oxides such as SnO2 and SnO are attracting special attention owing to their high theoretical reversible capacity, moderate operating voltage, good performance, and low cost. For example, Wang et al. reported SnO2 nanoparticles (5 nm) anchored on 2D rGO frameworks via a simple hydrothermal method, and the composite electrode delivered a stable capacity of 302 mA h g−1 over 100 cycles at a current density of 160 mA g−1.114 More recently, Pei et al. demonstrated that 3D SnO2@G (SnO2@3DG) showed better sodium storage performance than its 2D equivalent (SnO2@2DG), which was attributed to the high surface area and 3D porous architecture.116 They designed and achieved ultra-small SnO2 nanoparticles embedded in 3D graphene (SnO2@3DG) as anode for SIBs via assembling ice-templated 3DG with a tin source in a hydrothermal process (Fig. 8f and g). Compared to the SnO2@2DG anode, the SnO2@3DG anode showed a higher reversible capacity of 432 mA h g−1 after 200 cycles at 100 mA g−1 (Fig. 8h).116 SnO nanosheets with a controlled number of atomic layers were chemically prepared on carbon cloth and systematically studied (Fig. 8i and j).117 The number of atomic layers in SnO nanosheets was found to significantly influence their physical and electrochemical properties. The thinnest SnO nanosheet anodes (two to six SnO monolayers) exhibited the best performance (Fig. 8k). Specifically, an initial discharge and charge capacity of 1072 and 848 mA h g−1 were observed, respectively, at 0.1 A g−1. In addition, an impressive reversible capacity of 665 mA h g−1 after 100 cycles at 0.1 A g−1 and 452 mA h g−1 after 1000 cycles at a high current density of 1.0 A g−1 was observed, with excellent rate performance. The results indicate that the large volume changes typically observed in SnO anodes can be significantly reduced by controlling the number of atomic layers in each SnO nanosheet.
4.3 Metal sulfides/selenides
Compared to the metal oxide anodes, metal sulfide anodes have several potential advantages: (1) greater gravimetric energy density due to the lower weight of the S atoms; (2) better structural stability due to the smaller volume changes; (3) higher initial cycling efficiency resulting from the better reversibility of the Na2S formed during sodiation than its counterpart oxide (Na2O).118,119 Various metal sulfides, such as FeS2,120–123 FeS,124 Ni3S2,125 CoS,126–128 MnS,129 ZnS,130 and Bi2S3,131 have shown attractive electrochemical performance in sodium batteries. Moreover, the discovery of the amazing properties of graphene has stimulated exploration of single- and few-layer structures of layered inorganic materials. Layered metal sulfides (MoS2,132–141 SnS2,142–152 SnS,153–158 Sb2S3,159–162 WS2,163–165 and VS2165,166) have been extensively investigated as anode materials for sodium ion batteries. These layered metal sulfides typically have a unique layered structure with a large interlayer spacing, which makes them suitable for the intercalation of Na ions and allows compensation of the alloying/de-alloying volume changes. Nevertheless, the bulk materials still suffer from sluggish reaction kinetics and severe volume changes during the sodiation/desodiation process. Several strategies have been investigated to overcome these problems, including obtaining few-layer nanosheets and making hybrids with graphene flakes.
One way to overcome the drawbacks of layered metal sulfides is to obtain few-layer nanosheets or nanosheet assemblies, which facilitates the high-rate transportation of sodium ions due to the short diffusion paths provided by the ultrathin thickness. For example, ultrathin few-layer MoS2 nanosheets (∼10 nm in thickness) have been prepared through a simple and scalable exfoliation technique (Fig. 9a).134 When applied as anode material in SIBs, the MoS2 nanosheet electrode demonstrated a high reversible specific capacity of 530 mA h g−1, good cyclability, and high-rate performance (Fig. 9b). 2D SnS2 nanosheets (3–4 nm in thickness) were synthesized via a facile refluxing process and evaluated as an anode material for SIBs (Fig. 9c).142 The SnS2 nanosheets delivered a high reversible specific capacity of 733 mA h g−1 at 0.1 A g−1, and still exhibited high capacity retention of 647 mA h g−1 during the 50th cycle at 0.1 A g−1 (Fig. 9d). Moreover, Zhou et al. prepared VS2 nanosheet assemblies via a facile solvothermal method, which consisted of assemblies of ultrathin nanosheets with few-atomic layer thickness (Fig. 9e).165 Electrochemical measurements showed that the VS2 nanosheet assemblies enabled the rapid and durable storage of Na+ ions. Noteworthy capacities of 550 and 400 mA h g−1 were attained at 1 and 2 A g−1, respectively (Fig. 9f). The excellent electrochemical properties observed for the few-layer nanosheets could be ascribed to the ultrathin nanosheet architecture, which is beneficial for achieving fast sodiation/desodiation reaction kinetics and accommodating electrode volume changes.
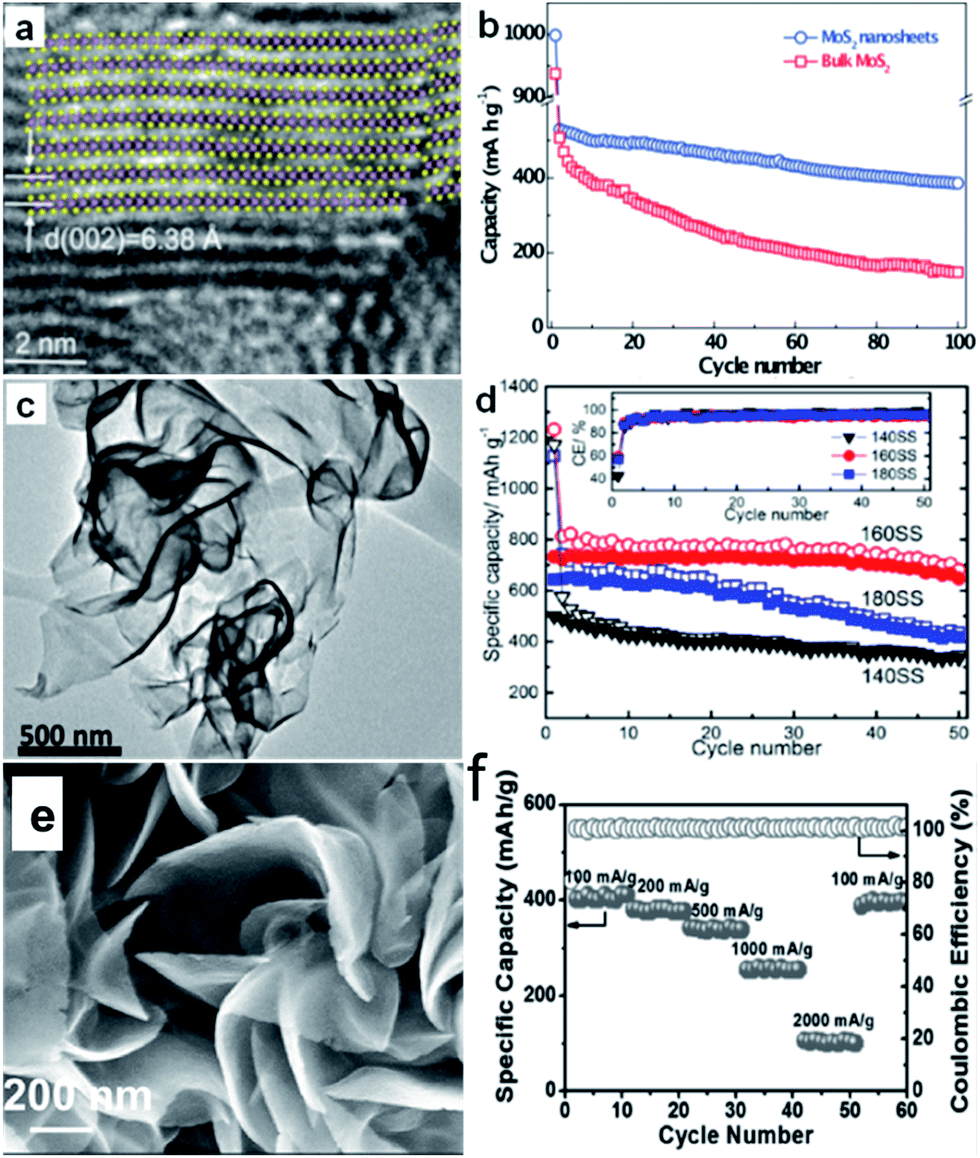 | ||
| Fig. 9 (a) Lattice resolved HRTEM image of MoS2 nanosheet; (b) comparison of cycling performances of bulk and exfoliated MoS2 nanosheet electrodes at 40 mA g−1 current density. Inset of (a) is the crystal structure of MoS2 viewed along the b-axis. Reprinted with permission from ref. 134, Copyright 2014, WILEY-VCH Verlag GmbH & Co. (c) HRTEM image of SnS2 synthesized at 160 °C, (d) cycling performances of the cells with SnS2 samples synthesized at 140 °C (140SS), 160 °C (160SS), and 180 °C (180SS), respectively, at 0.1 A g−1. Inset of (d) is the coulombic efficiency (CE) of the three cells. Reprinted with permission from ref. 142. Copyright 2015, American Chemical Society. (e) SEM images of VS2 nanosheet assemblies (NSA); (f) rate capability of VS2 NSA from 100 to 2000 mA g−1 and the corresponding coulombic efficiency. Reprinted with permission from ref. 165, Copyright 2017, WILEY-VCH Verlag GmbH & Co. | ||
Furthermore, nanocomposites of few-layer sulfide and graphene or reduced graphene oxide flakes for use as SIB anodes have been developed. Normally, the excellent performance of a 2D sulfide/graphene hybrid can be attributed to the 2D conductive channels provided by the graphene, the small lateral size and ultrathin nature of the layered sulfide, the unique hybrid structure with enhanced electrolyte penetration, and the rapid Na-ion transport across the thin solid-electrolyte interphase (SEI) layer. For example, Zhang et al. fabricated a unique plate-on-sheet structured SnS2/rGO nanohybrid consisting of ultrafine (<10 nm), few-layered (≤7 layers) SnS2 and few-layered rGO (<6 layers) (Fig. 10a).143 The charge capacity at 100 mA g−1 increased from 178 mA h g−1 for bare SnS2 to 649 mA h g−1 for SnS2/rGO due to the dispersive and conductive effects of rGO. Moreover, the hybrid could yield high charge capacities of 524, 501, and 452 mA h g−1 at high current densities of 1.6, 3.2, and 6.4 A g−1, respectively. Even at 12.8 A g−1 (28C), it could still deliver a moderate charge capacity of 337 mA h g−1 (Fig. 10b). Our recent results showed that the hexagonal-SnS2 phase can also be transformed into the orthorhombic-SnS phase after an annealing step in argon atmosphere, and the thus transformed SnS shows sodium-ion storage performance that is enhanced over that of the original SnS2.153 As shown in Fig. 10c and d, the SnS@graphene hybrid nanostructured composite, built from two-dimensional SnS and graphene nanosheets as complementary building blocks, delivers an excellent specific capacity of 940 mA h g−1 and impressive rate capability of 492 and 308 mA h g−1 after 250 cycles at the current densities of 810 and 7290 mA g−1, respectively, which is much better than the performance of the SnS2@graphene hybrid. Detailed investigation showed that the SnS experienced a two-structural-phase transformation mechanism (orthorhombic-SnS to cubic-Sn, and further to orthorhombic-Na3.75Sn), while the SnS2 experienced a three-structural-phase transformation mechanism (hexagonal-SnS2 to tetragonal-Sn, and further to orthorhombic-Na3.75Sn) during the sodiation process. Thus the lesser structural changes in SnS during the conversion are expected to lead to good structural stability and excellent cycling stability in its SIB performance. More recently, our group successfully designed ultrafine SnS/SnO2 heterostructures, where an interfacial amorphous carbon layer anchored the SnS/SnO2 heterostructures directly to graphene nanosheets (Fig. 10e).156 When evaluated as an anode material for sodium-ion batteries, the C@SnS/SnO2@Gr sample (where Gr is graphene), featured excellent performance and outstanding cycling stability at high rates, so that it had much superior performance compared to C@SnO2@Gr, C@SnS@Gr, or a mechanical mixture of them (Fig. 10f). The boosted charge transfer in SnS/SnO2 heterostructures is attributed to the heterostructure interface effect, which induces an electric field within the nanocrystals, giving them much lower ion-diffusion resistance and facilitating interfacial electron transport.
