
Preparation of highly dispersed carbon supported AuPt nanoparticles via a capping agent-free route for efficient methanol oxidation†
Lijuan
Lu
a,
Yao
Nie
a,
Yao
Wang
a,
Guangping
Wu
a,
Lingjie
Li
a,
Jing
Li
a,
Xueqiang
Qi
*b and
Zidong
Wei
 *a
*a
aChongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400040, China. E-mail: zdwei@cqu.edu.cn
bCollege of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing 400054, China. E-mail: xqqi@cqu.edu.cn
First published on 27th November 2017
Abstract
A facile synthesis of carbon-supported bimetallic AuPt catalysts via a capping agent-free route is reported. The synthesized AuPt nanoparticles are well dispersed with a narrow size distribution ranging from 2.50 to 4.50 nm. Compared with commercial PtRu/C and Pt/C nanoparticles, the present AuPt/C catalysts exhibit distinctly superior activity for the methanol oxidation reaction with notable CO tolerance. Moreover, the composition-dependent catalytic activity of AuPt/C catalysts is observed, which follows the order Au2Pt1/C > Au1Pt1/C > Au1Pt2/C.
1 Introduction
Direct methanol fuel cells (DMFCs) are a highly promising and well-developed energy technology that may substitute for conventional batteries in portable electronics due to their low operating temperature, high-energy conversion efficiency, and low pollutant production.1–3 The most popular anode electrocatalysts for the methanol oxidation reaction (MOR) are carbon-supported Pt alloy nanoparticles.4 However, state-of-the-art platinum-based DMFC anode electrocatalysts suffer from poor reaction kinetics and are easily contaminated by oxygen-containing carbon species, such as CO. Thus, a significant amount of work has been dedicated to improving the CO tolerance ability.5–10 Au NPs have been reported to be extraordinary CO oxidation catalysts,11,12 and their affinity to CO oxidation has created a strong research interest in Au–M bimetallic or alloy (where M is, e.g., Pt,13–15 Pd,16 and Ru17) catalysts. Particularly, bimetallic platinum–gold (PtAu) catalysts have demonstrated superior activity for the oxidation of methanol18–22 and formic acid.23–26 However, the synthesis of alloy particles of Au–Pt are mainly limited by the usage of stabilizing ligands, such as poly(vinylalcohol) (PVA)27,28 or poly(vinylpyrrolidone) (PVP).29–31 There is a major drawback in the use of such methods, because the extensive changes in the size and shape of the nanoparticles would occur during ligand removal, which partially undermines the motive of using the ligand.32–35To overcome the above problems, here, we report a facile capping agent-free method for the synthesis of AuPt nanoparticles. The observed AuPt nanoparticles are uniformly dispersed on Vulcan-X72 with a narrow size distribution. Compared with commercial PtRu/C and commercial Pt/C, the as-made Au–Pt/C nanoparticles are more active and durable for the MOR accompanied by CO tolerance ability due to their synergistic effect. And the structure of the AuPt nanoparticles can be easily modulated by elaborately controlling the ratio of Au to Pt. When the Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio is 1:1, the as-prepared AuPt possessed an obvious alloy structure compared with the bimetallic AuPt nanoparticles synthesized in other Au/Pt ratios and shows the highest methanol oxidation activity. And the synergistic effect driven by the excellent alloy structure is believed to be responsible for the ultrahigh activity of AuPt catalysts.
:Au ratio is 1:1, the as-prepared AuPt possessed an obvious alloy structure compared with the bimetallic AuPt nanoparticles synthesized in other Au/Pt ratios and shows the highest methanol oxidation activity. And the synergistic effect driven by the excellent alloy structure is believed to be responsible for the ultrahigh activity of AuPt catalysts.
