
N-rich carbon coated CoSnO3 derived from in situ construction of a Co–MOF with enhanced sodium storage performance†
Guoqiang
Zou
a,
Hongshuai
Hou
*a,
Ganggang
Zhao
a,
Peng
Ge
a,
Dulin
Yin
b and
Xiaobo
Ji
 a
a
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: hs-hou@csu.edu.cn; Tel: +86-731-88879616
bNational & Local United Engineering Laboratory for New Petrochemical Materials & Fine Utilization of Resources, Hunan Normal University, Changsha 410081, P. R. China
First published on 19th February 2018
Abstract
In this study, N-rich carbon coated interconnected CoSnO3 nanoboxes (CoSnO3–NCs) with controllable amounts of N-rich carbon ranging from 7.5% to 24.6% were firstly prepared by the carbonization of CoSnO3–MOFs. Impressively, the preparation of CoSnO3–MOFs has been realized for the first time by directly utilizing CoSnO3 as the precursor under solvent-free conditions, bridging between the cobalt(II) ion in the skeleton of CoSnO3 and the 2-methylimidazole. Interestingly, the growth of these CoSnO3–MOFs can be manipulated by changing the amount of 2-methylimidazole used, resulting in a tunable N-rich carbon content. Furthermore, the electrochemical storage behavior of these materials for SIBs was initially explored. Compared with the pure CoSnO3, the as-resulted CoSnO3–NC materials showed largely enhanced sodium storage performance with a reversible capacity of 493.9 mA h g−1 at a current density of 0.1 A g−1 after 100 cycles. Moreover, the as-prepared materials showed an excellent high rate storage performance with a remarkable capacity of 273.8 mA h g−1 at 1 A g−1 after 1000 cycles. This work provides a new approach for constructing Co–MOFs as well as providing an efficient N-rich carbon-coating route, which may be expanded to other Co-based oxides and can greatly expand the development of new species of Co-based MOFs.
1. Introduction
In the last few decades, metal–organic frameworks (MOFs), a new family of crystalline porous materials made up of metal ions and interconnected organic linkers, have been widely investigated for their tunable porosities, various structural topologies, and functionalities.1–6 Their metallic ions mainly include the divalent and trivalent ions of 3d transition metals (Zn, Co, Fe, Ti, Ni, etc.), and the organic linkers primarily incorporate the organic carboxylic acid class and imidazole class.7–16 Amongst these metals, cobalt is a very tempting candidate owing to the high catalytic activity of Co species.17 ZIF-67, one of the most widely used Co-based MOFs composed of Co2+ ions and 2-methylimidazole, is broadly utilized in many areas, such as gas separation, catalysis, and energy storage.17–20 Currently, the reactions for synthesizing Co-based MOFs are carried out in solution including N,N-dimethyl formamide, ethanol, and water.8,17,21,22 ZIF-67 thin films obtained by dissolving Co(NO3)2·6H2O and 2-methylimidazole in methanol solutions delivered exceptionally long-lived excited states, exhibiting excellent photocatalytic performance.23 However, if the synthetic process can be achieved without any solvent, the separation of the product will be greatly simplified, and the process can become more meaningful from the perspective of economic and environmental protection. Furthermore, the morphology characteristics of the Co-based MOFs are relatively unitary when synthesised through hydrothermal methods. The Co-based MOFs obtained from the traditional precursor (Co(NO3)2·6H2O) only possess a single metal element, Co, limiting their potential application. In order to solve this problem, recently, a core–shell ZIF-67@ZIF-8 containing both Zn/Co elements was obtained via the growth of ZIF-8 at the surface of a ZIF-67 seed in methanol.24 Although the material showed excellent performance for the semi-hydrogenation of acetylene, materials with a more simple preparation method and a homogeneous distribution of the bimetal element are urgently needed. Hence, it will be great to build the bridging between the Co-based double metal oxide and the organic ligands. Given this, new methods to construct Co-based MOFs and the development of new species of Co-based MOFs will be fascinating.Sodium-ion batteries (SIBs) have been deemed as the most hopeful alternative for potential application in large-scale energy storage by considering their low cost and low environmental impact.25–39 Up to now, most MOF materials could not be directly employed as an anode for SIBs because of their poor electronic conductivity. In order to better utilize their advantages for SIBs, MOFs were usually employed as precursors to prepare electrochemically active materials by in situ carbonization, such as MOF-based derivative porous carbons, metal oxides, and metal oxide/carbon.8,10,40–42 Notably, the volume expansion of the metal oxide during the discharge/charge processes can be effectively released and the electronic conductivity is also improved by introducing a carbon matrix.9,41,42 Hierarchical hollow NiO/Ni/graphene was prepared from the carbonization of an Ni–MOF, and a capacity of about 190 mA h g−1 at a current density of 0.2 A g−1 was obtained after 200 cycles.43 Although some progress has been achieved for MOF-based anode materials, the MOF-based derivative materials are mainly acquired by a simple carbonization of several classic MOFs, meaning that the types of MOF-based anode material are limited. Furthermore, the electrochemical performance of these materials for SIBs cannot be effectively further evaluated, showing that their application in power storage is still at an early stage in comparison to their colourful usage in the fields of catalysis and drug delivery. CoSnO3, a good anode material for LIBs, has not yet been explored as an electrode material for SIBs.44 Considering that the current approaches for the preparation of carbon coatings are relatively complex and the carbon amounts cannot be effectively regulated, a new approach was suggested as shown in Scheme 1.