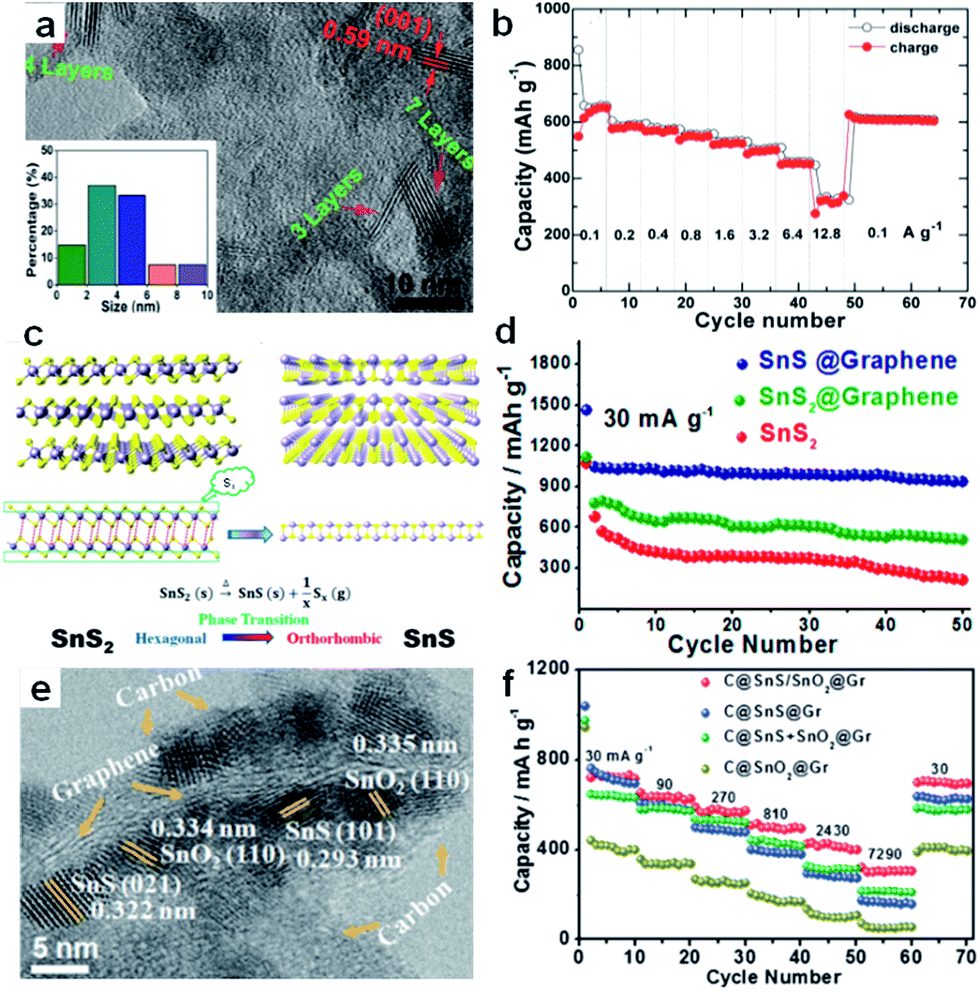 | ||
| Fig. 10 (a) HRTEM image, with the inset showing the particle size distribution, and (b) rate performance of SnS2/rGO. Reprinted with permission from ref. 143. Copyright 2014, WILEY-VCH Verlag GmbH & Co. (c) Schematic diagram illustrating the phase transition process between SnS2 and SnS, (d) cycling performances of SnS@graphene, SnS2@graphene, and SnS2 electrodes. Reprinted with permission from ref. 153. Copyright 2014, American Chemical Society. (e) TEM image of C@SnS/SnO2@Gr, (f) rate capabilities of C@SnS/SnO2@Gr, C@SnS@Gr, C@SnO2@Gr, and C@SnS + SnO2@Gr electrodes. Reprinted with permission from ref. 156. Copyright 2016, WILEY-VCH Verlag GmbH & Co. | ||
In additon to sulfides, 2D layer-structured selenides,167–172 such as MoSe2,168,169 Sb2Se3,170 SnSe2,171 and SnSe,172 have also attracted research attention for SIBs, owing to their similar physical and chemical properties to sulfides. In particular, compared with the sulfides, the selenides may possess larger interlayer spacing and higher conductivity, resulting from the smaller band gap. Such properties endow the selenides with considerable potential as anode for superior SIBs with better coulombic efficiency and rate capability. For example, MoSe2 was grown on N,P-co-doped carbon nanosheets by a solvothermal reaction followed by a high-temperature calcination.169 The obtained composite (MoSe2/N,P–rGO), consisted of loosely stacked carbon nanosheets and uniformly deposited MoSe2 with expanded interlayer spacing (Fig. 11a and b). This feature benefits electrolyte permeation to the composite, charge transportation throughout the electrode, and structural stability against volume changes during cycling. So, the composite exhibits outstanding electrochemical performance in SIBs. After 1000 cycles at 0.5 A, there is still a reversible capacity of 378 mA h g−1, which is ∼87% of the capacity at the second cycle (Fig. 11c). Ou et al. reported a hierarchically structured Sb2Se3/rGO composite as an excellent SIB anode. The uniquely structured Sb2Se3/rGO composite, in which 1D Sb2Se3 nanorods were uniformly wrapped by rGO, was synthesized by a facile solvothermal approach (Fig. 11d and e).170 The Sb2Se3/rGO anode exhibited a high reversible specific capacity of 682 mA h g−1 at 0.1 A g−1, and excellent stability for 500 cycles (Fig. 11f). Detailed mechanism investigations revealed that the high reversible capacity of Sb2Se3/rGO originated from a combined Na+ intercalation, a conversion reaction between Na+ and Se, and an alloying reaction between Na+ and Sb. Furthermore, the rGO nanosheets uniformly overcoating Sb2Se3 nanorods are enablers for high and stable performance by providing fast electron transport, easy accommodation to volume changes, and enhanced electron interaction. Moreover, pure SnSe2 nanosheets and SnSe2/RGO nanocomposites were synthesized via a facile hydrothermal method by using N2 saturated NaHSe solution as a safe and stable selenium source (Fig. 11g and h).171 The as-prepared SnSe2 has a layered structure, while the interlayer spacing can accommodate more sodium ions, and the insertion of RGO can improve the electrical conductivity and structural stability of the electrodes. When tested as anode material for SIBs, the SnSe2/RGO composite delivered a high initial capacity of 798 mA h g−1 and still exhibited a reversible capacity of 515 mA h g−1 at 0.1 A g−1 after 100 cycles (Fig. 11i).
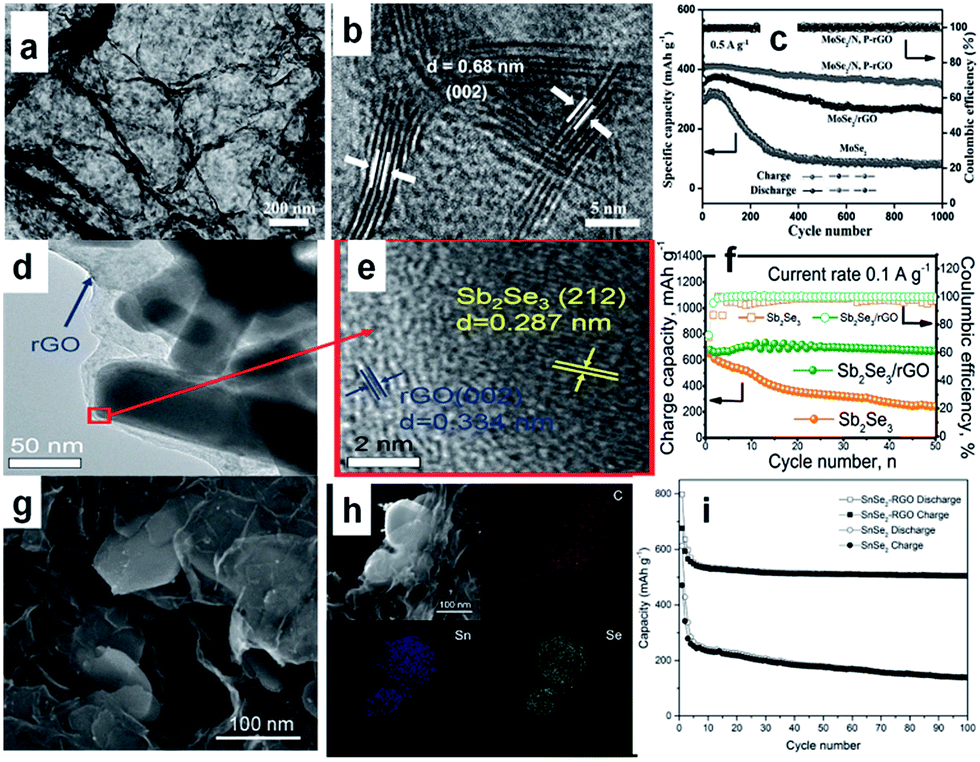 | ||
| Fig. 11 (a) TEM and (b) HRTEM images of MoSe2/N,P–rGO; (c) cycling stability of the electrodes based on MoSe2/N,P–rGO, MoSe2/rGO, and MoSe2. Reprinted with permission from ref. 169. Copyright 2017, WILEY-VCH Verlag GmbH & Co. (d) TEM and (e) HRTEM images of Sb2Se3/rGO; (f) cycling performance of Sb2Se3 and Sb2Se3/rGO at a current density of 0.1 A g−1. Reprinted with permission from ref. 170. Copyright 2017, WILEY-VCH Verlag GmbH & Co. (g) SEM image of SnSe2/RGO nanocomposites (h) energy dispersive X-ray spectroscopy (EDX) mapping images of C, Sn, and Se elements with corresponding SEM image; (i) cycling performances of SnSe2 and SnSe2/RGO electrodes at 0.1 A g−1 for 100 cycles. Reprinted with permission from ref. 171. Copyright 2016, WILEY-VCH Verlag GmbH & Co. | ||
5 Ti-based anode materials
With higher potentials for sodium insertion than carbon based materials, sodium plating can be avoided in Ti-based materials, which also makes them promising candidates for safe anodes in sodium-ion batteries. In the following sections, recent research achievements on titanium based anode materials are reviewed, with the focus on 2D architectures.5.1 Conventional Ti-based materials
Various TiO2 polymorphs, including amorphous TiO2,173 anatase,174–179 rutile,180,181 and TiO2(B),182–184 have been introduced as efficient anode materials for Na ion storage. For example, Xiong et al.173 investigated amorphous TiO2 grown on a 2D Ti substrate. Interestingly, the capacity increased gradually with charge–discharge cycling, presumably owing to the amorphous structure. A recent study, however, showed that anatase TiO2 presented better capacity than mixed anatase/rutile TiO2 or amorphous TiO2.178 Through crystallographic analysis, it was revealed that the anatase TiO2 crystal structure supplies two-dimensional diffusion paths for Na-ion intercalation and more accommodation sites. The TiO2(B) has an open-channel structure and has been regarded as the most promising polymorph of TiO2 for LIBs due to its outstanding Li-ion mobility and superior electrochemical performance.184 The reported electrochemical performance of TiO2(B) in SIBs is not as good as in LIBs, however, which is possibly due to the poor electrical and ionic conductivity of TiO2.182–184 Enhancing the electrical conductivity of TiO2 will most likely lead to improved electrochemical performance in SIBs.175–190 Recently, Chen et al. reported Na+ intercalation pseudocapacitance in a TiO2 (consisting of 76.9% TiO2(B) and 23.1% anatase TiO2)/graphene nanocomposite, which enabled high-rate capability and long cycle life in a sodium-ion battery.186 In their study, the hybridization of graphene with TiO2 nanocrystals provided a more feasible channel at the graphene–TiO2 interface for sodium intercalation/de-intercalation with a much lower energy barrier, thus leading to fast charge storage and long-term cyclability (Fig. 12a–d).186 More recently, Le et al. reported a microwave-assisted synthesis of single-crystal-like anatase TiO2 mesocages anchored on graphene as a sodium storage material, which delivered a high capacity of 268 mA h g−1 at 0.2C and retained 126 mA h g−1 at 10C for over 18000 cycles (Fig. 12e–g).190 The excellent performance can be attributed to the robust architecture of the nanocomposite. The mesoporous texture and small primary building blocks of TiO2 mesocages offer fast ion insertion/deinsertion and a short diffusion distance, while the graphene sheets provide a continuous electron conducting network.