2 Experimental
2.1 Materials
Vulcan XC-72 carbon, chloroplatinic acid (H2PtCl6·6H2O, 99.9%), chloroauric acid (HAuCl3·3H2O, 99.9%), and sodium borohydride (NaBH4, 99%) were purchased from Sigma-Aldrich. Anhydrous sodium carbonate (NaCO3), absolute ethyl alcohol and THF are analytically pure. All other chemicals are of reagent grade, and Milli-Q water with a resistivity greater than 18.0 MΩ cm is used in the preparation of aqueous solutions.2.2 Synthesis of the Au–Pt/C catalyst
Vulcan XC-72 carbon black, purchased from American Cabot, was firstly purified with nitric acid at 160 °C for 4 h. Then the XC-72 was dispersed in a solution of THF/H2O(1:1), and exposed to ultrasonic treatment for 30 min. After that, H2PtCl6 (20 mM) and HAuCl3 (20 Mm) were added, and the obtained solution was mechanically stirred for 24 h. Finally, a solution containing 100 ml of NaBH4 and Na2CO3 was added dropwise to the above solution in an ice-bath, and the reaction was continued for 12 h to form the carbon supported Au–Pt NPs. The achieved product was filtered, washed with absolute ethyl alcohol and distilled water in consecutive washing/centrifugation cycles, and dried at 60 °C.
2.3 Electrochemical tests
All potentials were given relative to the reversible hydrogen electrode (RHE). To prepare the AuPt/C working electrode, the electrocatalyst was dispersed in the solution containing isopropanol and water in a volume ratio of 3:1, and ultrasounded for 10 min to form a uniform catalyst ink. The obtained slurry (10 μl) was loaded on the surface of a glassy carbon electrode with a 5 mm diameter. A drop of 0.1 wt% Nafion solution was applied to the surface of the catalyst layer to form a thin protective film. The electrolyte was purged with high-purity nitrogen for at least 15 min to remove dissolved oxygen. The methanol oxidation experiment was conducted in a mixed solution of 0.1 M HClO4 and 1.0 M CH3OH at a scan rate of 10 mV s−1. Noble metal loading on the working electrode was 10 μg. CV accelerated durability tests (ADTs) were performed by potential cycling between 0 and 1.2 V (vs. the RHE) at a scan rate of 100 mV s−1 in N2-saturated 0.1 M HClO4 + 1 M CH3OH solution at room temperature for 500 cycles.
In order to monitor the electrochemical surface area (ECSA), a CO-stripping experiment was performed. In detail, the catalyst surface was firstly saturated with CO by bubbling CO through a 0.1 M HClO4 solution for 15 min. The remaining CO was purged by flowing N2 for 30 min before the measurements were taken.
The ECSA was calculated according to the following equation:
| ECSA = QCO/(Qref × m) |
2.4 Characterization
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were conducted on a Zeiss LIBRA 200 FEG TEM instrument operated at 200 kV. The number-average diameter dn and the volume-surface area average diameter dv/a are calculated as follows: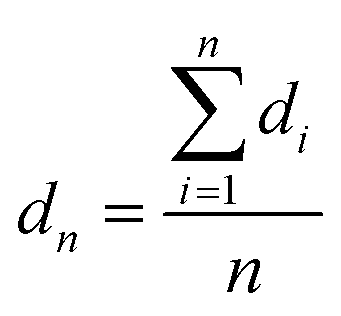 | (1) |
 | (2) |
The metal loading and atomic ratio of the Au–Pt/C catalyst were measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES). To obtain complete dissolution, the samples were dispersed in aqua regia solutions for a long time. The product composition was confirmed using the automated X-ray diffraction equipment (XRD, Rigaku D/MaXIIIA, Japan). X-ray photoelectron spectra were obtained on a Cratos XSAM800 spectrometer equipped with a monochromatic Al X-ray source (Al KR, 1.4866 keV), and the binding energy was calibrated by adopting 285 eV as the C 1s peak energy.