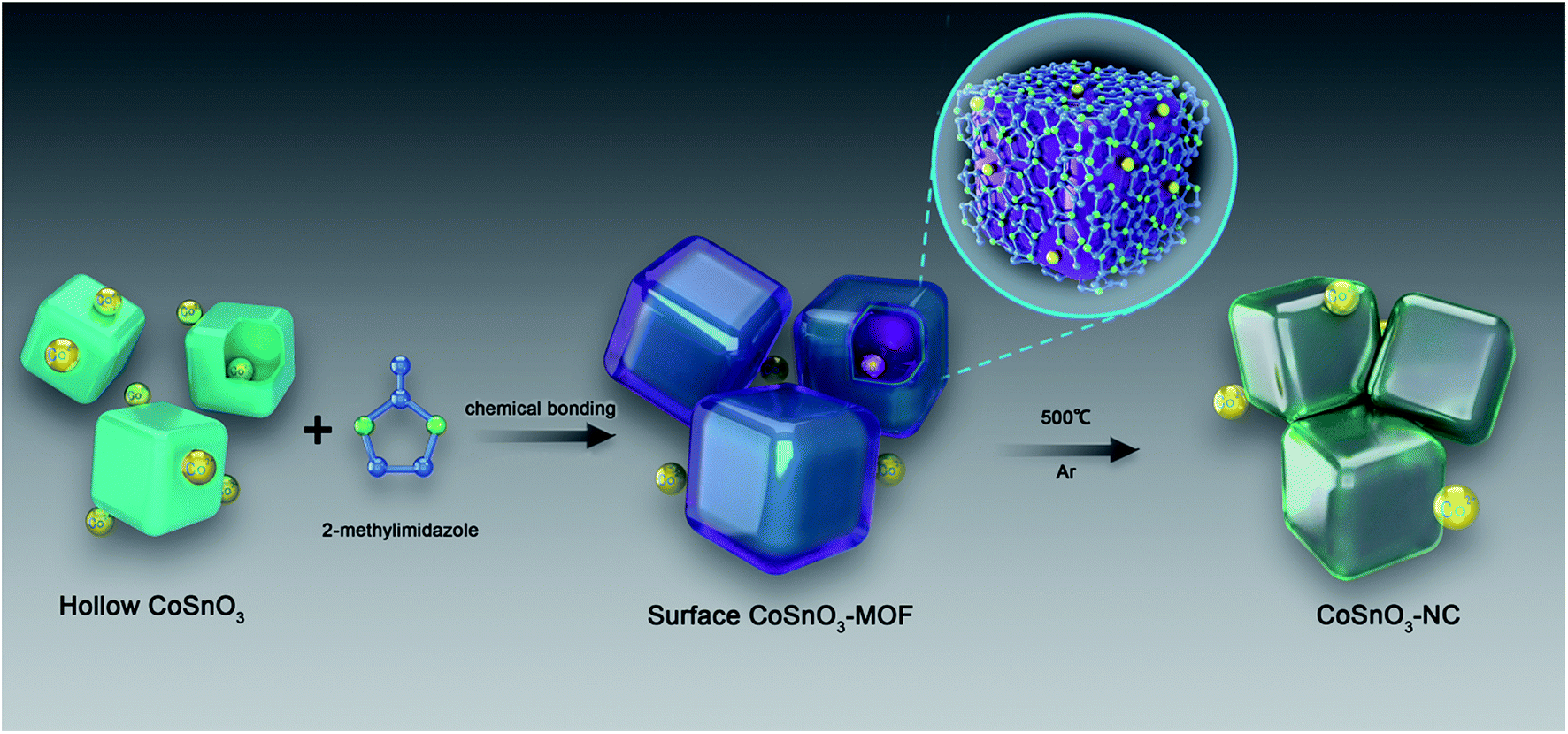 | ||
| Scheme 1 The design process for the preparation of CoSnO3–MOFs and CoSnO3–NCs. | ||
In this paper, a series of CoSnO3–NC nanobox materials with controllable carbon content were acquired by the in situ carbonization of CoSnO3–MOFs, showing a controllable N-rich carbon content ranging from 7.5% to 24.6%. The CoSnO3–MOFs were prepared for the first time via the chemical bonding method using the bridge role of Co2+ under solvent-free conditions (Scheme 1). Also, the storage behavior of the CoSnO3–NC material for SIBs was firstly explored. Overall, our work provides a new approach to construct Co-based MOFs and offers a carbon coating method for the Co-based oxides, greatly enhancing the electrochemical storage performance for SIBs with a high reversible capacity of 493.9 mA h g−1 at 0.1 A g−1 after 100 cycles.
2. Experimental section
2.1 Materials synthesis
2.2 Materials characterization
The crystalline phases of the above series of materials were detected using an X-ray diffractometer (XRD, Rigaku, Rint-2000) with a 0.154 nm Cu-Kα radiation. The morphology and composition of the as-obtained samples were recorded using scanning electron microscopy (SEM) (JSM 6400), transmission electron microscopy (TEM) (JEOL) and X-ray photoelectron spectroscopy (XPS, PerkinElmer, USA). The BET surface area and pore size distributions were measured using a Micromeritics ASAP 2020 analyzer. Thermogravimetric analysis (TGA) data were obtained using a thermal analysis instrument (NETZSCH STA449F3) with a heating rate of 5 °C min−1 from room temperature to 600 °C in air. The electronic conductivities were collected using the standard four probe method at room temperature with pellet samples (FOUR-POINT PROBE METER, SDY-4D, China).2.3 Electrochemical measurements
All the electrochemical measurements were tested using the half-cells (CR2016-type). The electrodes were prepared by uniformly dispersing a water slurry made up of active material, Super P (conductive carbon, TIMCAL), and binder carboxymethyl cellulose (CMC, Alfa Aesar) with a mass ratio of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3 onto Cu foil. After being dried at 90 °C for 12 h under vacuum, the CR2016-type half-cells were assembled in a Braun-glovebox in argon atmosphere. The mass loading of the active material is about 1.0–1.3 mg cm−2. The electrolyte was composed of NaClO4 (1 M) (Alfa Aesar, AR) and propylene carbonate (PC, Alfa Aesar, AR). The polypropylene film (Celgard 2400) and metallic sodium (Sinopharm Chemical Reagent Co., Ltd, 99.9%) were separately utilized as the separator and counter electrode for the SIBs. Cyclic voltammetry (CV) plots were recorded using a Solartron 1470 Multistat system and the galvanostatic charge/discharge tests were measured using an Arbin BT2000 instrument in the voltage range of 0.01 to 3.0 V (vs. Na/Na+). Electrochemical impedance spectroscopy (EIS) results were also obtained using a Solartron Analytical instrument under an AC voltage of 5 mV amplitude between 0.01 Hz and 100 kHz. Besides this, an equivalent circuit was utilized to fit the EIS results.