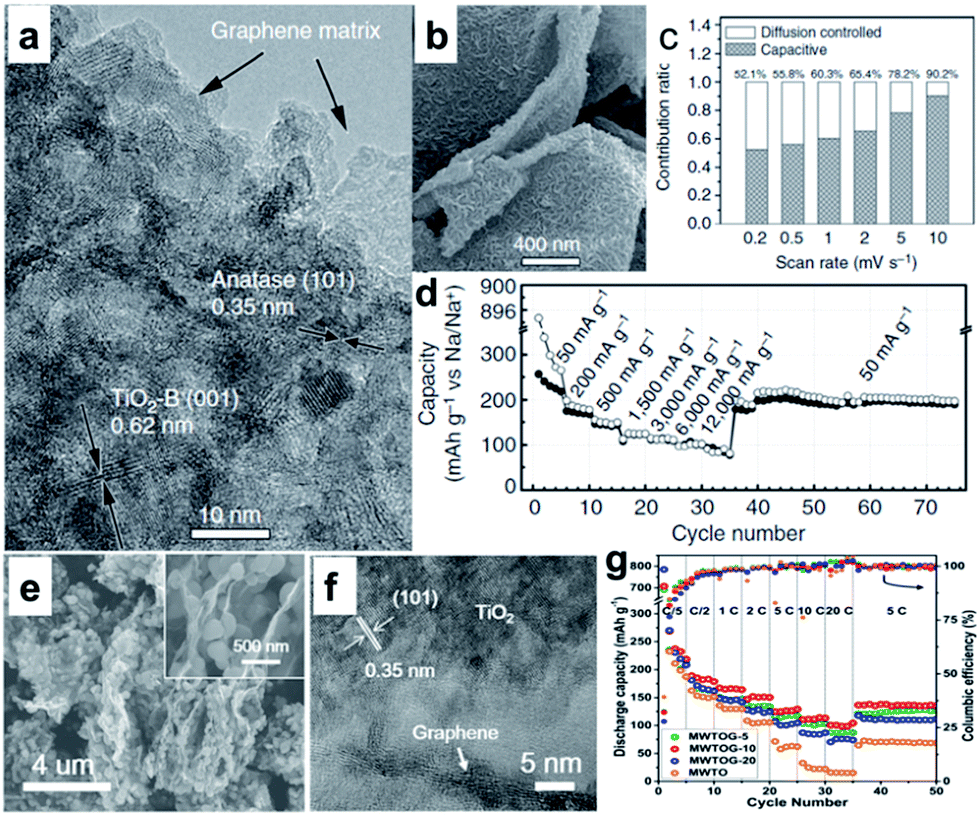 | ||
| Fig. 12 (a) High-resolution TEM image of TiO2/graphene composite, clear lattices with spacings of 0.62 and 0.35 nm are assigned to the (001) planes of TiO2-B and (101) planes of anatase, respectively. (b) High-magnification SEM image of G-TiO2, revealing the structural detail of an individual microsheet. (c) Contribution ratios of the capacitive and diffusion-controlled charge from CV curves at various scan rates. (d) Rate performance at various current densities from 50 to 12000 mA g−1. Reproduced with permission from ref. 186. Copyright 2015, Springer Nature. (e) SEM and (f) HRTEM images of MWTOG nanocomposite. (g) Rate performance of MWTOG (microwave-assisted TiO2 mesocage-graphene nanocomposite) and MWTO (microwave-assisted pure TiO2 mesocages) electrodes, ramping from 0.2 to 20C and back to 5C. Reprinted with permission from ref. 190. Copyright 2017, American Chemical Society. | ||
Spinel-type Li4Ti5O12 (LTO) is well known as a ‘zero-strain’ anode material for long-life stationary lithium-ion batteries.191 Recently, the suitability of Li4Ti5O12 as an anode for Na-ion batteries has been revealed.192–197 The intercalation mechanism has been proposed to involve three-phase separation, which is very different from that observed in LIBs.192 It was also reported that the sodium storage properties of LTO in SIBs are size-dependent, demonstrating that nanostructuring could lead to a significant increase in the specific capacity.193 Recently, LTO nanosheets were fabricated via a simple hydrothermal reaction and found to deliver a reversible capacity of up to 145 mA h g−1 at 1C, with 91% capacity retention after 400 cycles.195 Based on a scan rate-dependent cyclic voltammetry test, a pseudocapacitive charge storage mechanism has been proposed for Na-ion storage in pristine LTO electrode, which contributes to the excellent rate capacity and high cycling stability of LTO electrodes for SIBs. More recently, ultra-small MoS2 quantum dots (QDs) were exploited as surface sensitizers to boost the electrochemical properties of 2D LTO nanosheets.197 The LTO/MoS2 composite was prepared by anchoring ultra-small MoS2 QDs on 2D LTO nanosheets by using a simple and effective assembly technique (Fig. 13a). Impressively, such zero-dimensional (0D)/2D heterostructured composites possess enhanced surface-controlled Na storage behavior. During sodiation, a high capacity of 91 mA h g−1 at the rate of 5C and superior capacity retention, with 101 mA h g−1 after 200 cycles at the rate of 2C, were achieved (Fig. 13b and c).
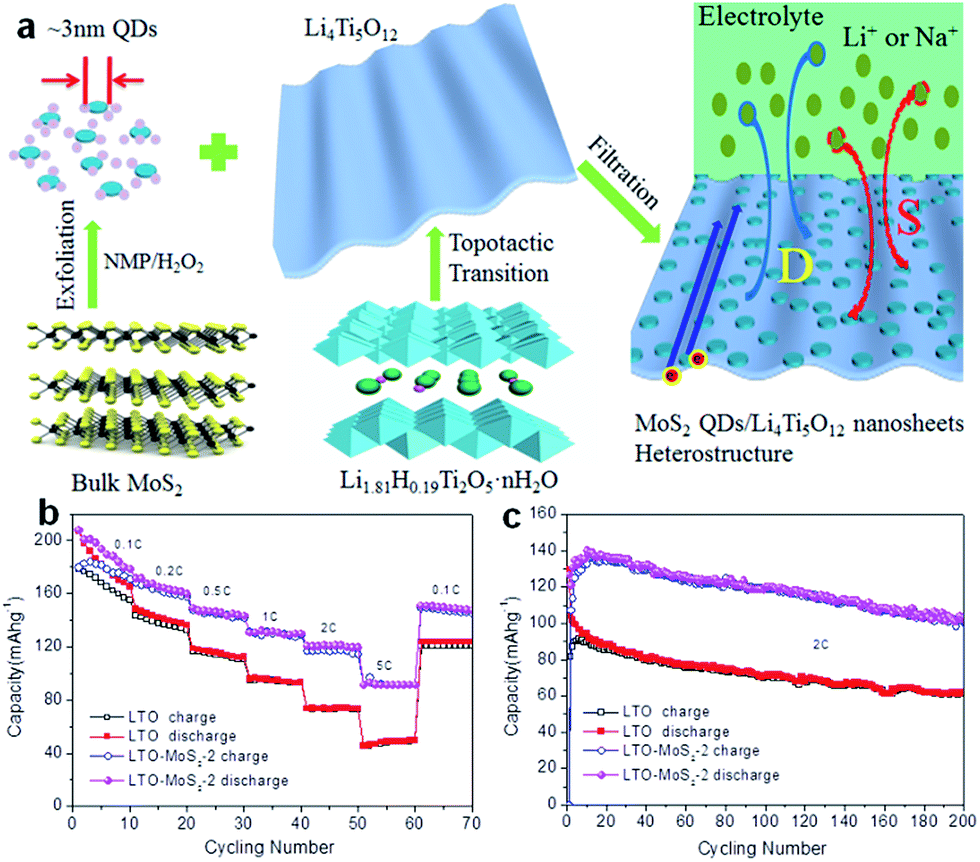 | ||
| Fig. 13 (a) Schematic illustration showing the synthesis of the heterostructured composite composed of ultrasmall MoS2 QDs and ultrathin LTO nanosheets. (b) Corresponding rate capabilities at different current rates from 0.1C to 5C for the LTO-MoS2-2 composite and LTO nanosheets electrodes. (c) Long cycling test of the LTO-MoS2-2 composite and LTO nanosheets as SIB electrodes at a rate of 2C. Reprinted with permission from ref. 197. Copyright 2016, WILEY-VCH Verlag GmbH & Co. | ||
Sodium titanate (Na2Ti3O7), with a lower discharge plateau and abundant raw material resources, is another promising titanium-based anode material for SIBs.198–203 The Na2Ti3O7 structure consists of zigzag layers of titanium and oxygen octahedra, in which up to 3.5 Na ions per formula unit can be intercalated into the interlayer space and easily exchanged, leading to a capacity of 310 mA h g−1. Pan et al.199 found that layered Na2Ti3O7 shows a reversible capacity of 85 mA h g−1 (0.5C) after 100 cycles. At the same time, the Na2Ti3O7 also suffers from structural distortion, which may account for the low coulombic efficiency and continuous capacity fading of Na2Ti3O7 electrodes. One strategy is to engineer Na2Ti3O7 nanostructures to improve the conductivity and stability issues.200–203 Dunn et al. reported that the electrochemical properties of Na2Ti3O7 can be improved by exfoliating its layered structure to forming 2D nanoscale morphologies such as nanoplatelets and nanosheets (Fig. 14a–c).202 Exfoliation of Na2Ti3O7 was carried out by controlling the amount of proton exchange for Na+ and then proceeding with the intercalation of larger cations such as methylammonium and propylammonium. The charge-storage kinetics for the nanosheets is capacitor-like in nature while that of the nanoplatelets is diffusion-controlled and resembles that of bulk Na2Ti3O7 (Fig. 14d). Materials that combined both nanosheets and nanoplatelets exhibited far better charge-storage properties (110 mA h g−1 at 10C) and more stable cycling behavior than that of the corresponding bulk materials, underscoring the importance of the 2D nanoscale morphology. A free-standing and sandwich-structured Na2Ti3O7 nanowires@carbon nanotube (CNT)@reduced graphene oxide (NTO NW@CNT@rGO) was prepared by hydrothermal-assisted modified vacuum filtration and investigated for sodium ion batteries (Fig. 14e–h).203 The as-obtained S–NTO NW@CNT@rGO flexible electrodes are not only binder-free and current-collector-free but also exhibit excellent electrochemical performances. The S–NTO NW@CNT@rGO electrode delivered discharge capacities of 204.8, 162, 115.5, 86.5, 56, and 31.9 mA h g−1 at current densities of 0.2C, 0.5C, 1C, 2C, 5C, and 10C, respectively. The electrode was even continuously cycled for 100 cycles at a current density of 0.2C, which showed durable cycling capacity, with a high reversible capacity of 114.1 mA h g−1 retained over 100 cycles (Fig. 14i). The superior electrochemical performance of the electrodes is believed to be related to the unique sandwich architecture formed by the ultralong Na2Ti3O7 nanowires@CNT layer and highly conductive graphene layer.