3 Results and discussion
As shown in Fig. 1a, AuPt nanoparticles are well dispersed on carbon supports with a narrow size distribution ranging from 2.50 to 4.50 nm, and the average diameter is around 3.40 nm (Fig. 1a–c). The atomic ratios of Au:Pt are 2/1, 1/1 and 1/2, respectively, confirmed by ICP-OES analyses (Fig. S1, Table S1†). The HRTEM images in Fig. 1d–f reveal that the Au2Pt1 and Au1Pt2 nanoparticles tend to take a twinned structure since the Au (111) plane with a d-spacing of 0.235 nm and the Pt (111) plane with a d-spacing of 0.224 nm are equal to those of Au and Pt, respectively. But in the Au1Pt1 nanoparticles the d-spacing of (111) is 0.230 nm which exactly locates between those of Au (111) and Pt (111).36 Moreover, a (200) plane with a d-spacing value of 0.200 nm and a (220) plane with a d-spacing of 0.140 nm have been determined, which are different from those for monometallic Pt and Au. Thus, it is believed that an alloyed structure has formed in the Au1Pt1 nanoparticles.
 | ||
| Fig. 1 TEM images of (a) Au2Pt1/C, (b) Au1Pt1/C and (c) Au1Pt2/C and HRTEM images of (d) Au2Pt1/C, (e) Au1Pt1/C and (f) Au1Pt2/C. | ||
To further verify the crystal structure, the XRD patterns of AuPt/C catalysts are presented in Fig. 2. Three diffraction peaks located at 2θ values of 38.8°, 44.9° and 65.5° can be well ascribed to (111), (200) and (220) planes, respectively. Particularly, they can neither be assigned to monometallic Pt nor can correspond to monometallic Au.
 | ||
| Fig. 2 The XRD patterns of the Au/C, Pt/C, Au2Pt1/C, Au1Pt1/C and Au1Pt2/C. | ||
The observation that the characteristic peaks located between those of monometallic Pt and Au could be attributed to the formation of the Au1Pt1/C alloy.37–39 Besides, there is no indication for the formation of alloyed Au2Pt1/C and Au1Pt2/C nanoparticles, since only characteristic base peaks of Pt and Au crystals could be observed. And it could be thought that the bimetallic structures are formed in the Au2Pt1/C and Au1Pt2/C nanoparticles.
In order to investigate the oxidation states and surface species together with the surface composition of Pt and Au in the nanocomposites, X-ray photoelectron spectroscopy (XPS) analysis was performed. Fig. 3 shows the XPS spectra for the Pt 4f and Au 4f of all the nanoparticles. It can be found that Au retains its zero valent chemical state with a binding energy of 84.0 ± 0.1 eV. The negative shift of the binding energy implies electron gaining of Au, presumably due to electron transfer from Pt to Au. Au atoms in the vicinity of Pt atoms must be responsible for modifying either or both the local electronic properties (alloyed character) of the bimetallic system and the morphology of the resultant nanoparticles. Pt contains more than one oxidation state in the nanoparticles since three peaks can be found in all bimetallic nanocomposites. And the Pt 4f spectra can be deconvoluted to Pt0 in the zero valent state with a binding energy of 71.5 eV and Pt2+ and Pt4+ in oxidized states with the binding energy of 72.3 and 73.9 eV, respectively.40
 | ||
| Fig. 3 XPS spectra for Pt 4f regions of (a) Pt/C, (b) Au2Pt1/C, (c) Au1Pt1/C and (d) Au1Pt2/C. | ||
Table 1 shows that the Au:Pt surface ratios obtained from the XPS analysis for bimetallic compositions are 7:93 for Au2Pt1/C, 1:97 for Au1Pt1/C and 1:99 for Au1Pt2/C, indicating that the surface composition of Pt is remarkably higher than the nominal composition. A previous study pointed out that the chemical bonding state of Pt0 can provide more active sites for the methanol oxidation reaction than those of the Pt2+ and Pt4+ species.41
| Catalyst | (ICP) Au:Pt |
(XPS) Au:Pt |
Pt0/Pt (%) |
|---|---|---|---|
| Au/C | 100:0 |
— | — |
| Pt/C | 0:100 |
— | 37 |
| Au2Pt1/C | 2:1 |
7:93 |
49 |
| Au1Pt1/C | 1:1 |
3:97 |
51 |
| Au1Pt2/C | 1:2 |
1:99 |
54 |
With the increment of the Au concentration, Pt has a greater tendency to lose electrons to Au. The increased instability of the d-electron system in AuPt, particularly Pt, causes the possibility of the presence of Pt atoms on the surface of AuPt. Generally, in multi-metallic materials, the metals having the highest affinity for the adsorbates will preferentially segregate to the surface. It has been examined by Chandler et al.42 that in the presence of ligands such as CO, the surfaces of Pt–Au bimetallic particles become enriched with Pt. It is reasonable that Pt has a higher affinity towards oxygen and forms a more stable oxide in the present case, which well explains the enrichment of Pt on the alloy surface.