:3:3 onto Cu foil. After being dried at 90 °C for 12 h under vacuum, the CR2016-type half-cells were assembled in a Braun-glovebox in argon atmosphere. The mass loading of the active material is about 1.0–1.3 mg cm−2. The electrolyte was composed of NaClO4 (1 M) (Alfa Aesar, AR) and propylene carbonate (PC, Alfa Aesar, AR). The polypropylene film (Celgard 2400) and metallic sodium (Sinopharm Chemical Reagent Co., Ltd, 99.9%) were separately utilized as the separator and counter electrode for the SIBs. Cyclic voltammetry (CV) plots were recorded using a Solartron 1470 Multistat system and the galvanostatic charge/discharge tests were measured using an Arbin BT2000 instrument in the voltage range of 0.01 to 3.0 V (vs. Na/Na+). Electrochemical impedance spectroscopy (EIS) results were also obtained using a Solartron Analytical instrument under an AC voltage of 5 mV amplitude between 0.01 Hz and 100 kHz. Besides this, an equivalent circuit was utilized to fit the EIS results.
3. Results and discussion
3.1 Solvent-free synthesis and characterization of the CoSnO3–MOF
As shown in Scheme 1, CoSnO3 nanoboxes were chosen as the precursor of the Co2+-source, and 2-methylimidazole was utilized as the organic ligand. When heating up to 200 °C, 2-methylimidazole first melted and permeated the surface of CoSnO3, and then was chemically bonded with Co2+, resulting in the formation of a surface Co-based MOF at the internal and external surface of the CoSnO3 nanoboxes. The formation of the CoSnO3–MOFs was confirmed by the X-ray diffraction (XRD) characterization presented in Fig. 1a. Apparently, unlike the amorphous structure of CoSnO3, CoSnO3–MOF-60, CoSnO3–MOF-120, and CoSnO3–MOF-240 exhibit the same characteristic peaks as the standard Co–MOF(ZIF-67).17 Note that the characteristic peaks in CoSnO3–MOF-60 corresponding to ZIF-67 are not obvious, which was attributed to the incomplete growth of CoSnO3–MOF resulting from the insufficient amount of 2-methylimidazole. Remarkably, the characteristic peak intensity of CoSnO3–MOF-60, CoSnO3–MOF-120 and CoSnO3–MOF-240 became stronger with the higher amounts of 2-methylimidazole, showing that the growth degree of CoSnO3–MOFs can be manipulated by the amount of 2-methylimidazole used. This also convincingly demonstrates the formation of the surface CoSnO3–MOF, and this method may be superior to the traditionally uncontrolled hydrothermal preparation method. The N2 adsorption–desorption isotherms and pore distributions of CoSnO3 and CoSnO3–MOF-120 are depicted in Fig. 1b and c. The surface area of CoSnO3–MOF-120 is 526 m2 g−1, about ten times larger than that of CoSnO3 (48.5 m2 g−1). Besides, the pore distribution of CoSnO3–MOF-120 is centered at 1.16 nm, rather than at 9.33 nm like CoSnO3, which demonstrates the formation of CoSnO3–MOF from another point.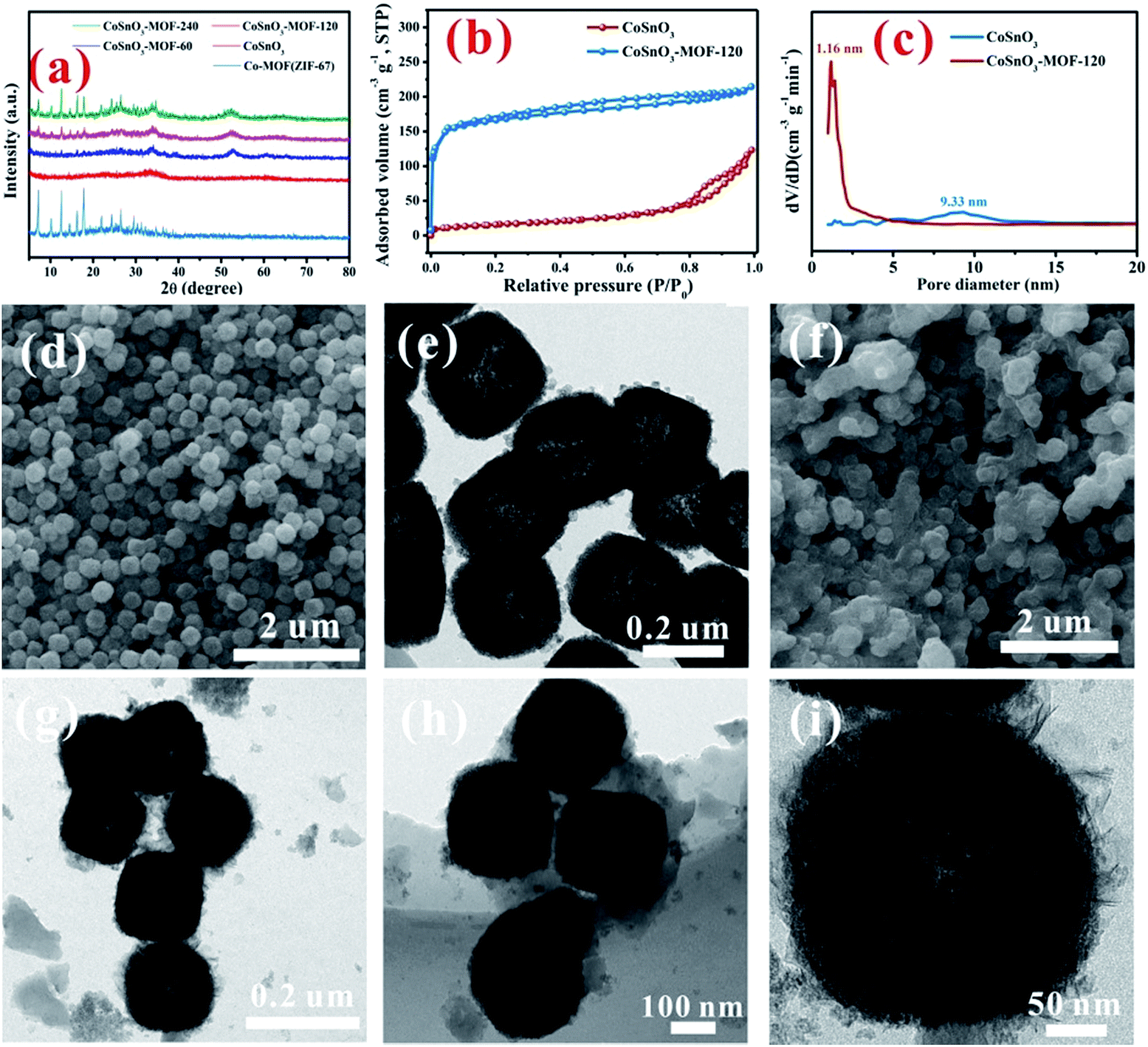 | ||
| Fig. 1 (a) The XRD patterns of the obtained samples. (b) and (c) N2 adsorption–desorption isotherms and pore size distribution of the related samples. The SEM (d) and TEM (e) images of the CoSnO3 nanoboxes. The SEM (f) and TEM (g–i) images of the CoSnO3–MOF-120 nanoboxes. | ||
SEM and TEM were employed to explore the morphology during the formation of CoSnO3–MOF-120. As shown in Fig. 1d and e, the SEM and TEM images show that CoSnO3 exhibits a nanobox shape with a shell width of about 30–40 nm, in which the inner cavities are obviously exposed by the sharp difference between the shells and their hollow interiors. Interestingly, as depicted in Fig. 1f–i, CoSnO3–MOF-120 presents a very uniformly distributed nanobox shape, suggesting that the reaction between Co2+ and 2-methylimidazole does not destroy the original structure of CoSnO3. Besides, a layer of membrane-like substance is clearly observed and well covers the surface of CoSnO3, connecting the CoSnO3 nanoboxes together. Note that the inner cavities are not apparent following the formation of the surface film.