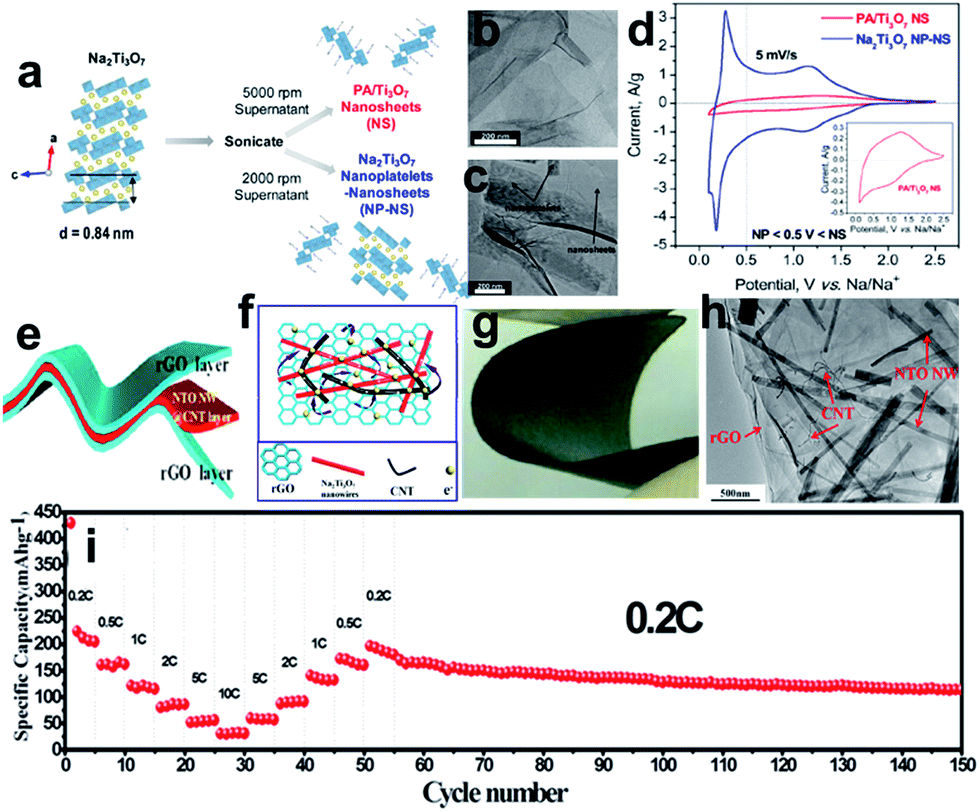 | ||
| Fig. 14 (a) Exfoliation flowchart for obtaining propylammonium Ti3O7 nanosheets (PA/Ti3O7 NS) and mixed nanoplatelets and nanosheets of Na2Ti3O7 (Na2Ti3O7 NP-NS); TEM images of (b) PA/Ti3O7 nanosheets and (c) mixed nanoplatelets and nanosheets of Na2Ti3O7; (d) cyclic voltammograms of mixed nanoplatelets and nanosheets of Na2Ti3O7 and PA/Ti3O7 nanosheets cycled at 5 mV s−1 from 0.1 to 2.5 V vs. Na/Na+. Reprinted with permission from ref. 202. Copyright 2017, American Chemical Society. (e, f) Schematic illustration and ideal electron-transfer pathways of a free-standing and sandwich-structured flexible film electrode; (g) digital optical image of the bending state of the NTO NW@CNT@rGO film; (h) TEM image of the NTO NW@CNT@rGO film; (i) rate performance at various current densities from 0.2C to 10C and then cycling performance at 0.2C. Reprinted with permission from ref. 203. Copyright 2017, American Chemical Society. | ||
Transition metal phosphates, such as NaTi2(PO4)3 (NTP),204–209 have also been studied so far. NTP has an open three-dimensional sodium superionic conductor (NASICON)-type framework structure and high Na+ conductivity, which are decisive advantages for electrochemical storage kinetics. Its low electronic conductivity induces low capacity release and poor cycling stability, however. An effective conductive network created by combining NTP with electrically conductive materials will improve its performance, however. A novel architecture consisting of nanosized porous NTP particles embedded in a microsized 3D graphene network has been designed by Wu et al. (Fig. 15a–d).206 Such an architecture synergistically combines the advantages of a 3D graphene network and of 0D porous nanoparticles. It greatly increases the electron/ion transport kinetics and endows the electrode with structural integrity, leading to attractive electrochemical performance, as reflected by its high rate-capability (112 mA h g−1 at 1C, 105 mA h g−1 at 5C, 96 mA h g−1 at 10C, 67 mA h g−1 at 50C) and long cycle life (capacity retention of 80% after 1000 cycles at 10C) (Fig. 15e). It also should be noted that NTP has a relatively high voltage plateau at ∼2.1 V vs. Na+/Na, which sacrifices energy density to some extent when compared to other anodes (e.g., hard carbon). This redox reaction takes place, however, at potentials that are much more positive than the H2 evolution potential of water in the Na2SO4 electrolyte (<−1.2 V vs. Ag/AgCl). Thus, this disadvantage is turned into a benefit in neutral aqueous solutions, excluding the decomposition of water.207–209
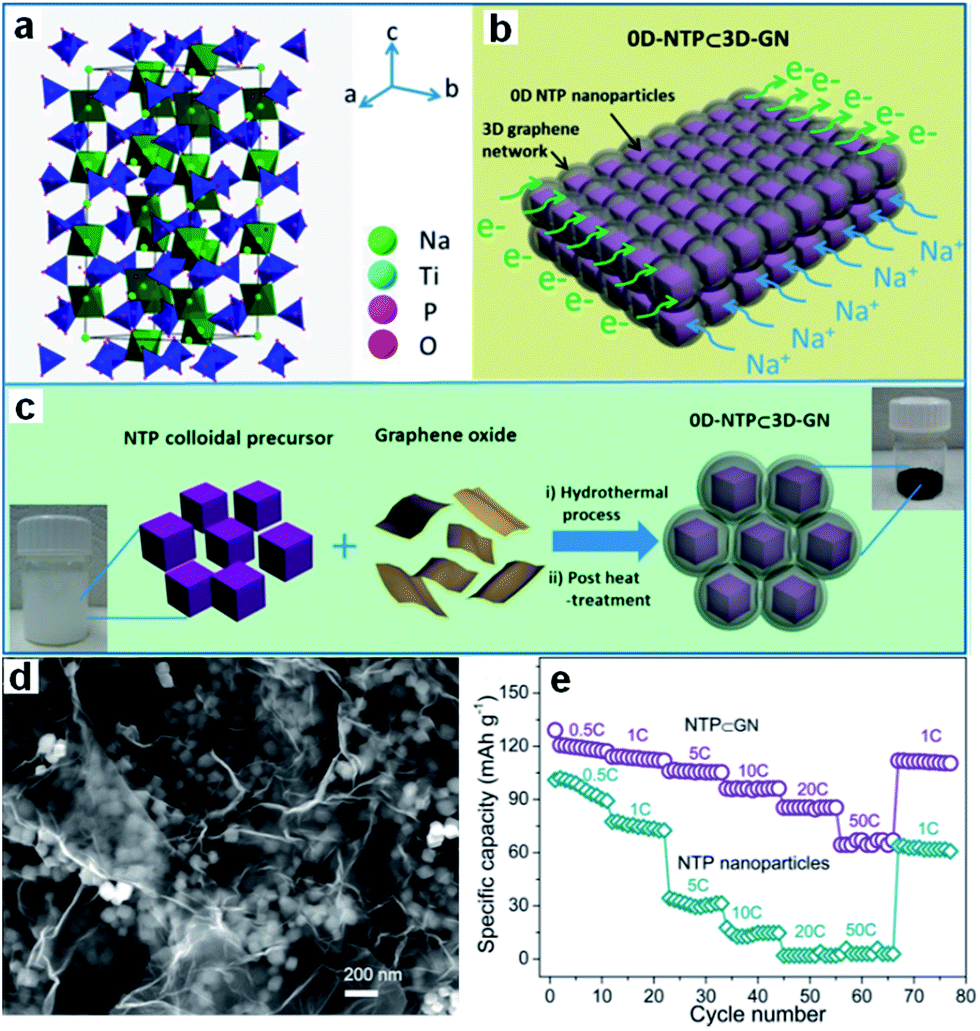 | ||
| Fig. 15 (a) Crystal structure of the NTP phase. (b) Schematic illustration of 0D-NTP⊂3D-GN, showing that 0D porous NTP nanoparticles are embedded in a 3D graphene network. (c) Schematic illustration of synthesis of 0D-NTP⊂3D-GN, including the two steps of the hydrothermal process and the post heat treatment. (d) SEM image of NTP⊂GN, (e) rate performance and capacity retention capability of the NTP⊂GN and NTP particle electrodes. Reprinted with permission from ref. 206. Copyright 2015, American Chemical Society. | ||
5.2 New Ti-based materials: MXene nanosheets
Recently, a new group of 2D materials called MXenes (Mn+1XnTx: M = Ti, V, Nb, etc.; X = C, N; n = 1–3; Tx is the functional termination group) has been discovered, and their potential applications have been demonstrated in rechargeable batteries, including sodium ion batteries.210–218 MXenes are produced by selective etching of the A element from the MAX phases (Fig. 16a and b).210 The latter are so-called because of their composition: namely, Mn+1AXn, where M is an early transition metal, A is mainly a group IIIA or IVA (i.e., groups 13 or 14) element, X is C and/or N, and n = 1, 2, or 3. The MAX phase structure can be described as 2D layers of early transition metal carbides and/or nitrides “glued” together with an A element (Fig. 16b). For example, the SEM image of Ti2AlC after HF treatment (Fig. 16d) confirms the successful exfoliation of individual particles, which is similar to that of exfoliated Ti3AlC2 (Fig. 16e) or Ta4AlC3 (Fig. 16f), where the layers are clearly separated from each other compared to the unreacted powder (Fig. 16c).211 Sonication of the treated powders resulted in the separation of 2D sheets.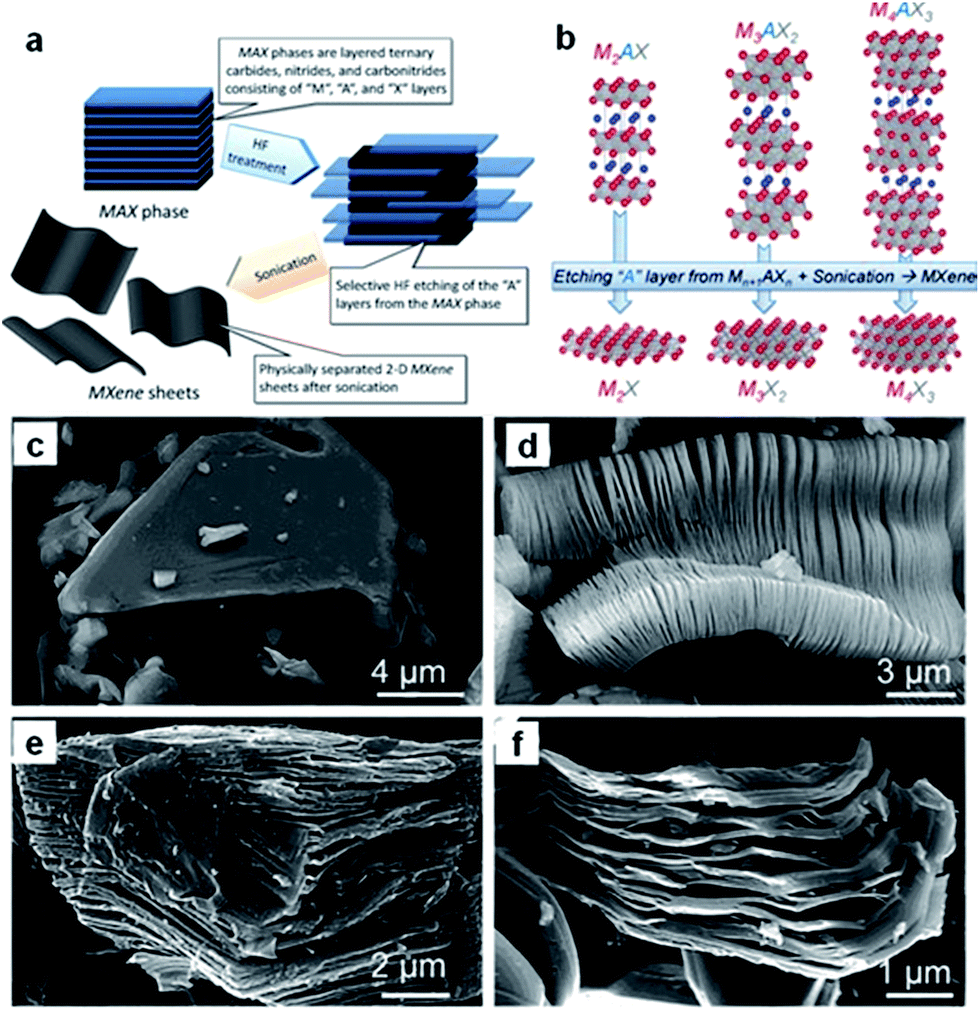 | ||
| Fig. 16 (a) Schematic illustration describing the synthesis process for MXenes from MAX phases. (b) Structure of MAX phases and the corresponding MXenes. Reprinted with permission from ref. 210. Copyright 2013, WILEY-VCH Verlag GmbH & Co. Secondary electron SEM micrographs for (c) Ti3AlC2 particle before treatment, which is typical of unreacted MAX phases, (d) Ti3AlC2 after HF treatment, (e) Ti2AlC after HF treatment, (f) Ta4AlC3 after HF treatment. Reprinted with permission from ref. 211. Copyright 2012, American Chemical Society. | ||
These MXene nanosheets have rich surface chemistries and high electronic conductivities, and they have exhibited outstanding performances in many applications that range from sensors and electronic device materials to catalysts in the chemical industry and electrochemical energy storage materials.212–218 In particular, the family of MXene nanosheets represents an emerging class of electrode materials that are capable of electrochemical reactions with various ions, including sodium.212–218 The screened MXene materials can provide a theoretical capacity of 190–288 mA h g−1 by accommodating two alkali ions per formula unit. They also exhibit an activation barrier of 0.1–0.2 eV for ionic motion, suggesting that this kind of material is promising for high-power applications.211,214
In particular, the MXene Ti3C2Tx has been demonstrated as an anode material for sodium-ion batteries and found to show good capacity retention over 1000 cycles as well as excellent rate capability.215,216 Detailed analyses demonstrated that Ti3C2Tx undergoes expansion of the interlayer distance during the first sodiation, whereby desolvated Na+ is intercalated/deintercalated reversibly. Meanwhile, the Na+ intercalation/deintercalation during the electrochemical reaction is not accompanied by any substantial structural change.215,216 The electrochemical performance of MXenes is expected to be further improved by delaminating the multilayered MXenes into few-layered flakes and combining them with carbon nanomaterials to form MXene/carbon composites, which not only prevents the aggregation and restacking of MXene sheets, but also improves the electrochemical performance.217,218 For example, porous Ti3C2 MXene/CNT composite paper electrodes have been fabricated by taking advantage of the electrostatic attraction between negatively charged 2D MXene nanosheets and positively charged 1D CNTs (Fig. 17a).218 This method efficiently prevented restacking of MXene nanosheets and produced a well-defined porous structure (Fig. 17b and c), thereby facilitating electrolyte transport and access of ions to the electrode. When applied as free-standing electrodes for sodium-ion storage, the built-to-order Ti3C2 MXene/CNTs porous films showed high volumetric capacity of 421 mA h cm−3 at 20 mA g−1, good rate performance, and excellent cycling stability (Fig. 17d). Thus the MXene Ti2CTx is very promising as SIB anode due to its structural stability and excellent performance.