These highlight the combined roles of the particle size, phase separation and collation of particles in the distribution of metals. Pt enrichment was also supported by electrochemical characterisation and catalytic activity. Since the catalytic properties are essentially dependent on the characteristics of the surface, the results obtained above reveal that the surface species and compositions are most likely responsible for the observed catalytic activity.
The catalytic activity of the AuPt/C toward the methanol oxidation is investigated as shown in Fig. 4. For comparison, commercial Pt/C and PtRu/C electrocatalysts are also studied. It can be found that the current densities of the MOR at 0.45 V are 0.0231 A mg(Au+Pt)−1 (0.023 A mgPt−1), 0.06 A mg(Au+Pt)−1 (0.12 A mgPt−1), 0.75 A mg(Au+Pt)−1 (2.25 A mgPt−1), 0.71 A mg(Au+Pt)−1 (1.42 A mgPt−1) and 0.58 A mg(Au+Pt)−1 (0.87 A mgPt−1) for commercial Pt/C, commercial PtRu/C, Au2Pt1/C, Au1Pt1/C and Au1Pt2/C, respectively. Moreover, as listed in Table 2, the series of AuPt/C nanoparticles gives a larger ratio of positive peak current/reverse peak current (If/Ib) than that of commercial PtRu/C and Pt/C. This observation implies that methanol molecules can be more effectively oxidized on the AuPt/C during the forward potential scan, generating relatively less poisoning species as compared to the commercial Pt electrocatalyst.43 Among all the catalysts, Au2Pt1/C shows the highest MOR activity considering its highest current density at 0.45 V. For all the tested catalysts, their activities follow the order Au2Pt1/C > Au1Pt1/C > Au1Pt2/C > PtRu/C > Pt/C (Fig. 4c).
 | ||
| Fig. 4 Cyclic voltammograms for commercial Pt/C, commercial PtRu/C, Au2Pt1/C, Au1Pt1/C and Au1Pt2/C in N2-saturated mixed solution of 0.1 M HClO4 and 1 M CH3OH at room temperature at a sweep rate of 10 mV s−1. | ||
| Name | Current density (at 0.45 V) (A mg−1) | Positive peak current density (A mg−1) | Reverse peak current density (A mg−1) | Positive peak current density/reverse peak current density (If/Ib) |
|---|---|---|---|---|
| Pt/C | 0.02 | 15.50 | 26.00 | 0.60 |
| PtRu/C | 0.06 | 9.00 | 6.20 | 1.45 |