The impact of different amounts of 2-methylimidazole on the morphology was also explored as shown in Fig. 2. A very thin layer of the membrane can be seen at the surface of CoSnO3–MOF-60, when observed from the SEM and TEM images displayed in Fig. 2b, compared with CoSnO3 in Fig. 2a. The thicknesses of the surface membrane in CoSnO3–MOF-60 (Fig. 2b), CoSnO3–MOF-120 (Fig. 2c), and CoSnO3–MOF-240 (Fig. 2d) became thicker with the incremental amounts of 2-methylimidazole, indicating that the growth of the MOFs increased with more 2-methylimidazole. It should be noted that the CoSnO3 nanoboxes are totally coated by the 2-methylimidazole layer in the CoSnO3–MOF-120 and CoSnO3–MOF-240 samples. Besides, unlike the relatively smooth surface of the CoSnO3, the surface of CoSnO3–MOFs becomes rougher and the inner cavities become less obvious. Entertainingly, considering the colourful colour features of the Co-based compounds, the colours of CoSnO3–MOFs are displayed in Fig. S1,† exhibiting a change from dark grey (CoSnO3–MOF-60) to purple (CoSnO3–MOF-120), and lastly to bright purple (CoSnO3–MOF-240), which means the MOFs are closer to the colour of ZIF-67 when the higher amounts of 2-methylimidazole are used. Furthermore, the BET specific areas of CoSnO3, CoSnO3–MOF-60, CoSnO3–MOF-120, and CoSnO3–MOF-240 are 48.5, 202.6, 526, and 806 m2 g−1 (Fig. S2†), respectively, indicating an increasing trend. The elemental mapping under TEM has been conducted to confirm the formation of ZIF-67. As shown in Fig. S3,† the C, N, Co, Sn, and O elements are uniformly distributed, proving that the formation of ZIF-67 is consistent throughout all the particles.
 | ||
| Fig. 2 The SEM and TEM images of CoSnO3 (a), CoSnO3–MOF-60 (b), CoSnO3–MOF-120 (c), and CoSnO3–MOF-240 (d). | ||
3.2 Characterization of the CoSnO3–NC materials
After carbonization in Ar atmosphere, a series of CoSnO3–NC materials were obtained. As shown in Fig. S4,† CoSnO3–NC-120 exhibits the characteristic peaks of amorphous CoSnO3 (between 30 and 40°) and amorphous carbon (around 24° and 43°) resulting from the carbonization of the organic linkers.44 The nitrogen sorption isotherm of CoSnO3–NC-120 (Fig. 3a) shows a typical type IV hysteresis loop, proving the coexistence of the mesopores and micropores,45 which can allow better electrolyte wetting. The surface area of CoSnO3–NC-120 is 58.84 m2 g−1, which is far less than that of CoSnO3–MOF-120, and this was attributed to the carbonization of 2-methylimidazole. Moreover, the surface areas of CoSnO3–NC-60 and CoSnO3–NC-240 are 50.2 and 62.8 m2 g−1 (Fig. S5†), respectively, which might be ascribed to their different carbon content derived from the organic precursor. The mesopores and micropores of CoSnO3–NC-120 are mainly centered at 3.9 nm and 1.4 nm, respectively (Fig. 3b). Besides, as shown in Fig. S5,† CoSnO3–NC-60 and CoSnO3–NC-240 present a similar nitrogen sorption isotherm to CoSnO3–NC-120 (type IV), indicating that they possess similar pore structures. Furthermore, the carbon content of the CoSnO3–NC materials was measured using the TGA method. The carbon content of CoSnO3–NC-60 (Fig. 3c), CoSnO3–NC-120 (Fig. 3d), and CoSnO3–NC-240 (Fig. 3e) was 7.5%, 14.6%, and 24.6%, respectively, which can effectively improve the electronic conductivity of the CoSnO3 nanoboxes. Furthermore, the conductivity value of CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 was 8.6 × 10−7, 8.8 × 10−4, 7.5 × 10−3 and 2.6 × 10−3 S cm−1, respectively, also showing an elevation in conductivity.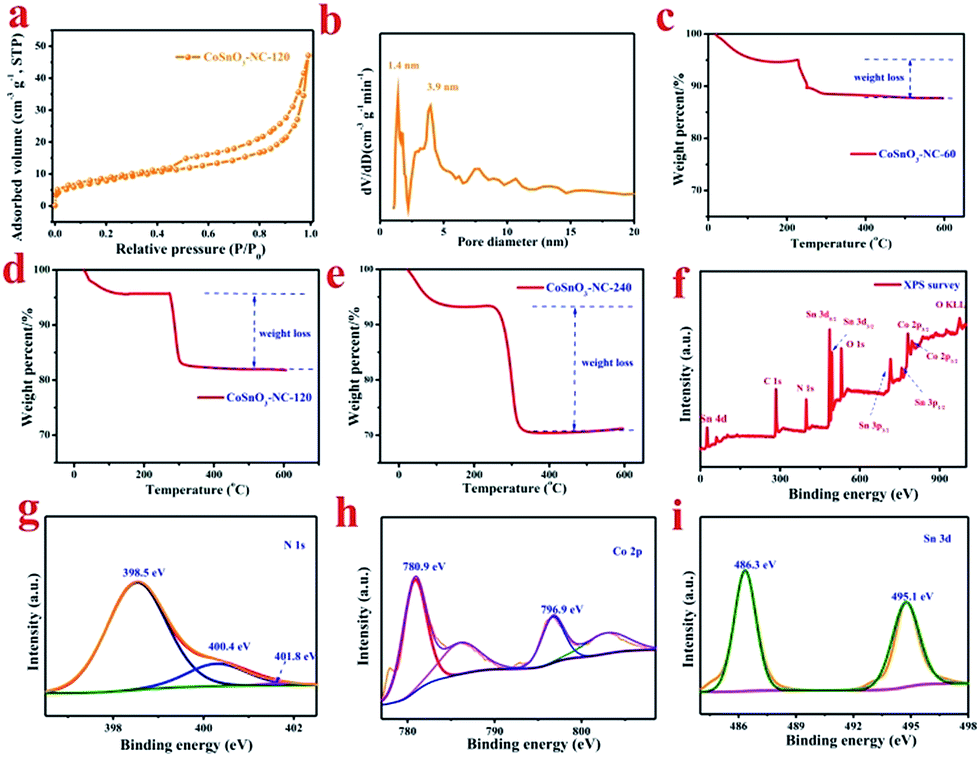 | ||
| Fig. 3 (a) The N2 adsorption–desorption isotherms of the obtained sample, and (b) the pore size distribution of CoSnO3–NC-120. TGA curves of (c) CoSnO3–NC-60, (d) CoSnO3–NC-120, and (e) CoSnO3–NC-240. (f) to (i) are the XPS survey and N1s, Co 2p, and Sn 3d spectra of the CoSnO3–NC-120 sample, respectively. | ||