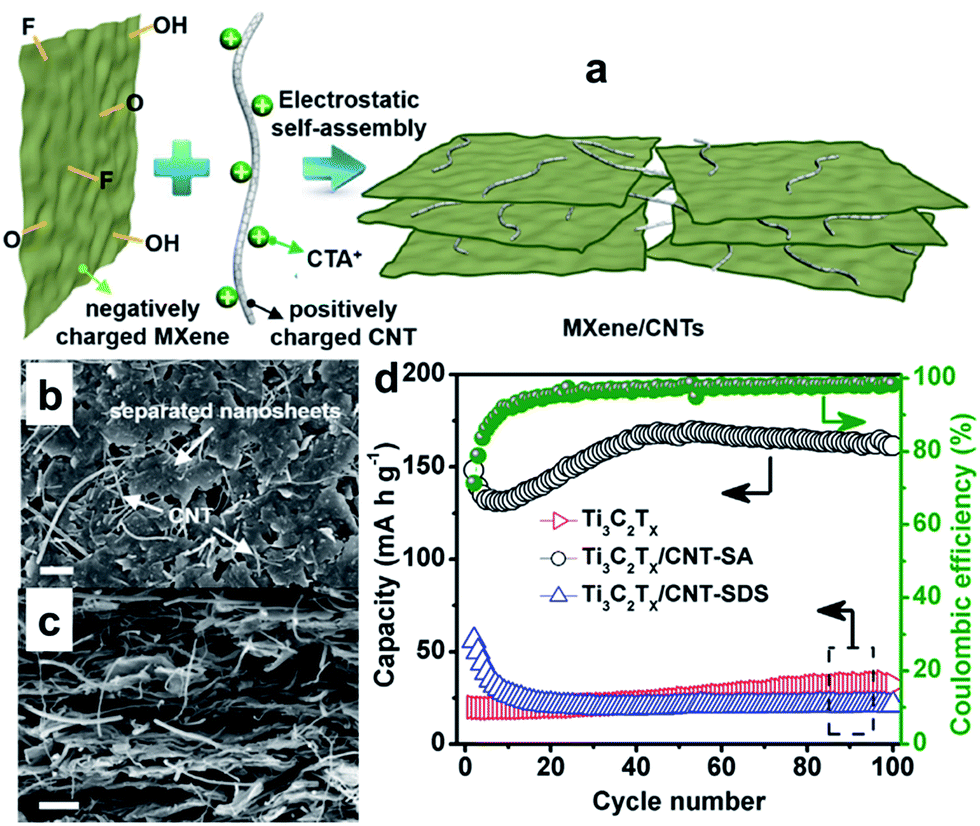 | ||
| Fig. 17 (a) Schematic illustration showing preparation of a porous MXene/CNT electrode by the self-assembly method; top-view (b) and cross-sectional (c) SEM images of porous self-assembled Ti3C2Tx/CNT-SA; (d) cycling performance of pure Ti3C2Tx, Ti3C2Tx/CNT-SA, and Ti3C2Tx/CNT-SDS papers at a current density of 20 mA g−1 from the second cycle, with the coulombic efficiency of Ti3C2Tx/CNT-SA electrode also shown. Ti3C2Tx/CNT-SDS film was prepared in a similar way, but sodium dodecylsulphate instead of cetyltrimethylammonium bromide (CTAB) was used to modify the CNTs. Reprinted with permission from ref. 218. Copyright 2016, Elsevier. | ||
6 Electrochemical properties of 2D nanostructures in full batteries
It should be noted that the electrochemical performance of 2D nanostructures mentioned above are based on half cells, the materials should be assembled and tested in full-cells for evaluating the practicability, however. Several papers tried to assemble full-cell batteries, and reported their electrochemical performance.73,142,157,168,172,200,218 For example, Sb nanospheres bonded with 2D rGO nanosheets deliver an energy density of 160 W h kg−1 at C/5 and power density up to 7.1 kW kg−1 at 120C when coupled with Na3V2(PO4)3 cathode.73 Carbon-stabilized interlayer-expanded few-layer MoSe2 nanosheets show an impressively high reversible capacity of 421 mA h g−1 at 0.2 A g−1 after 100 cycles when assembled into sodium ion full batteries with Na3V2(PO4)3 as cathode materials.168 2D Ti3C2Tx/CNT papers deliver a volumetric discharge capacity of 286 mA h cm−3 when coupled with Na0.44MnO2. The full cell had a high coulombic efficiency (99%) and relatively stable cycling performance, retaining a volumetric discharge capacity of 242 mA h cm−3 after 60 cycles.218 The preliminary results demonstrate that 2D nanostructured materials have potential as anodes for the practical SIBs, although further optimization is needed to improve the performance to meet the demand of practical applications.7 Summary and future prospects
Undoubtedly, the most difficult issues facing the commercialization of Na-ion batteries is how to achieve acceptable energy density, good cycle life and high safety with relatively low cost. With the ability to prepare various 2D nanostructures, combined with advanced nanofabrication techniques, current research on the preparation of SIB anode materials depends on not only the choice of functional active material components, crystal phase, and structure, but also on the spatial organization/assembly, surface exposure, interactions between the individual components, and geometric properties of the composites. In this review, we have summarized the recently developed strategies for 2D electrode materials and their application in SIBs. Various 2D nanomaterials with unique properties, planar hybrids, porous hierarchical architectures, and vertically stacked heterostructures have been prepared, which have been classified into four classes: carbonaceous materials (graphene and carbon nanosheets), alloy based materials (Sn, Sb, and P), conversion materials (phosphides/oxides/sulfides/selenides), and intercalation materials (Ti-based compounds), based on the manner of sodium storage. Although promising results have been achieved on 2D electrode materials for SIBs due to their unique features such as high active surface and shortened path for sodium ion diffusion and insertion, there are still many tough challenges to overcome before high performance SIBs based on 2D materials can be realized in the future.Firstly, it should be admitted that the study of 2D-based functional composite anodes for the sodium-ion battery is still in its infant stage. The underlying mechanisms of Na storage in 2D materials are still unclear, so more understanding with respect to the surface functionalities/defects, the hierarchical electrode structures, and kinetic transport at the electrode/electrolyte interface is needed to achieve better electrochemical performance. For example, the high surface area of graphene and its composites may lead to large irreversible capacity loss in the first cycle, and porous graphene-based composites exhibit small tap densities and consequently, an unfavorably low volumetric capacity. Moreover, a comparative study of such 2D nanostructures for LIBs and SIBs shows more enhanced specific capacity, rate capability, and cycling stability in LIBs than in SIBs, which is due to more sluggish kinetics, higher charge transfer resistance, and higher volume change occurring in insertion/extraction of Na ions than that of Li ions.63,89,91,101,103,164,165,197 In this regards, design of 2D materials with open and larger ion diffusion channels, sufficiently large interstitial sites, large effective surface area and good mechanical properties for SIBs are highly required. Secondly, the compatibility of each of the components in the 2D materials needs to be further improved, and heterostructure interface problems still exist due to the weak interaction forces in the hybrid structure, although not in heterostructures with crystalline bonding. Meanwhile, comparisons between amorphous and crystalline materials might prove to be an interesting area. The charge storage mechanism of such amorphous materials is still vague, and explanations are leaning more towards pseudocapacitive behavior instead. Thirdly, some special conditions are often needed to obtain novel 2D structures, which often lead to a high-cost synthesis. Thus, controlled low-cost and large-scale synthesis methods for novel 2D nanostructures need to be developed by integrating various advanced technologies, since the high cost of electrode materials significantly restricts scalable production and application. Fourthly, design of new and old electrode materials with high theoretical capacity (e.g. alloy materials) in 2D form is likely to be the next performance growth point for SIB anode materials. Since the discovery of graphene, many other 2D materials have been coming into the spotlight. They form a very large family that includes hexagonal boron nitride, transition metal dichalcogenides, metal oxides, common clays, and the recently discovered transition metal carbides, as well as silicene, germanene, phosphorene, antimonene, and stanene. All of them have great promise and will inevitably cause a new peak in SIB research. On the other hand, most of the materials used for hybridization with 2D anode materials are inorganic nanomaterials, such as alloy metals, metal oxides, and carbonaceous nanomaterials. One of the most promising future directions lies in the combination of 2D anode materials with polymers to create 2D-based polymer composites for a wide range of applications. Finally, in addition to the materials, further studies are needed on electrolytes, additives, and binders, which can greatly influence the electrode performance. With continuous endeavors by all parties concerned, it is expected that high-performance 2D materials and composites can be applied in commercialized high-performance SIBs in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by the Australian Research Council (No. FT150100109) and the National Natural Science Foundation of China (No. 21476063) are gratefully acknowledged. The authors would like to thank Dr T. Silver for critical reading of the manuscript.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef CAS PubMed.
- J. Liu, J. G. Zhang, Z. G. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Y. Li, J. Z. Hu, C. M. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. L. Li, Y. Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929 CrossRef CAS.