| Au2Pt1/C | 0.75 | 14.69 | 7.97 | 1.84 |
| Au1Pt1/C | 0.71 | 19.07 | 10.97 | 1.74 |
| Au1Pt2/C | 0.58 | 14.43 | 9.00 | 1.60 |
The CO stripping cyclic voltammograms of the catalysts are comparatively studied as shown in Fig. 5. The CO stripping on Pt/C indicated by a peak potential at 0.77 V can be determined. However, the CO anodic oxidation on the AuPt/C catalyst starts much earlier than that on the commercial Pt/C and the same is the case with that on commercial PtRu/C, which suggests that AuPt/C nanoparticles show a greater ease of CO removal and a possibility of improved CO tolerance in practice.44 To further understand the CO oxidation ability of the three AuPt catalysts, the electrochemically active surface area (ECSA) was calculated by measuring the coulombic charge in CO stripping. Au1Pt1/C has the minimal ECSA. The specific values of the ECSA based on the precious metal mass for Au1Pt1/C, commercial PtRu/C and commercial Pt/C catalysts are estimated to be 28.66, 33.43 and 49.02 cm2 mg−1, respectively. The results suggest that the enhanced CO electrooxidation activity diminishes the poisoning of the Au2Pt1/C catalyst which has the minimum peak potential at 0.54 V and leads to a higher methanol oxidation activity.45
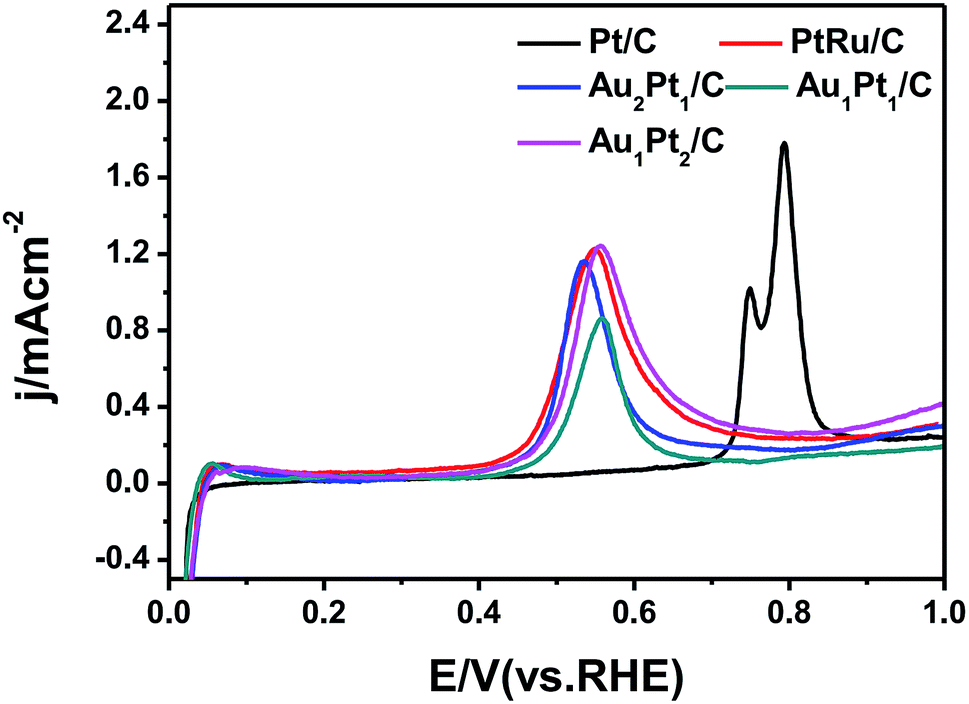 | ||
| Fig. 5 CO stripping cyclic voltammograms of the commercial Pt/C, commercial PtRu/C, Au2Pt1/C, Au1Pt1/C and Au1Pt2/C catalysts in N2-saturated 0.1 M HClO4 solution at a scan rate of 10 mV s−1. | ||
 | ||
| Fig. 6 The ADT of the (a) Pt/C, (b) PtRu/C, and (c) Au2Pt1/C by potential cycling between 0 and 1.2 V for 500 cycles in N2-saturated 0.1 M HClO4 + 1 M CH3OH solution at room temperature at a sweep rate of 100 mV s−1. | ||
The stability of methanol oxidation was critical for the application of PEMFCs. The ADTs for the Pt/C (Fig. 6a), PtRu/C (Fig. 6b) and Au2Pt1/C (Fig. 6c) were thus carried out in N2-saturated 0.1 M HClO4 + 1 M CH3OH solution by applying continuous potential sweeps between 0.0 and 1.2 V (vs. the RHE) for 500 cycles at a scan rate of 100 mV s−1. The Au2Pt1/C catalyst exhibited negligible changes for a 4.6% drop in the electric current density after 500 cycles.