To further characterize the element species and the element valences of the as-obtained samples, XPS experiments were implemented and the results are depicted in Fig. 3f–i. The XPS survey of CoSnO3–NC-120 (Fig. 3f) showed that the sample consisted of Co, Sn, C, N, and O elements. For comparison, the XPS surveys of CoSnO3, CoSnO3–NC-60, and CoSnO3–NC-240 are displayed in Fig. S6a.† According to the XPS results, CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 show a surface nitrogen content of 0.0%, 2.4%, 4.56% and 7.6%, respectively, exhibiting a controllable and high surface nitrogen content, and this can enhance the electronic conductivity of the as-obtained materials and further optimize their electrochemical performance.46 Fig. S6b† shows the C 1s XPS spectrum, and the three peaks located at 284.8, 286.5, and 288.3 eV can be attributed to the sp2 or sp3 hybridized carbon (C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), the C–O or C–N bond, and the CO bond, respectively.47 From the N1s spectrum depicted in Fig. 3g, the three peaks centered at 398.5, 400.4, and 400.8 eV may be separately related to pyridinic N, pyrrolic N, and graphitic N, respectively.19 The high-resolution XPS spectrum of Co 2p (Fig. 3h) exhibits two major peaks around 780.9 and 796.9 eV accompanied by a broad satellite peak, and these were assigned to Co 2p3/2 and 2p1/2 derived from Co2+,48,49 which indicates that the valence of Co2+ almost remains unchanged during the carbonization process. The Sn 3d peak in the survey scan can be allocated into two parts (Fig. 3i), and the peaks located at 486.3 and 495.1 eV are separately attributed to Sn 3d5/2 and Sn 3d3/2 which is well in accordance with the electronic state of Sn4+.49
C), the C–O or C–N bond, and the CO bond, respectively.47 From the N1s spectrum depicted in Fig. 3g, the three peaks centered at 398.5, 400.4, and 400.8 eV may be separately related to pyridinic N, pyrrolic N, and graphitic N, respectively.19 The high-resolution XPS spectrum of Co 2p (Fig. 3h) exhibits two major peaks around 780.9 and 796.9 eV accompanied by a broad satellite peak, and these were assigned to Co 2p3/2 and 2p1/2 derived from Co2+,48,49 which indicates that the valence of Co2+ almost remains unchanged during the carbonization process. The Sn 3d peak in the survey scan can be allocated into two parts (Fig. 3i), and the peaks located at 486.3 and 495.1 eV are separately attributed to Sn 3d5/2 and Sn 3d3/2 which is well in accordance with the electronic state of Sn4+.49
The morphologies of the CoSnO3–NC materials were also marked by SEM and TEM characterization, and the results are depicted in Fig. 4. It is clearly observed that the nanobox shape of CoSnO3–MOF is well kept after carbonization at 500 °C. Compared to the unobvious carbon layer in CoSnO3–NC-60 (Fig. 4a), the carbon layer in CoSnO3–NC-120 (Fig. 4b) can be easily noted. Moreover, the carbon layer is too thick to easily observe the CoSnO3 nanoboxes in CoSnO3–NC-240 (Fig. 4c) according to the SEM images, and these observations are similar to those of the corresponding CoSnO3–MOFs. These nitrogen-rich carbon coated structures can largely enhance the electronic conductivity, effectively ease the volume expansion, and thus avoid the structure collapse during the charge/discharge processes. Besides, note that the CoSnO3–NC nanoboxes are stacked by countless nanoparticles with a carbon layer covering their surface according to the TEM images. Also, note that the carbon layers are obviously observed in the HRTEM images and become thicker with the more 2-methylimidazole used, showing controllable carbon content of the CoSnO3–NC material.