- S. W. Kim, D. H. Seo, X. H. Ma, G. Cede and K. Kang, Adv. Energy Mater., 2012, 2, 710 CrossRef CAS.
- H. L. Pan, Y. S. Hu and L. Q. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef CAS PubMed.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529 RSC.
- W. K. Pang, S. Kalluri, V. Peterson, N. Sharma, J. Kimpton, B. Johannessen, H. K. Liu, S. X. Dou and Z. P. Guo, Chem. Mater., 2015, 27, 3150 CrossRef CAS.
- E. Pomerantseva and Y. Gogotsi, Nat. Energy, 2017, 2, 17089 CrossRef.
- C. L. Tan, X. H. Cao, X. J. Wu, Q. Y. He, J. Yang, X. Zhang, J. Z. Chen, W. Zhao, S. K. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225 CrossRef CAS PubMed.
- J. L. Liu, C. X. Guo, A. Vasileff and S. Z. Qiao, Small Methods, 2017, 1, 1600006 CrossRef.
- M. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709 CAS.
- S. Chen, J. J. Duan, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 3804 CrossRef CAS PubMed.
- M. Zhou, X. W. Lou and Y. Xie, Nano Today, 2013, 8, 598 CrossRef CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271 CrossRef CAS PubMed.
- R. C. Asher, J. Inorg. Nucl. Chem., 1959, 10, 238 CrossRef CAS.
- P. Ge and M. Fouletier, Solid State Ionics, 1988, 28–30, 1172 CrossRef.
- M. M. Doeff, Y. Ma, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1993, 140, L169 CrossRef CAS.
- I. A. Udod, H. B. Orman and V. K. Genchel, Carbon, 1994, 32, 101 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169 CrossRef CAS PubMed.
- H. Kim, J. Hong, Y. U. Park, J. Kim, I. Hwang and K. Kang, Adv. Funct. Mater., 2014, 25, 534 CrossRef.
- Y. Wen, K. He, Y. J. Zhu, F. D. Han, Y. H. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. S. Wang, Nat. Commun., 2014, 5, 4033 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS.
- R. Alcántara, J. M. Jiménez-Mateos, P. Lavela and J. L. Tirado, Electrochem. Commun., 2001, 3, 639 CrossRef.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859 CrossRef CAS.
- K. L. Hong, L. Qie, R. Zeng, Z. Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q. J. Fan, W. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 12733 CAS.
- Y. L. Cao, L. F. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. M. Nie, L. V. Saraf, Z. G. Yang and J. Liu, Nano Lett., 2012, 12, 3783 CrossRef CAS PubMed.
- X. F. Luo, C. H. Yang, Y. Y. Peng, N. W. Pu, M. D. Ger, C. T. Hsieh and J. K. Chang, J. Mater. Chem. A, 2015, 3, 10320 CAS.
- L. L. Peng, Y. Zhu, D. H. Chen, R. S. Ruoff and G. H. Yu, Adv. Energy Mater., 2016, 6, 1600025 CrossRef.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Carbon, 2013, 57, 202 CrossRef CAS.
- X. S. Zhou, X. S. Zhu, X. Liu, Y. Xu, Y. X. Liu, Z. H. Dai and J. C. Bao, J. Phys. Chem. C, 2014, 118, 22426 CAS.
- D. Datta, J. W. Li and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 1788 CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 CAS.
- H. G. Wang, Z. Wu, F. L. Meng, D. L. Ma, X. L. Huang, L. M. Wang and X. B. Zhang, ChemSusChem, 2013, 6, 56 CrossRef CAS PubMed.
- T. Z. Yang, T. Qian, M. F. Wang, X. W. Shen, N. Xu, Z. Z. Sun and C. L. Yan, Adv. Mater., 2016, 28, 539 CrossRef CAS PubMed; H. W. Song, N. Li, H. Cui and C. X. Wang, Nano Energy, 2014, 4, 81 CrossRef.
- H. W. Song, N. Li, H. Cui and C. X. Wang, Nano Energy, 2014, 4, 81 CrossRef CAS.
- F. H. Yang, Z. A. Zhang, K. Du, X. X. Zhao, W. Chen, Y. Q. Lai and J. Li, Carbon, 2015, 91, 88 CrossRef CAS.
- X. L. Wang, G. Li, F. M. Hassan, J. D. Li, X. Y. Fan, R. Batmaz, X. C. Xiao and Z. W. Chen, Nano Energy, 2015, 15, 746 CrossRef CAS.
- J. Q. Yang, X. L. Zhou, D. H. Wu, X. D. Zhao and Z. Zhou, Adv. Mater., 2017, 29, 1604108 CrossRef PubMed.
- K. Tang, L. Fu, R. J. White, L. Yu, M. M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873 CrossRef CAS.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342 CAS.
- Z. Guan, H. Liu, B. Xu, X. Hao, Z. X. Wang and L. Q. Chen, J. Mater. Chem. A, 2015, 3, 7849 CAS.
- J. T. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. X. Dou and L. M. Dai, Adv. Mater., 2015, 27, 2042 CrossRef CAS PubMed.
- J. Ding, H. L. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. W. Xu, B. Zahiri, X. H. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004 CrossRef CAS PubMed.
- Y. Yan, Y. X. Yin, Y. G. Guo and L. J. Wan, Adv. Energy Mater., 2014, 4, 1301584 CrossRef.
- V. L. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, 158, A1011 CrossRef CAS.
- L. D. Ellis, B. N. Wilkes, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A416 CrossRef CAS.
- L. Baggetto, J. K. Keum, J. F. Browning and G. M. Veith, Electrochem. Commun., 2013, 34, 41 CrossRef CAS.
- A. Kohandehghan, K. Cui, M. Kupsta, J. Ding, E. M. Lotfabad, W. P. Kalisvaart and D. Mitlin, Nano Lett., 2014, 14, 5873 CrossRef CAS PubMed.
- L. D. Ellis, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A1801 CrossRef CAS.
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897 CrossRef CAS PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805 CrossRef CAS PubMed.
- J. F. Qian, Y. Chen, L. Wu, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2012, 48, 7070 RSC.
- Z. M. Liu, X. Y. Yu, X. W. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 2314 CAS.
- J. F. Qian, X. Y. Wu, Y. L. Cao, X. P. Ai and H. X. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633 CrossRef CAS PubMed.
- Y. J. Kim, Y. Park, A. Choi, N. S. Choi, J. S. Kim, J. S. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045 CrossRef CAS PubMed.
- L. Baggetto, P. Ganesh, R. P. Meisner, R. R. Unocic, J.-C. Jumas, C. A. Bridges and G. M. Veith, J. Power Sources, 2013, 234, 48 CrossRef CAS.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255 CrossRef CAS PubMed.
- H. L. Zhu, Z. Jia, Y. C. Chen, N. Weadock, J. Y. Wan, O. Vaaland, X. G. Han, T. Li and L. B. Hu, Nano Lett., 2013, 13, 3093 CrossRef CAS PubMed.
- Y. H. Liu, Y. H. Xu, Y. J. Zhu, J. N. Culver, C. A. Lundgren, K. Xu and C. S. Wang, ACS Nano, 2013, 7, 3627 CrossRef CAS PubMed.
- B. Luo, T. F. Qiu, D. L. Ye, L. Z. Wang and L. J. Zhi, Nano Energy, 2016, 22, 232 CrossRef CAS.
- H. S. Hou, M. J. Jing, Y. C. Yang, Y. Zhang, W. X. Song, X. M. Yang, J. Chen, Q. Y. Chen and X. B. Ji, J. Power Sources, 2015, 284, 227 CrossRef CAS.
- L. Li, K. H. Seng, Y. Y. Xia, H. K. Liu and Z. P. Guo, Nano Res., 2014, 7, 1466 CrossRef CAS.
- C. Nithya and S. Gopukumar, J. Mater. Chem. A, 2014, 2, 10516 CAS.
- T.-H. Kim, K.-S. Hong, D. Sohn, M. Kim, D.-H. Nam, E. Cho and H. Kwon, J. Mater. Chem. A, 2017, 5, 20304 CAS.
- Y. Jeon, X. G. Han, K. Fu, J. Q. Dai, J. H. Kim, L. B. Hu, T. Song and U. Paik, J. Mater. Chem. A, 2016, 4, 18306 CAS.
- L. Y. Hu, X. S. Zhu, Y. Du, Y. F. Li, X. S. Zhou and J. C. Bao, Chem. Mater., 2015, 27, 8138 CrossRef CAS.
- X. L. Zhou, Y. R. Zhong, M. Yang, M. Hu, J. P. Wei and Z. Zhou, Chem. Commun., 2014, 50, 12888 RSC.
- H. S. Hou, G. Q. Zou, P. Ge, G. G. Zhao, W. F. Wei, X. B. Ji and L. P. Huang, New J. Chem., 2017, 41, 13724 RSC.
- B. Kong, L. H. Zu, C. X. Peng, Y. Zhang, W. Zhang, J. Tang, C. Selomulya, L. D. Zhang, H. X. Chen, Y. Wang, Y. Liu, H. L. He, J. Wei, X. C. Lin, W. Luo, J. P. Yang, Z. W. Zhao, Y. Liu, J. H. Yang and D. Y. Zhao, J. Am. Chem. Soc., 2016, 138, 16533 CrossRef CAS PubMed.
- F. Wan, J. Z. Guo, X. H. Zhang, J. P. Zhang, H. Z. Sun, Q. Y. Yan, D. X. Han, L. Niu and X. L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 7790 CAS.
- C. Hwang, S. Choi, G. Y. Jung, J. Yang, S. K. Kwak, S. Park and H.-K. Song, Electrochim. Acta, 2017, 252, 25 CrossRef CAS.
- X. W. Liu, M. Gao, H. Yang, X. W. Zhong and Y. Yu, Nano Res., 2017, 10, 4360 CrossRef CAS.
- J. N. Gu, Z. G. Du, C. Zhang, J. G. Ma, B. Li and S. B. Yang, Adv. Energy Mater., 2017, 1700447 CrossRef.
- Y. J. Zhu, Y. Wen, X. L. Fan, T. Gao, F. D. Han, C. Luo, S. C. Liou and C. S. Wang, ACS Nano, 2015, 9, 3254 CrossRef CAS PubMed.
- J. X. Song, Z. X. Yu, M. L. Gordin, S. Hu, R. Yi, D. H. Tang, T. Walter, M. Regula, D. Choi, X. L. Li, A. Manivannan and D. H. Wang, Nano Lett., 2014, 14, 6329 CrossRef CAS PubMed.
- L. K. Pei, Q. Zhao, C. C. Chen, J. Liang and J. Chen, ChemElectroChem, 2015, 2, 1652 CrossRef CAS.
- C. Zhang, X. Wang, Q. F. Liang, X. Z. Liu, Q. H. Weng, J. W. Liu, Y. J. Yang, Z. H. Dai, K. J. Ding, Y. Bando, J. Tang and D. Golberg, Nano Lett., 2016, 16, 2054 CrossRef CAS PubMed.
- G. Zeng, X. Hu, B. L. Zhou, J. X. Chen, C. S. Cao and Z. H. Wen, Nanoscale, 2017, 9, 14722 RSC.
- Y. H. Liu, A. Y. Zhang, C. F. Shen, Q. Z. Liu, X. Cao, Y. Q. Ma, L. Chen, C. Lau, T. C. Chen, F. Wei and C. W. Zhou, ACS Nano, 2017, 11, 5530 CrossRef CAS PubMed.
- H. Gao, T. F. Zhou, Y. Zheng, Y. Q. Liu, J. Chen, H. K. Liu and Z. P. Guo, Adv. Energy Mater., 2016, 6, 1601037 CrossRef.
- L. Z. Kou, C. F. Chen and S. C. Smith, J. Phys. Chem. Lett., 2015, 6, 2794 CrossRef CAS PubMed.
- T. Ramireddy, T. Xing, M. M. Rahman, Y. Chen, Q. Dutercq, D. Gunzelmann and A. M. Glushenkov, J. Mater. Chem. A, 2015, 3, 5572 CAS.