4 Conclusions
In summary, highly dispersed AuPt/C catalysts have been prepared via a capping agent-free route. This method is rather simple compared to traditional thermal decomposition routes with high-boiling point organic solvents and strict conditions without oxygen. The physical characterization proved that the AuPt nanoparticles are uniformly dispersed on Vulcan-X72 carbon with an average particle size of 3.4 nm. The synthesized Au2Pt1/C nanoparticles show superior stability and activity for methanol oxidation with a significant resistance to CO poisoning in comparison to commercial PtRu/C. The electron transfer from Pt to Au leads to a synergistic effect due to their excellent catalytic activity. This efficient approach is highly favourable for the facile synthesis of alloy electrocatalysts for electrochemical applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the China Nation 973 Program (2012CB720300 and 2012CB215500) and by the NSFC of China (Grants No. 21376283 and 21436003).Notes and references
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1871 CrossRef CAS PubMed.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2011, 132, 13636–13640 CrossRef PubMed.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, Appl. Catal., B, 2006, 63, 137–149 CrossRef CAS.
- L. Kuai, S. Wand and B. Geng, Chem. Commun., 2011, 47, 6093–6095 RSC.
- D. J. Chen, S. G. Sun and Y. Y. J. Jong, Chem. Commun., 2014, 50, 12963–12965 RSC.
- M. LM Xiao, L. G. Feng, J. B. Zhu, C. P. Liu and W. Xing, Nanoscale, 2015, 7, 9467–9471 RSC.
- Z. M. Cui, C. M. Li and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 16349–16357 RSC.
- M. A. Matin, E. Lee, H. Kim, W. Yoon and Y. Kwon, J. Mater. Chem. A, 2015, 3, 17154–17164 CAS.
- A. N. Gavrilov, E. R. Savinova, P. A. Simonov, V. I. Zalkovskii, S. V. Cherepanova, G. A. Tsirlina and V. N. Parmon, Phys. Chem. Chem. Phys., 2007, 9, 5476–5489 RSC.
- Y. J. Jang, J. Jang, J. Lee, J. H. Kim, H. Kumagai, T. Minegishi, J. Kubota, K. Domen and J. S. Lee, Energy Environ. Sci., 2016, 9, 1114 CAS.
- L. Guo, S. Chen, L. Li and Z. Wei, J. Power Sources, 2014, 247, 360–364 CrossRef CAS.
- M. Eyrich, T. Diemant, H. Hartmann, J. Bansmann and R. J. Behm, J. Phys. Chem. C, 2016, 116, 11154–11165 Search PubMed.
- W. C. Ye, H. H. Kou, Q. Z. Liu, J. F. Yan, F. Zhou and C. M. Wang, Int. J. Hydrogen Energy, 2012, 37, 4088–4097 CrossRef CAS.
- F. Terzi, C. Zanardi, S. Daolio, M. Fabrizio and R. Seeber, Electrochim. Acta, 2011, 56, 3673–3678 CrossRef CAS.
- M. Khalid, N. Wasio, T. Chase and K. Bandyopadhyay, Nanoscale Res. Lett., 2010, 5, 61–67 CrossRef CAS PubMed.
- X. Y. Lang, H. Guo, L. Y. Chen, A. Kudo, J. S. Yu, W. Zhang, A. Inoue and M. W. Chen, J. Phys. Chem. C, 2010, 114, 2600–2603 CAS.
- R. Imbeault, D. Reyter, S. Garbarino, L. Roué and D. Guay, J. Phys. Chem. C, 2012, 116, 5262–5269 CAS.
- L. Zhao, J. P. Thomas, N. F. Heinig, M. Abd-Ellah, X. Wang and K. T. Leung, J. Mater. Chem. C, 2014, 2, 2707–2714 RSC.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Auard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horm, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS PubMed.
- P. Hernandez-Fernandez, S. Rojas, P. Ocon, A. de Frutos, J. M. Figueroa, P. Terreros, M. A. Pena and J. L. G. J. Fierro, J. Power Sources, 2008, 177, 9–16 CrossRef CAS.
- L. X. Yang, W. Y. Yang and Q. Y. Cai, J. Phys. Chem. C, 2007, 111, 16613–16617 CAS.