 | ||
| Fig. 4 The SEM and TEM images of CoSnO3–NC-60 (a), CoSnO3–NC-120 (b), and CoSnO3–NC-240 (c). | ||
3.3 Electrochemical performance tests
Fig. 5a/S7a† show the charge/discharge cycling performances of the as-obtained samples for SIBs. The first discharge/charge capacities of CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 were 927.2/558.5, 899.2/526, 879.2/494.7, and 733.8/442.9 mA h g−1 at a current density of 100 mA g−1 within a voltage range of 0.01 to 3.0 V. The capacity loss in the initial cycle might be attributed to the unavoidable formation of a solid electrolyte interface (SEI) layer, some irreversible side reactions between the electrolyte and the sodium species, electrolyte decomposition, and the irreversible insertion process, which is a common phenomenon for most of the SIB anode materials.44,46,47,50,51 Moreover, CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 separately displayed a capacity of 218.3, 293.1, 493.9, and 399.9 mA h g−1 at 100 mA g−1 after 100 cycles. The capacity of CoSnO3–NC-60 gradually decreased with the cycling numbers, and this may be attributed to the fact that there was insufficient carbon to completely coat the CoSnO3 particles. CoSnO3–NC-120 and CoSnO3–NC-240 were stable after 100 cycles. However, the capacity of CoSnO3–NC-240 was lower than that of CoSnO3–NC-120 due to the high carbon content, showing that a carbon content of 14.6% is the optimal coating. Overall, the big difference in cycling performance after 100 cycles is attributed to the different amounts of carbon element, which can effectively enhance the electronic conductivity and maintain the structural stability through the carbon-wrapped and interconnected carbon structures. | ||
| Fig. 5 (a) The discharge capacities of the corresponding samples, and (b) the rate capability of the as-obtained products. (c) The first five CV curves and (d) the discharge/charge profiles of the half-cells utilizing CoSnO3–NC-120 as the electrode, and (e) the long cycling life tests of the half-cells utilizing CoSnO3–NC-120 as the electrode at a high current density of 1 A g−1. | ||
Importantly, no noticeable morphology changes were detected for the CoSnO3–NC-120 anode after 100 cycles at a current density of 100 mA h g−1 (Fig. 6), which confirms that the carbon layer can effectively stabilize the structure and remit the large volume change during the insertion/extraction of sodium ions.
 | ||
| Fig. 6 (a) and (b) The SEM images of CoSnO3–NC-120 after cycling for 100 cycles. | ||
Fig. 5b presents the rate performance of the as-obtained anode materials measured at current densities of 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, and 5.0 A g−1. As expected, CoSnO3–NC-120 delivers a superior rate performance with a reversible capacity of 519.2, 472.8, 403.7, 322.8, 249.1, 197.3, and 168.1 mA h g−1 at 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, and 5.0 A g−1, respectively. Furthermore, CoSnO3–NC-240 also shows a relatively good rate performance when compared with that of CoSnO3and CoSnO3–NC-60. The capacities of CoSnO3–NC-120 and CoSnO3–NC-240 can recover to 512.6 and 421.7 mA h g−1, respectively, nearly equaling their initial capacities, which indicates the high stability of the material under high-rate charge/discharge processes. Comparing with the poor performance of CoSnO3, the excellent rate performance might be assigned to the carbon layer on the surface which helps to maintain structural integrity, increasing the contact area between the active material and the electrolyte, and buffering the volume expansion.
The first five cyclic voltammetry (CV) curves of the CoSnO3–NC-120 electrode were plotted in the voltage range of 0.01 to 3.0 V under 0.1 mV s−1 for sodium-ion storage. As depicted in Fig. 5c, the reduction of the peak intensities between the initial cycle and the subsequent cycles suggests an irreversible capacity loss in the first discharge process, which might be connected to the formation of a solid–electrolyte interface (SEI) film on the surface of the electrodes, and to some irreversible side reactions. Importantly, after the second cycle, the CV curves of the CoSnO3–NC-120 active material virtually overlap with the succeeding cycles, which demonstrates a good stability and cycling performance of CoSnO3–NC-120 during the uptake and release of sodium ions. Fig. 5d shows the galvanostatic charge–discharge profiles of CoSnO3–NC-120 under the 1st, 5th, 20th and 50th cycles at 100 mA g−1. CoSnO3–NC-120 exhibited a discharge capacity of 879.2, 506.3, 499.2, and 490.1 mA h g−1 under the 1st, 5th, 20th and 50th cycles at 100 mA g−1, respectively, which is far better than the performance of CoSnO3 with corresponding discharge capacities of 927.2, 461.7, 394.2, and 296.4 mA h g−1 (Fig. S7b†). Besides, like in most metal-oxide electrodes,42,52,53 no obvious plateau-like features were observed in any of the discharge/charge profiles and the discharge profiles of CoSnO3–NC-120 reflect a long slope below 1.5 V. More importantly, CoSnO3–NC-120 delivered a superior long-term cycling stability with a high capacity of 273.8 mA h g−1 after 1000 cycles at 1 A g−1 (Fig. 5e), and this can be attributed to the stable structure derived from the interconnected carbon layer. This value is better than the capacities of most of the reported metal-oxide electrodes for sodium ion storage.
The electrochemical impedance spectroscopy (EIS) of the half-cells employed CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120 and CoSnO3–NC-240 as anodes, and were measured after 5 cycles at a certain voltage of 0.8 V. As shown in Fig. 7a, the semicircle in the high frequency region and the inclined line in the low frequency region of the Nyquist plots can be separately related to the charge transfer resistance and the sodium-ion diffusion in the electrode.54 The charge transfer impedances (R3) of CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 after 5 discharge/charge processes are 4860, 2640, 1380, and 1180 Ω, separately, which may be attributed to the N-rich carbon coated and interconnected structure. Besides, the charge transfer resistance of CoSnO3–NC-120 under different cycles is depicted in Fig. 7b. The charge transfer impedance of the fresh cells was 920 Ω. The charge transfer impedance increased to 1380 Ω after 5 cycles, which may be attributed to the formation of the SEI film. Subsequently, the charge transfer impedance decreased from 1380 to 326 Ω when the cycling numbers increased from 5 to 50 cycles, and this can be ascribed to the remnant Na+ stemming from the irreversible process and the enhanced wettability of the electrode.9,55 The comparison of CoSnO3–NC-120 to other metal oxides for sodium storage performance reported in previous reports is provided in Table 1. The outstanding electrochemical performances should be ascribed to the N-rich carbon wrapped and interconnected structures, which can efficiently modify the electronic conductivity, increase the mechanical stabilities, and relieve the volume expansion.
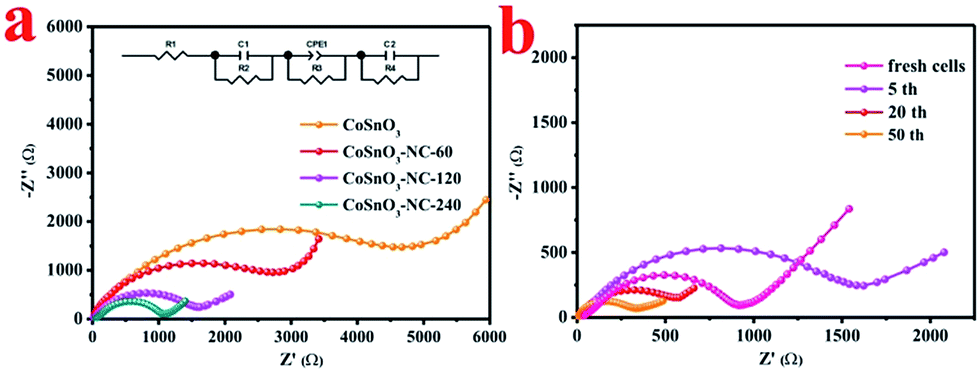 | ||
| Fig. 7 (a) The Nyquist plots of CoSnO3, CoSnO3–NC-60, CoSnO3–NC-120, and CoSnO3–NC-240 after five cycles and the inset is the equivalent circuit, and (b) the Nyquist plots of CoSnO3–NC-120 with different cycle numbers. | ||
| Anodes | Cycling performance (mA h g−1) | Rate capability (mA h g−1) | References |
|---|---|---|---|
| Carbon-confined SnO2 | 374 at 50 mA g−1 after 100 cycles | ∼307 at 0.2 A g−1 | 56 |
| ∼184 at 0.8 A g−1 | |||
| Nanostructured Co3O4 | ∼447 at 0.05 A g−1 after 50 cycles | Not given | 57 |
| Co3O4 sheets/3D graphene | ∼523.5 at 25 mA g−1 after 50 cycles | ∼82.3 at 0.5 A g−1 | 58 |
| CuO/Cu2O | ∼415 at 0.05 A g−1 after 50 cycles | ∼273 at 0.5 A g−1 | 42 |
| 212 at 1 A g−1 after 400 cycles | ∼153.8 at 2.5 A g−1 | ||
| SnO2/reduced graphene oxide | ∼333 at 0.1 A g−1 after 150 cycles | ∼125 at 1 A g−1 | 59 |
| Carbon coated rutile TiO2 | ∼175 at 84 mA g−1 after 200 cycles | ∼86.8 at 1.68 A g−1 | 9 |
| ∼70.6 at 3.36 A g−1 | |||
| Anatase TiO2@C | 167.4 at 100 mA g−1 after 110 cycles | 125.5 at 0.5 A g−1 | 60 |
| 111.1 at 1 A g−1 | |||
| 88.9 at 2.5 A g−1 | |||
| MgFe2O4 hollow microboxes | 135 at 50 mA g−1 after 150 cycles | 172 at 0.1 A g−1 | 61 |
| 85 at 1 A g−1 | |||
| Porous CoFe2O4 nanocubes | 350 at 50 mA g−1 after 50 cycles | 329.7 at 0.2 A g−1 | 62 |
| 224 at 1 A g−1 | |||
| 175.5 at 2.5 A g−1 | |||
| CoSnO3–NCs | ∼493.9 at 0.1 A g−1 after 100 cycles | ∼249.1 at 1.6 A g−1 | This work |
| ∼273.8 at 1 A g−1 after 1000 cycles | ∼197.3 at 3.2 A g−1 | ||
| ∼168.1 at 5 A g−1 |
4. Conclusion
In summary, a series of CoSnO3–NC nanobox materials with controllable amounts of N-rich carbon have been prepared by regulating the growth of the CoSnO3–MOFs under different amounts of 2-methylimidazole, in which the CoSnO3 materials are well wrapped and interconnected by a N-rich carbon layer. Significantly, a simple and solvent-free approach for the synthesis of novel CoSnO3–MOFs was provided through the chemical bonding between Co2+ and 2-methylimidazole. The CoSnO3–MOFs present a nanobox shape with a layer substance consisting of a CoSnO3–MOF covering it, and a high specific surface area. Furthermore, a superior sodium-ion storage performance of 493.9 mA h g−1 at 0.1 A g−1 can be acquired after 100 cycles and a reversible capacity of 273.8 mA h g−1 is reserved at 1 A g−1 after 1000 cycles. This work offers a new method to prepare Co–MOFs and a good N-rich carbon-coating route for Co-based oxides, which may also be suitable for use with other Co-based oxides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), the National Postdoctoral Program for Innovative Talents (BX201600192), the National Natural Science Foundation of China (51622406, 21673298 and 21473258), the Innovation-Driven Project of Central South University (No. 2017CX004), the Fundamental Research Funds for the Central Universities of Central South University (2017zzts115 and 2017zzts454), the China Postdoctoral Science Foundation (2017M6203552), the National Key Research and Development Program of China (2017YFB0102000), and the Hunan Provincial Science and Technology Plan (2017TP1001).References
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- G. Korotcenkov, Metal-Organic Frameworks, Springer, New York, 2014, vol. 32, pp. 276–288 Search PubMed.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- D. Yuan, D. Zhao, D. Sun and H. C. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5357–5361 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- F. Meng, Z. Fang, Z. Li, W. Xu, M. Wang, Y. Liu, J. Zhang, W. Wang, D. Zhao and X. Guo, J. Mater. Chem. A, 2013, 1, 7235–7241 CAS.
- G. Zou, J. Chen, Y. Zhang, C. Wang, Z. Huang, S. Li, H. Liao, J. Wang and X. Ji, J. Power Sources, 2016, 325, 25–34 CrossRef CAS.
- F. Zou, Y. M. Chen, K. Liu, Z. Yu, W. Liang, S. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2015, 10, 377 CrossRef PubMed.
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520–524 CrossRef CAS.
- W. Meng, W. Chen, L. Zhao, Y. Huang, M. Zhu, Y. Huang, Y. Fu, F. Geng, J. Yu and X. Chen, Nano Energy, 2014, 8, 133–140 CrossRef CAS.
- R. Wu, X. Qian, X. Rui, H. Liu, B. Yadian, K. Zhou, J. Wei, Q. Yan, X. Q. Feng and Y. Long, Small, 2014, 10, 1932–1938 CrossRef CAS PubMed.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590 CrossRef CAS PubMed.
- X. Yu, Y. Feng, B. Guan, X. W. D. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- B. W. Hao, Y. X. Bao, Y. Le, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef PubMed.
- R. R. Kuruppathparambil, T. Jose, R. Babu, G. Y. Hwang, A. C. Kathalikkattil, D. W. Kim and D. W. Park, Appl. Catal., B, 2016, 182, 562–569 CrossRef CAS.
- H. An, S. Park, H. T. Kwon, H. K. Jeong and J. S. Lee, J. Membr. Sci., 2017, 526, 367–376 CrossRef CAS.
- Y. Hou, J. Li, Z. Wen, S. Cui, C. Yuan and J. Chen, Nano Energy, 2015, 12, 1–8 CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- X. Han, W. M. Chen, X. Han, Y. Z. Tan and D. Sun, J. Mater. Chem. A, 2016, 4, 13040–13045 CAS.
- H. Y. Cho, D. A. Yang, J. Kim, S. Y. Jeong and W. S. Ahn, Catal. Today, 2012, 185, 35–40 CrossRef CAS.
- B. Pattengale, S. Yang, J. Ludwig, Z. Huang, X. Zhang and J. Huang, J. Am. Chem. Soc., 2016, 138, 8072 CrossRef CAS PubMed.
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., 2015, 54, 10889–10893 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033–5033 CAS.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- K. Zhang, M. Park, L. Zhou, G. H. Lee, J. Shin, Z. Hu, S. L. Chou, J. Chen and Y. M. Kang, Angew. Chem., Int. Ed., 2016, 55, 12822–12826 CrossRef CAS PubMed.
- L. Wei, S. Fei, C. Bommier, H. Zhu, X. Ji and L. Hu, Acc. Chem. Res., 2016, 49, 231 CrossRef PubMed.
- W. Luo, J. Schardt, C. Bommier, B. Wang, J. Razink, J. Simonsen and X. Ji, J. Mater. Chem. A, 2013, 1, 10662–10666 CAS.
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed.
- X. Xie, T. Makaryan, M. Zhao, K. L. Van Aken, Y. Gogotsi and G. Wang, Adv. Energy Mater., 2015, 6, 1502161 CrossRef.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- Y. Yan, Y. X. Yin, Y. G. Guo and L. J. Wan, Adv. Energy Mater., 2014, 4, 1301584 CrossRef.
- F. Xie, L. Zhang, D. Su, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1700989 CrossRef PubMed.
- L. Xia, S. Wang, G. Liu, L. Ding, D. Li, H. Wang and S. Qiao, Small, 2016, 12, 853–859 CrossRef CAS PubMed.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- L. Fu, K. Tang, K. Song, P. A. van Aken, Y. Yu and J. Maier, Nanoscale, 2014, 6, 1384–1389 RSC.
- G. Zou, H. Hou, P. Ge, Z. Huang, G. Zhao, D. Yin and X. Ji, Small, 2018, 14, 1702648 CrossRef PubMed.
- G. Zou, X. Jia, Z. Huang, S. Li, H. Liao, H. Hou, L. Huang and X. Ji, Electrochim. Acta, 2016, 196, 413–421 CrossRef CAS.
- Y. Wang, C. Wang, Y. Wang, H. Liu and Z. Huang, J. Mater. Chem. A, 2016, 4, 5428–5435 CAS.
- X. Zhang, W. Qin, D. Li, D. Yan, B. Hu, Z. Sun and L. Pan, Chem. Commun., 2015, 51, 16413–16416 RSC.
- F. Zou, Y. M. Chen, K. Liu, Z. Yu, W. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, W. Liu, W. Xiao and X. W. Lou, Energy Environ. Sci., 2012, 6, 87–91 Search PubMed.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS.
- Z. Zhu, S. Wang, J. Du, Q. Jin, T. Zhang, F. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed.
- H. Hou, C. E. Banks, M. Jing, Y. Zhang and X. Ji, Adv. Mater., 2015, 27, 7861–7866 CrossRef CAS PubMed.
- C. Guan, X. Li, H. Yu, L. Mao, L. H. Wong, Q. Yan and J. Wang, Nanoscale, 2014, 6, 13824–13830 RSC.
- M. W. Nydegger, G. Couderc and M. A. Langell, Appl. Surf. Sci., 1999, 147, 58–66 CrossRef CAS.
- Y. Zhu, Y. Wen, X. Fan, T. Gao, F. Han, C. Luo, S.-C. Liou and C. Wang, ACS Nano, 2015, 9, 3254–3264 CrossRef CAS PubMed.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- D. Su, S. Dou and G. Wang, Chem. Mater., 2015, 27, 6022–6029 CrossRef CAS.
- M. Hu, Y. Jiang, W. Sun, H. Wang, C. Jin and M. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 19449 CAS.
- K. Tang, L. Fu, R. J. White, L. Yu, M. M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS.
- K. Xi, S. Cao, X. Peng, C. Ducati, R. V. Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192–2194 RSC.
- M. Dirican, Y. Lu, Y. Ge, O. Yildiz and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 18387–18396 CAS.
- M. M. Rahman, A. M. Glushenkov, T. Ramireddy and Y. Chen, Chem. Commun., 2014, 50, 5057–5060 RSC.
- Y. Liu, Z. Cheng, H. Sun, H. Arandiyan, J. Li and M. Ahmad, J. Power Sources, 2015, 273, 878–884 CrossRef CAS.
- Y. X. Wang, Y. G. Lim, M. S. Park, S. L. Chou, J. Kim, H. K. Liu, S. X. Dou and Y. J. Kim, J. Mater. Chem. A, 2013, 2, 529–534 Search PubMed.
- X. Shi, Z. Zhang, K. Du, Y. Lai, J. Fang and J. Li, J. Power Sources, 2016, 330, 1–6 CrossRef CAS.
- Y. Guo, Y. Zhu, C. Yuan and C. Wang, Mater. Lett., 2017, 199, 101–104 CrossRef CAS.
- X. Zhang, D. Li, G. Zhu, T. Lu and L. Pan, J. Colloid Interface Sci., 2017, 499, 145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta10448k |
| This journal is © The Royal Society of Chemistry 2018 |