- J. Sun, H. W. Lee, M. Pasta, H. T. Yuan, G. Y. Zheng, Y. M. Sun, Y. Z. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980 CrossRef CAS PubMed.
- Z. D. Huang, H. S. Hou, Y. Zhang, C. Wang, X. Q. Qiu and X. B. Ji, Adv. Mater., 2017, 29, 1702372 CrossRef PubMed.
- F. H. Yang, H. Gao, J. Chen and Z. P. Guo, Small Methods, 2017, 1, 1700216 CrossRef.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef PubMed.
- X. L. Ge, Z. Q. Li and L. W. Yin, Nano Energy, 2017, 32, 117 CrossRef CAS.
- X. J. Wang, K. Chen, G. Wang, X. J. Liu and H. Wang, ACS Nano, 2017, 11, 11602 CrossRef CAS PubMed.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139 CrossRef CAS PubMed.
- J. F. Qian, Y. Xiong, Y. L. Cao, X. P. Ai and H. X. Yang, Nano Lett., 2014, 14, 1865 CrossRef CAS PubMed.
- J. F. Mao, X. L. Fan, C. Luo and C. S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7147 CAS.
- W. C. Zhang, J. F. Mao, W. K. Pang, Z. P. Guo and Z. X. Chen, Electrochim. Acta, 2017, 235, 107 CrossRef CAS.
- X. L. Fan, J. F. Mao, Y. J. Zhu, C. Luo, L. M. Suo, T. Gao, F. D. Han, S. C. Liou and C. S. Wang, Adv. Energy Mater., 2015, 5, 1500174 CrossRef.
- Q. Li, Zh. Q. Li, Z. W. Zhang, C. X. Li, J. Y. Ma, C. X. Wang, X. L. Ge, Sh. H. Dong and L. W. Yin, Adv. Energy Mater., 2016, 6, 1600376 CrossRef.
- Y. L. Xu, B. Peng and F. M. Mulder, Adv. Energy Mater., 2017, 1701847 Search PubMed.
- W. C. Zhang, J. F. Mao, S. Li, Z. X. Chen and Z. P. Guo, J. Am. Chem. Soc., 2017, 139, 3316 CrossRef CAS PubMed.
- J. P. Yang, T. F. Zhou, R. Zhu, X. Q. Chen, Z. P. Guo, J. W. Fan, H. K. Liu and W. X. Zhang, Adv. Mater. Interfaces, 2016, 3, 1500464 CrossRef.
- W. P. Sun, X. H. Rui, J. X. Zhu, L. H. Yu, Y. Zhang, Z. C. Xu, S. Madhavi and Q. Y. Yan, J. Power Sources, 2015, 274, 755 CrossRef CAS.
- L. L. Peng, P. Xiong, L. Ma, Y. F. Yuan, Y. Zhu, D. H. Chen, X. Y. Luo, J. Lu, K. Amine and G. H. Yu, Nat. Commun., 2017, 8, 15139 CrossRef PubMed.
- D. H. Chen, L. L. Peng, Y. F. Yuan, Y. Zhu, Z. W. Fang, C. S. Yan, G. Chen, R. Shahbazian-Yassar, J. Lu, K. Amine and G. H. Yu, Nano Lett., 2017, 17, 3907–3913 CrossRef CAS PubMed.
- Z. L. Jian, B. Zhao, P. Liu, F. J. Li, M. B. Zheng, M. W. Chen, Y. Shi and H. S. Zhou, Chem. Commun., 2014, 50, 1215 RSC.
- X. J. Liu, T. Q. Chen, H. P. Chu, L. Y. Niu, Z. Sun, L. K. Pan and C. Q. Sun, Electrochim. Acta, 2015, 166, 12 CrossRef CAS.
- Y. Q. Fu, Q. L. Wei, X. Y. Wang, G. X. Zhang, H. B. Shu, X. K. Yang, A. C. Tavares and S. H. Sun, RSC Adv., 2016, 6, 16624 RSC.
- Y. G. Liu, Z. Y. Cheng, H. Y. Sun, H. Arandiyan, J. P. Li and M. Ahmad, J. Power Sources, 2015, 273, 878 CrossRef CAS.
- Y. H. Dou, Y. X. Wang, D. L. Tian, J. T. Xu, Z. J. Zhang, Q. N. Liu, B. Y. Ruan, J. M. Ma, Z. Q. Sun and S. X. Dou, 2D Mater., 2017, 4, 015022 CrossRef.
- F. Zou, Y. M. Chen, K. W. Liu, Z. T. Yu, W. F. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377 CrossRef CAS PubMed.
- X. J. Li, A. L. Hector and J. R. Owen, J. Phys. Chem. C, 2014, 11, 29568 Search PubMed.
- J. F. Huang, Z. W. Xu, L. Y. Cao, Q. L. Zhang, H. B. Ouyang and J. Y. Li, Energy Technol., 2015, 3, 1108 CrossRef CAS.
- Y. Z. Jiang, M. J. Hu, D. Zhang, T. Z. Yuan, W. P. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60 CrossRef CAS.
- S. Li, Y. Z. Wang, J. X. Qiu, M. Ling, H. H. Wang, W. Martens and S. Q. Zhang, RSC Adv., 2014, 4, 50148 RSC.
- Y. X. Wang, Y. G. Lim, M. S. Park, S. L. Chou, J. H. Kim, H. K. Liu, S. X. Dou and Y. J. Kim, J. Mater. Chem. A, 2014, 2, 529 CAS.
- Y. D. Zhang, J. Xie, S. C. Zhang, P. Y. Zhu, G. S. Cao and X. B. Zhao, Electrochim. Acta, 2015, 151, 8 CrossRef CAS.
- L. K. Pei, Q. Jin, Z. Q. Zhu, Q. Zhao, J. Liang and J. Chen, Nano Res., 2015, 8, 184 CrossRef CAS.
- F. Zhang, J. J. Zhu, D. L. Zhang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2017, 17, 1302 CrossRef CAS PubMed.
- Y. Zhao, L. P. Wang, M. T. Sougrati, Z. Feng, Y. Leconte, A. Fisher, M. Srinivasan and Z. Xu, Adv. Energy Mater., 2017, 7, 1601424 CrossRef.
- X. Y. Yu and X. W. Lou, Adv. Energy Mater., 2018, 8, 1701592 CrossRef.
- Z. Hu, Z. Q. Zhu, F. Y. Cheng, K. Zhang, J. B. Wang, C. C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309 CAS.
- Y. J. Zhu, X. L. Fan, L. M. Suo, C. Luo, T. Gao and C. S. Wang, ACS Nano, 2016, 10, 1529 CrossRef CAS PubMed.
- W. H. Chen, S. H. Qi, M. M. Yu, X. M. Feng, S. Z. Cui, J. M. Zhang and L. W. Mi, Electrochim. Acta, 2017, 230, 1 CrossRef CAS.
- Z. M. Liu, T. C. Liu, T. Song, X. Y. Yu, X. W. Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576 CAS.
- Q. H. Wang, We. C. Zhang, C. Guo, Y. J. Liu, C. Wang and Z. P. Guo, Adv. Funct. Mater., 2017, 27, 1703390 CrossRef.
- T. S. Wang, P. Hu, C. J. Zhang, H. P. Du, Z. H. Zhang, X. G. Wang, S. G. Chen, J. W. Xiong and G. L. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 7811 CAS.
- S. J. Peng, X. P. Han, L. L. Li, Z. Q. Zhu, F. Y. Cheng, M. Srinivansan, S. Adams and S. Ramakrishna, Small, 2016, 12, 1359 CrossRef CAS PubMed.
- T. T. Chen, Y. F. Ma, Q. B. Guo, M. Yang and H. Xia, J. Mater. Chem. A, 2017, 5, 3179 CAS.
- H. Gao, T. Zhou, Y. Zheng, Q. Zhang, Y. Liu, J. Chen, H. Liu and Z. Guo, Adv. Funct. Mater., 2017, 27, 1702634 CrossRef.
- X. J. Xu, S. M. Ji, M. Z. Gu and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20957 CAS.
- W. Qin, D. S. Li, X. J. Zhang, D. Yan, B. W. Hu and L. K. Pan, Electrochim. Acta, 2016, 191, 435 CrossRef CAS.
- W. P. Sun, X. H. Rui, D. Zhang, Y. Z. Jiang, Z. Q. Sun, H. K. Liu and S. X. Dou, J. Power Sources, 2016, 309, 135 CrossRef CAS.
- Z. Hu, L. X. Wang, K. Zhang, J. B. Wang, F. Y. Cheng, Z. L. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794 CrossRef CAS PubMed.
- Y. F. Li, Y. L. Liang, F. C. R. Hernandez, H. D. Yoo, Q. Y. An and Y. Yao, Nano Energy, 2015, 15, 453 CrossRef CAS.
- D. W. Su, S. X. Dou and G. X. Wang, Adv. Energy Mater., 2015, 5, 1401205 CrossRef.
- M. W. Xu, F. L. Yi, Y. B. Niu, J. L. Xie, J. K. Hou, S. G. Liu, W. H. Hu, Y. T. Li and C. M. Li, J. Mater. Chem. A, 2015, 3, 9932 CAS.
- S. H. Choi, Y. N. Ko, J. K. Lee and Y. C. Kang, Adv. Funct. Mater., 2015, 25, 1780 CrossRef CAS.
- S. Kalluri, K. H. Seng, Z. P. Guo, A. J. Du, K. Konstantinov, H. K. Liu and S. X. Dou, Sci. Rep., 2015, 5, 11989 CrossRef CAS PubMed.
- Y. P. Liu, X. Y. He, D. Hanlon, A. Harvey, J. N. Coleman and Y. G. Li, ACS Nano, 2016, 10, 8821–8828 CrossRef CAS PubMed.
- W. N. Ren, H. F. Zhang, C. Guan and C. W. Cheng, Adv. Funct. Mater., 2017, 27, 1702116 CrossRef.
- T. S. Sahu, Q. Q. Li, J. S. Wu, V. P. Dravid and S. Mitra, J. Mater. Chem. A, 2017, 5, 355 CAS.
- S.-K. Park, J. Lee, S. Bong, B. Jang, K.-D. Seong and Y. Z. Piao, ACS Appl. Mater. Interfaces, 2016, 8, 19456 CAS.
- W. P. Sun, X. H. Rui, D. Yang, Z. Q. Sun, B. Li, W. Y. Zhang, Y. Zong, S. Madhavi, S. X. Dou and Q. Y. Yan, ACS Nano, 2015, 9, 11371 CrossRef CAS PubMed.
- Y. D. Zhang, P. Y. Zhu, L. L. Huang, J. Xie, S. C. Zhang, G. S. Cao and X. B. Zhao, Adv. Funct. Mater., 2015, 25, 481 CrossRef CAS.
- C. Liu, H. Y. Kang, L. F. Jiao, C. C. Chen, K. Z. Cao, Y. J. Wang and H. T. Yuan, Nanoscale, 2015, 7, 1325 RSC.
- B. H. Qu, C. Z. Ma, G. Ji, C. H. Xu, J. Xu, Y. S. Meng, T. H. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854 CrossRef CAS PubMed.
- P. V. Prikhodchenko, D. Y. W. Yu, S. K. Batabyal, V. Uvarov, J. Gun, S. Sladkevich, A. A. Mikhaylov, A. G. Medvedev and O. Lev, J. Mater. Chem. A, 2014, 2, 8431 CAS.
- Y. Jiang, M. Wei, J. K. Feng, Y. C. Ma and S. L. Xiong, Energy Environ. Sci., 2016, 9, 1430 Search PubMed.
- J. J. Wang, C. Luo, J. F. Mao, Y. J. Zhu, X. L. Fan, T. Gao, A. C. Mignerey and C. S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11476 CAS.
- D. L. Chao, P. Liang, Z. Chen, L. Y. Bai, H. Shen, X. X. Liu, X. H. Xia, Y. L. Zhao, S. V. Savilov, J. Y. Lin and Z. X. Shen, ACS Nano, 2016, 10, 10211 CrossRef CAS PubMed.