- D. Zhao and B. Q. Xu, Phys. Chem. Chem. Phys., 2006, 8, 5106–5114 RSC.
- J. B. Xu, T. S. Zhao and Z. X. J. Liang, J. Power Sources, 2008, 185, 857–861 CrossRef CAS.
- J. P. Wang, D. F. Thomas and A. C. Chen, Chem. Commun., 2008, 5010–5012 RSC.
- L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. B. Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS PubMed.
- I. P-Santos and L. M. Liz-Marzán, Langmuir, 2002, 18, 2888–2894 CrossRef.
- T. F. Jaramillo, S.-H. Baeck, B. R. Cuenya and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 7148–7149 CrossRef CAS PubMed.
- D. Xu, Z. P. Liu, H. Z. Yang, Q. S. Liu, J. Zhang, J. Y. Fang, S. Z. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217 CrossRef CAS PubMed.
- J. H. Zeng, J. Y. Lee and W. J. J. Zhou, J. Power Sources, 2006, 159, 509–513 CrossRef CAS.
- D. Bin, F. F. Ren, H. W. Wang, K. Zhang, B. B. Yang, C. Y. Zhai, M. S. Zhu, P. Yang and Y. K. Du, RSC Adv., 2014, 4, 39612–39618 RSC.
- S. Mukherjee, D. Das and P. K. Chakrabarti, J. Phys. Chem. C, 2010, 114, 14763–14771 CAS.
- H. Tsunoyama and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 18216–18217 CrossRef CAS PubMed.
- Y. Borodko, P. Ercius, D. Zherebetskyy, Y. Wang, Y. Sun and G. Somorjai, J. Phys. Chem. C, 2013, 117, 26667–26674 CAS.
- G. Collins, M. Schmidt, G. P. McGlacken, C. O'Dwyer and J. D. Holmes, J. Phys. Chem. C, 2014, 118, 6522–6530 CAS.
- Z. Yin, M. Chi, Q. Zhu, D. Ma, J. Sun and X. Bao, J. Mater. Chem. A, 2013, 1, 9157–9163 CAS.
- D. Mott, J. Luo, A. Smith, P. N. Njoki, L. Wang and C. J. Zhong, Nanoscale Res. Lett., 2007, 2, 12–16 CrossRef CAS.
- D. Mott, J. Luo, P. N. Njoki, Y. Lin, L. Wang and C. J. Zhong, Catal. Today, 2007, 122, 378–385 CrossRef CAS.
- W. Tian, M. Ge, F. Gu, T. Yamada and Y. Aoki, J. Phys. Chem. A, 2006, 110, 6285–6293 CrossRef CAS PubMed.
- NIST-XPS database, http://srdata.nist.gov/xps/, version 3.5.
- F. Sen and G. Gökağac, J. Phys. Chem. C, 2007, 111, 5715–5720 CAS.
- H. Lang, S. Maldonado, K. J. Stevenson and B. D. Chandler, J. Am. Chem. Soc., 2004, 126, 12949–12956 CrossRef CAS PubMed.
- S. Guo, S. Zhang, X. Sun and S. Sun, J. Am. Chem. Soc., 2011, 133, 15354–15357 CrossRef CAS PubMed.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177–181 CAS.
- J. Zheng, D. A. Cullen, R. V. Forest, J. A. Wittkopf, Z. Zhuang, W. Sheng, J. G. Chen and Y. Yan, ACS Catal., 2015, 5, 1468–1474 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows the ICP correction element parameters of Au (a) and Pt (b). Fig. S2 shows the XPS survey spectra of Au2Pt1/C, Au1Pt1/C and Au1Pt2/C (a), Au 4f of Au2Pt1/C (b), Au1Pt1/C (c) and Au1Pt2/C (d). Table S1 shows the ICP analysis results of Au2Pt1/C, Au1Pt1/C and Au1Pt2/C. See DOI: 10.1039/c7ta08343b |
| This journal is © The Royal Society of Chemistry 2018 |