- D. L. Chao, C. R. Zhu, P. H. Yang, X. H. Xia, J. L. Liu, J. Wang, X. F. Fan, S. V. Savilov, J. Y. Lin, H. J. Fan and Z. X. Shen, Nat. Commun., 2016, 7, 12122 CrossRef CAS PubMed.
- F. Z. Tu, X. Xu, P. Z. Wang, L. Si, X. S. Zhou and J. C. Bao, J. Phys. Chem. C, 2017, 121, 3261 CAS.
- P. Zhou, X. Wang, W. H. Guan, D. Zhang, L. B. Fang and Y. Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 6979 CAS.
- T. F. Zhou, W. K. Pang, C. F. Zhang, J. P. Yang, Z. X. Chen, H. K. Liu and Z. P. Guo, ACS Nano, 2014, 8, 8323 CrossRef CAS PubMed.
- P. L. He, Y. J. Fang, X. Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 12202 CrossRef CAS PubMed.
- S. H. Choi and Y. C. Kang, Nano Res., 2015, 8, 1595 CrossRef CAS.
- Y. Zheng, T. F. Zhou, C. F. Zhang, J. F. Mao, H. K. Liu and Z. P. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408 CrossRef CAS PubMed.
- X. H. Xiong, C. H. Yang, G. H. Wang, Y. W. Lin, X. Ou, J.-H. Wang, B. Zhao, Me. L. Liu, Z. Lin and K. Huang, Energy Environ. Sci., 2017, 10, 1757 CAS.
- C. Xia, F. Zhang, H. F. Liang and H. N. Alshareef, Nano Res., 2017, 10, 4368 CrossRef CAS.
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 Search PubMed.
- Y. B. Zhao and A. Manthiram, Chem. Commun., 2015, 51, 13205 RSC.
- H. S. Hou, M. J. Jing, Z. D. Huang, Y. C. Yang, Y. Zhang, J. Chen, Z. B. Wu and X. B. Ji, ACS Appl. Mater. Interfaces, 2015, 7, 19362 CAS.
- X. H. Xiong, G. H. Wang, Y. W. Lin, Y. Wang, X. Ou, F. H. Zheng, C. H. Yang, J.-H. Wang and M. L. Liu, ACS Nano, 2016, 10, 10953 CrossRef CAS PubMed.
- S. H. Choi and Y. C. Kang, Nanoscale, 2015, 7, 3965 RSC.
- Y. Wang, D. Z. Kong, W. H. Shi, B. Liu, G. J. Sim, Q. Ge and H. Y. Yang, Adv. Energy Mater., 2016, 6, 1601057 CrossRef.
- J. H. Zhou, L. Wang, M. Y. Yang, J. H. Wu, F. J. Chen, W. J. Huang, N. Han, H. L. Ye, F. P. Zhao, Y. Y. Li and Y. G. Li, Adv. Mater., 2017, 29, 1702061 CrossRef PubMed.
- D. X. Yu, Q. Pang, Y. Gao, Y. J. Wei, C. Z. Wang, G. Chen and F. Du, Energy Storage Materials, 2018, 11, 1 CrossRef.
- D. Zhang, G. Q. Zhao, P. Li, Y. Zhang, W. B. Qiu, J. Shu, Y. Z. Jiang, S. X. Dou and W. P. Sun, Chem.–Eur. J., 2017, 23, 1 CrossRef CAS.
- Y. C. Tang, Z. B. Zhao, Y. W. Wang, Y. F. Dong, Y. Liu, X. Z. Wang and J. S. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 32324 CAS.
- F. E. Niu, J. Yang, N. N. Wang, D. P. Zhang, W. L. Fan, J. Yang and Y. T. Qian, Adv. Funct. Mater., 2017, 1700522 CrossRef.
- X. Ou, C. H. Yang, X. H. Xiong, F. H. Zheng, Q. C. Pan, C. Jin, M. L. Liu and K. Huang, Adv. Funct. Mater., 2017, 27, 1606242 CrossRef.
- F. Zhang, C. Xia, J. J. Zhu, B. Ahmed, H. F. Liang, D. B. Velusamy, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601188 CrossRef.
- S. Yuan, Y. H. Zhu, W. Li, S. Wang, D. Xu, L. Li, Y. Zhang and X. B. Zhang, Adv. Mater., 2017, 29, 1602469 CrossRef PubMed.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560 CrossRef CAS.
- L. M. Wu, D. Buchholz, D. Bresser, L. G. Chagas and S. Passerini, J. Power Sources, 2014, 251, 379 CrossRef CAS.
- K. T. Kim, G. Ali, K. Y. Chung, C. S. Yoon, H. Yashiro, Y. K. Sun, J. Lu, K. Amine and S. T. Myung, Nano Lett., 2014, 14, 416 CrossRef CAS PubMed.
- Y. Xu, E. M. Lotfabad, H. L. Wang, B. Farbod, Z. W. Xu, A. Kohandehghan and D. Mitlin, Chem. Commun., 2013, 49, 8973 RSC.
- S. M. Oh, J. Y. Hwang, C. S. Yoon, J. Lu, K. Amine, I. Belharouak and Y. K. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 11295 CAS.
- D. W. Su, S. X. Dou and G. X. Wang, Chem. Mater., 2015, 27, 6022 CrossRef CAS.
- L. M. Wu, D. Bresser, D. Buchholz, G. A. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy Mater., 2015, 5, 1401142 CrossRef.
- Y. Zhang, X. L. Pu, Y. C. Yang, Y. R. Zhu, H. S. Hou, M. J. Jing, X. M. Yang, J. Chen and X. B. Ji, Phys. Chem. Chem. Phys., 2015, 17, 15764 RSC.
- H. Usui, S. Yoshioka, K. Wasada, M. Shimizu and H. Sakaguchi, ACS Appl. Mater. Interfaces, 2015, 7, 6567 CAS.
- L. M. Wu, D. Bresser, D. Buchholz and S. Passerini, J. Electrochem. Soc., 2015, 16, A3052 Search PubMed.
- J. P. Huang, D. D. Yuan, H. Z. Zhang, Y. L. Cao, G. R. Li, H. X. Yang and X. P. Gao, RSC Adv., 2013, 3, 12539 Search PubMed.
- M. Fehse and E. Ventosa, ChemPlusChem, 2015, 80, 785 CrossRef CAS.
- T. F. Zhou, Y. Zheng, H. Gao, S. Min, S. Li, H. K. Liu and Z. P. Guo, Adv. Sci., 2015, 2, 1500027 CrossRef PubMed.
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. H. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed.
- Y. Zhang, C. W. Foster, C. E. Banks, L. D. Shao, H. S. Hou, G. Q. Zou, J. Chen, Z. D. Huang and X. B. Ji, Adv. Mater., 2016, 28, 9391 CrossRef CAS PubMed.
- Y. F. Wen, J.-H. Yun, B. Luo, M. Q. Lyu and L. Z. Wang, Electrochim. Acta, 2016, 219, 163 CrossRef CAS.
- A. Shoaib, Y. X. Huang, J. Liu, J. J. Liu, M. Xu, Z. H. Wang, R. J. Chen, J. T. Zhang and F. Wu, J. Power Sources, 2017, 342, 405 CrossRef CAS.
- Z. Y. Le, F. Liu, P. Nie, X. R. Li, X. Y. Liu, Z. F. Bian, G. Chen, H. B. Wu and Y. F. Lu, ACS Nano, 2017, 11, 2952 CrossRef CAS PubMed.
- T. F. Yi, S. Y. Yang and Y. Xie, J. Mater. Chem. A, 2015, 3, 5750 CAS.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- X. Q. Yu, H. L. Pan, W. Wan, C. Ma, J. M. Bai, Q. P. Meng, S. N. Ehrlich, Y. S. Hu and X. Q. Yang, Nano Lett., 2013, 13, 4721 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, K. Nakanishi and T. Abe, Adv. Energy Mater., 2015, 5, 1400730 CrossRef.
- L. Y. Yang, H. Z. Li, J. Liu, S. S. Tang, Y. K. Lu, S. T. Li, J. Min, N. Yan and M. Lei, J. Mater. Chem. A, 2015, 3, 24446 CAS.
- C. J. Chen, H. H. Xu, T. F. Zhou, Z. P. Guo, L. N. Chen, M. Y. Yan, L. Q. Mai, P. Hu, S. J. Cheng, Y. H. Huang and J. Xie, Adv. Energy Mater., 2016, 6, 1600322 CrossRef.
- G. B. Xu, L. W. Yang, X. L. Wei, J. W. Ding, J. X. Zhong and P. K. Chu, Adv. Funct. Mater., 2016, 26, 3349 CrossRef CAS.
- P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109 CrossRef CAS; A. Rudola, K. Saravanan, C. W. Mason and P. Balaya, J. Mater. Chem. A, 2013, 1, 2653 Search PubMed.
- H. L. Pan, X. Lu, X. Q. Yu, Y. S. Hu, H. Li, X. Q. Yang and L. Q. Chen, Adv. Energy Mater., 2013, 3, 1186 CrossRef CAS.
- J. F. Ni, S. D. Fu, C. Wu, Y. Zhao, J. Maier, Y. Yu and L. Li, Adv. Energy Mater., 2016, 6, 1502568 CrossRef.
- F. X. Xie, L. Zhang, D. W. Su, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1700989 CrossRef PubMed.
- J. S. Ko, V. V. T. Doan-Nguyen, H.-S. Kim, G. A. Muller, A. C. Serino, P. S. Weiss and B. S. Dunn, ACS Appl. Mater. Interfaces, 2017, 9, 1416 CAS.
- Z. H. Li, S. C. Ye, W. Wang, Q. J. Xu, H. M. Liu, Y. G. Wang and Y. Y. Xia, ACS Omega, 2017, 2, 5726 CrossRef CAS.
- P. Senguttuvan, G. Rousse, M. E. A. Y. de Dompablo, H. Vezin, J. M. Tarascon and M. R. Palacin, J. Am. Chem. Soc., 2013, 135, 3897 CrossRef CAS PubMed.
- W. Wu, J. Yan, A. Wise, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2014, 161, A561 CrossRef CAS.
- C. Wu, P. Kopold, Y. L. Ding, P. A. van Aken, J. Maier and Y. Yu, ACS Nano, 2015, 9, 6610–6661 CrossRef CAS PubMed.
- S. I. Park, I. Gocheva, S. Okada and J.-i. Yamaki, J. Electrochem. Soc., 2011, 158, A1067 CrossRef CAS.
- X. Y. Wu, Y. L. Cao, X. P. Ai, J. F. Qian and H. X. Yang, Electrochem. Commun., 2013, 31, 145 CrossRef CAS.
- X. N. Li, X. B. Zhu, J. W. Liang, Z. G. Hou, Y. Wang, N. Lin, Y. C. Zhu and Y. T. Qian, J. Electrochem. Soc., 2014, 161, A1181 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322 CrossRef CAS PubMed.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- S.-M. Bak, R. M. Qiao, W. L. Yang, S. Lee, X. Q. Yu, B. Anasori, H. Lee, Y. Gogotsi and X.-Q. Yang, Adv. Energy Mater., 2017, 7, 1700959 CrossRef.
- A. N. Enyashin and A. L. Ivanovskii, Comput. Theor. Chem., 2012, 989, 27 CrossRef CAS.
- X. F. Wang, X. Shen, Y. R. Gao, Z. X. Wang, R. C. Yu and L. Q. Chen, J. Am. Chem. Soc., 2015, 137, 2715 CrossRef CAS PubMed.
- S. Kajiyama, L. Szabova, K. Sodeyama, H. Iinuma, R. Morita, K. Gotoh, Y. Tateyama, M. Okubo and A. Yamada, ACS Nano, 2016, 10, 3334 CrossRef CAS PubMed.
- M.-Q. Zhao, C. E. Ren, Z. Ling, M. R. Lukatskaya, C. Zhang, K. L. VanAken, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 339 CrossRef CAS PubMed.
- X. Q. Xie, M. Q. Zhao, B. k. Anasori, K. Maleski, C. E. Ren, J. W. Li, B. W. Byles, E. Pomerantseva, G. X. Wang and Y. Gogotsi, Nano Energy, 2016, 26, 513 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |