
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisualization of diffusion limited antimicrobial peptide attack on supported lipid membranes†
George R.
Heath
 a,
Patrick L.
Harrison
b,
Peter N.
Strong
b,
Stephen D.
Evans
*a and
Keith
Miller
*b
a,
Patrick L.
Harrison
b,
Peter N.
Strong
b,
Stephen D.
Evans
*a and
Keith
Miller
*b
aDepartment of Physics and Astronomy, Leeds University, Leeds, LS2 9JT, UK. E-mail: k.miller@shu.ac.uk; s.d.evans@leeds.ac.uk
bBiomolecular Research Centre, Biosciences Division, Sheffield Hallam University, Sheffield, S1 1WB, UK
First published on 12th July 2018
Abstract
Understanding the mechanism of action of antimicrobial peptides (AMP) is fundamental to the development and design of peptide based antimicrobials. Utilizing fast-scan atomic force microscopy (AFM) we detail the attack of an AMP on both prototypical prokaryotic (DOPC:DOPG) and eukaryotic (DOPC:DOPE) model lipid membranes on the nanoscale and in real time. Previously shown to have a favourable therapeutic index, we study Smp43, an AMP with a helical-hinge-helical topology isolated from the venom of the North African scorpion Scorpio maurus palmatus. We observe the dynamic formation of highly branched defects being supported by 2D diffusion models and further experimental data from liposome leakage assays and quartz crystal microbalance-dissipation (QCM-D) analysis, we propose that Smp43 disrupts these membranes via a common mechanism, which we have termed ‘diffusion limited disruption’ that encompasses elements of both the carpet model and the expanding pore mechanism.
Introduction
Antimicrobial peptides (AMPs) are essential contributors to the innate immune system and are found among all biological classes. In addition to their native biological importance they offer a potential novel therapeutic class of broad spectrum antibiotics1,2 and have been shown to exhibit selective inhibition against several types of cancerous cells.3,4 Generally targeting lipid membranes, AMPs are selective to fundamental differences between microbial and host membranes. Due to the fundamental role that cellular membranes play in the regulation and function of cells, it is critical for the design and clinical introduction of AMPs that we understand the mechanisms by which they interact with both prokaryotic and eukaryotic membranes.Over one thousand AMPs have been discovered and their antimicrobial profiles characterized to date.5 However, the mechanisms by which the vast majority of these peptides function is not fully understood. Of those characterized, the membrane-disruptive effects of AMPs have been proposed to generally fall into three main mechanistic categories, namely the barrel stave, toroidal pore and carpet models.6 While these mechanisms are still the most frequently proposed, a number of variants on these basic mechanisms have emerged including the interfacial, leaky slit, aggregate, electroporation, sinking raft, and lipid aggregate models (see ref. 1, 2 and 7 for reviews). Although highly diverse, all are thought to involve a similar pathway. Initially, the unfolded peptide in solution binds to the membrane surface, typically driven by electrostatic forces, and adopts an amphipathic helical secondary structure. Below a certain bound peptide to lipid ratio (P/L*) threshold, the helical axis of the peptide is oriented parallel to the plane of membrane in a so called S-state (surface).3,4,8 When the peptide concentration increases so as to exceed this threshold value, a fraction of the peptide monomers change to the I-state (inserted), corresponding to the helical axis oriented perpendicular to the plane of the bilayer.6,9–11 In this I-state the peptide is able to destabilize the membrane where the local peptide concentration is high enough to cause membrane disruption via a variety of mechanisms. However, due to the highly dynamic nature and small size-scales involved, visualization and study of these processes, which are both lipid and peptide specific, is difficult to observe directly without ensemble averaging.
Atomic force microscopy offers the ability to interrogate biological membranes at the nanoscale under physiological conditions and has been previously used to obtain images of peptide disruption over 5–10 min time intervals.11–14 However, membrane binding of AMPs in cells has been shown to occur over microseconds to minutes and thus the dynamics of membrane disruption are often missed via conventional AFM. Recent advances in AFM have resulted in a significant increase in temporal resolution making it now possible to capture nanoscale molecular processes in real time. Such time resolution has allowed AFM to leap forward into new areas, from studying the diffusion of membrane proteins to visualizing the walking of myosins along actin filaments.15–19 Fantner et. al. showed how high speed AFM can be used to image whole bacterial cells whilst under attack by AMPs.20 Although whole cell investigations provide a real life system, the complexity of the membrane often makes it difficult to determine the molecular processes at play, whereas employing model membrane systems allows these processes to be isolated and characterized.
In this study we use fast-scanning AFM to examine antimicrobial peptide model membrane interactions. We investigate a 43 amino acid AMP (Smp43) derived from the venom of the North African scorpion Scorpio maurus palmatus.21 Smp43 has been previously shown to have good broad spectrum antimicrobial activity with a fast-acting membrane disruptive mechanism and a favorable cytotoxicity profile.22 By performing fast-scanning AFM studies on planar supported lipid bilayers composed of lipid mixtures containing major lipid components to model bacterial (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DOPG:DOPC) and mammalian (1:1 DOPE:DOPC) membranes,23 we are able to study peptide attack with previously unseen time and spatial resolution. This work provides new insights into the processes of peptide–lipid interactions. The growth of membrane defects that we observe are consistent with diffusion-limited aggregation processes leading to a mechanism we describe as ‘diffusion limited disruption’. This mechanism lies between a carpet model and an expanding pore, where we have detergent effects, as well as peptide molecules partly stabilizing defective membrane edges.
:1 DOPG:DOPC) and mammalian (1:1 DOPE:DOPC) membranes,23 we are able to study peptide attack with previously unseen time and spatial resolution. This work provides new insights into the processes of peptide–lipid interactions. The growth of membrane defects that we observe are consistent with diffusion-limited aggregation processes leading to a mechanism we describe as ‘diffusion limited disruption’. This mechanism lies between a carpet model and an expanding pore, where we have detergent effects, as well as peptide molecules partly stabilizing defective membrane edges.
Materials and methods
Materials
Smp43 (MW – 4654.35, purity – 97.65%) was synthesised using solid-phase chemistry and was purchased from Think Peptides (Oxford, UK). Carboxyfluorescein was purchased from Sigma (Gillingham, UK) and all solvents other reagents were of the highest grade available and were obtained from Sigma (Gillingham, UK). Phospholipids 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3- phospho-(1′-rac-glycerol) (DOPG) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased from Avanti Polar Lipids (Alabaster, AL).Liposome leakage assays
Liposomes were made using an extruder method with lipids at a concentration of 0.5 mg mL−1. Liposome release assays using carboxyfluorescein (CF) were performed as follows: peptide sample (1 μM at 20 μL), liposomes (10 μL) and buffer (50 mM sodium phosphate, 10 mM NaCl and 1 mM of ethylenediaminetetraacetic acid (EDTA) pH 7.4) (170 μL) were incubated in the dark for 15 min. Fluorescence (480/520 nm) was measured on a Tecan infinite M200 plate reader (Tecan, UK), The reaction mix without CF was used as a negative control to normalise results and 10% Triton-X was used as a positive control to measure complete dye release. The rate of CF leakage was expressed as the percentage of dye released of the total encapsulated carboxyfluorescein and then normalised against the blank buffer signal. All samples were run in triplicate. Error is presented as standard deviation from the mean.Fast-scanning atomic force microscopy on hydrated bilayers
Supported planar lipid bilayers were produced on a freshly cleaved mica surface by vesicle rupture using tip sonicated vesicles incubated on the mica at a concentration of 0.5 mg mL−1. For bilayers containing negatively charged lipids 2 mM CaCl2 was added to the lipid mixture just before incubation on the surface. After 20 min incubation, the surface was rinsed at least 10 times to remove vesicles from the bulk phase. The presence of a uniform bilayer was confirmed by lateral scanning in tapping mode over a 64 μm2 area and force spectroscopy curves. Smp43 was injected in situ by directly pipetting a Smp43 solution into the imaging solution at 10% volume to achieve a imaging solution concentration of 1.0 μM Smp43, the sample volume was then pipette mixed six times with 33% sample volume to ensure rapid mixing and homogeneous peptide concentration. This process takes approximately 5 seconds and was performed while scanning. The time to disruption is then taken from the last pipette mix. Observations of the attack of Smp43 were performed in tapping mode using a Dimension FastScan Bio with Fast Scan D probes (Bruker) with a nominal spring constant of 0.25 N m−1. Scanning was typically performed using 256 scan lines per image at a rate of 10 seconds per image. The force applied by the tip on the sample was minimized by continually maximizing the set point whilst maintaining tracking of the surface. Each experiment was performed a minimum of six times to ensure reproducibility and the videos presented in ESI,† Movies 1 and 2 are representative of observed data.Quartz crystal microbalance-dissipation
QCM-D measurements were performed using a QSense E4 multifrequency QCM-D instrument (Q-Sense, Gothenburg, Sweden) in a flow through cell of 40 μL volume. Data from 15, 25, 35, 45, 55, and 65 MHz overtones (third, fifth, seventh, ninth, 11th, and 13th, respectively) were collected. Before use, all SiO2 crystals were cleaned by ultrasonication in 0.4% SDS for 15 min followed by copious rinsing and ultrasonication in MilliQ for 15 min. The crystals were then dried under nitrogen and UV–ozone cleaned for 30 min. After UV–ozone treatment, they were rinsed with Milli Q water, dried under nitrogen, and used immediately. Initially, the system was filled with buffer such that the resonant frequencies of the overtone to be used could be found. The vesicle solution was injected, under a constant flow of 70 μL min−1, at a concentration of 0.5 mg mL−1. Frequency and dissipation changed in a manner characteristic of vesicle adsorption and then rupture, to form a planar bilayer. Upon reduction of the dissipation peak to a minimum, indicating complete bilayer formation, the bilayer was rinsed with Milli Q water. After bilayer formation, Smp43 was added at 1 μM and the change in frequency and dissipation were monitored. Experiments were performed at a flow rate of 70 μL min−1 at a constant temperature of 22 °C throughout.Diffusion limited aggregation models
Simulations to model the morphology of aggregates for varying sticking probabilities were performed by modifying a previously developed diffusion limited aggregation in continuous 2D space model.24 The Java program based on aggregation of particles undergoing random walks was modified using the Easy Java Simulations (EJS) modeling tool. The simulation was modified by adding an “if” function which only permitted particles sticking if a randomly generated number was less than a user specified probability (held constant throughout a single run). The simulations were run for varying sticking probabilities from 0.5% to 100% with each simulation being run until the aggregate reached 10000 particles. All ± data presented is standard deviation from the mean.
Results
To determine the threshold concentrations at which peptide insertion and membrane disruption occurs for bacterial (DOPC:DOPG 1:1) and mammalian (DOPC:DOPE 1:1) model membranes, liposome leakage assays were performed (ESI,† Fig. S1). At Smp43 concentrations of 0.2 μM (P/L = 0.006), negligible leakage of carboxyfluorescein (CF) was observed from DOPC:DOPE (1:1) liposomes, whereas at the same peptide concentration DOPC:DOPG (1:1) liposomes showed 17 ± 4% release. Increasing the Smp43 concentration to 1.0 μM (P/L = 0.03) caused almost complete disruption of DOPC:DOPG liposomes with 97 ± 2% leakage of CF observed. Against the neutrally charged DOPC:DOPE (1:1) composition, CF release was less pronounced with 9.2% (±1.4) observed at 1.0 μM (P/L = 0.03) peptide concentration with a gradual increase in leakage to 46% (±2.0) at 2 μM (P/L = 0.06). This data shows Smp43 creates significant membrane disruption but preferentially to negatively charged model bacterial membranes.
Based on the results of the liposome leakage assays, the effect of 1 μM Smp43 on supported lipid bilayers was investigated using fast-scanning AFM. Fig. 1 shows the surface of a DOPC:DOPG lipid bilayer over time after the addition of 1 μM Smp43. Images were selected from a fast-scanning AFM imaging sequence obtained at an imaging rate of 10.7 seconds per frame (ESI,† Movie S1). After an initial lag period of 85 s, two independent defects, akin to nucleation sites, appear and rapidly grow in a branched and disordered manner. A third defect appears after 270 s and grows at a similar rate. The observed lag times suggest Smp43 first binds to the surface and after reaching a threshold concentration nucleates a defect allowing further membrane disruption. As shown in Fig. 2C the growth is initially rapid at 7.2% of the total membrane area per min but then slows considerably to 1.8% per min. This slowing of the growth rate occurs when the leading edges of two largest defects (which both initiated at 85 s) come into close proximity. In close proximity there is less available membrane to remove and thus growth rate slows eventually stopping whilst the 3rd defect continues to grow. Disruption continues until the majority of the membrane area (58 ± 1%) under observation is covered in branched defects. To check that continuous AFM scanning was not causing the membrane disruption that we observed, the area was imaged for at several minutes before the peptide was injected into the flow cell, resulting in no visible changes to the membrane. Furthermore, imaging of the surrounding area showed the same features that were observed in the continuous scan area of 1 μm2, demonstrating that the area under observation was representative of the rest of the bilayer (ESI,† Fig. S2).
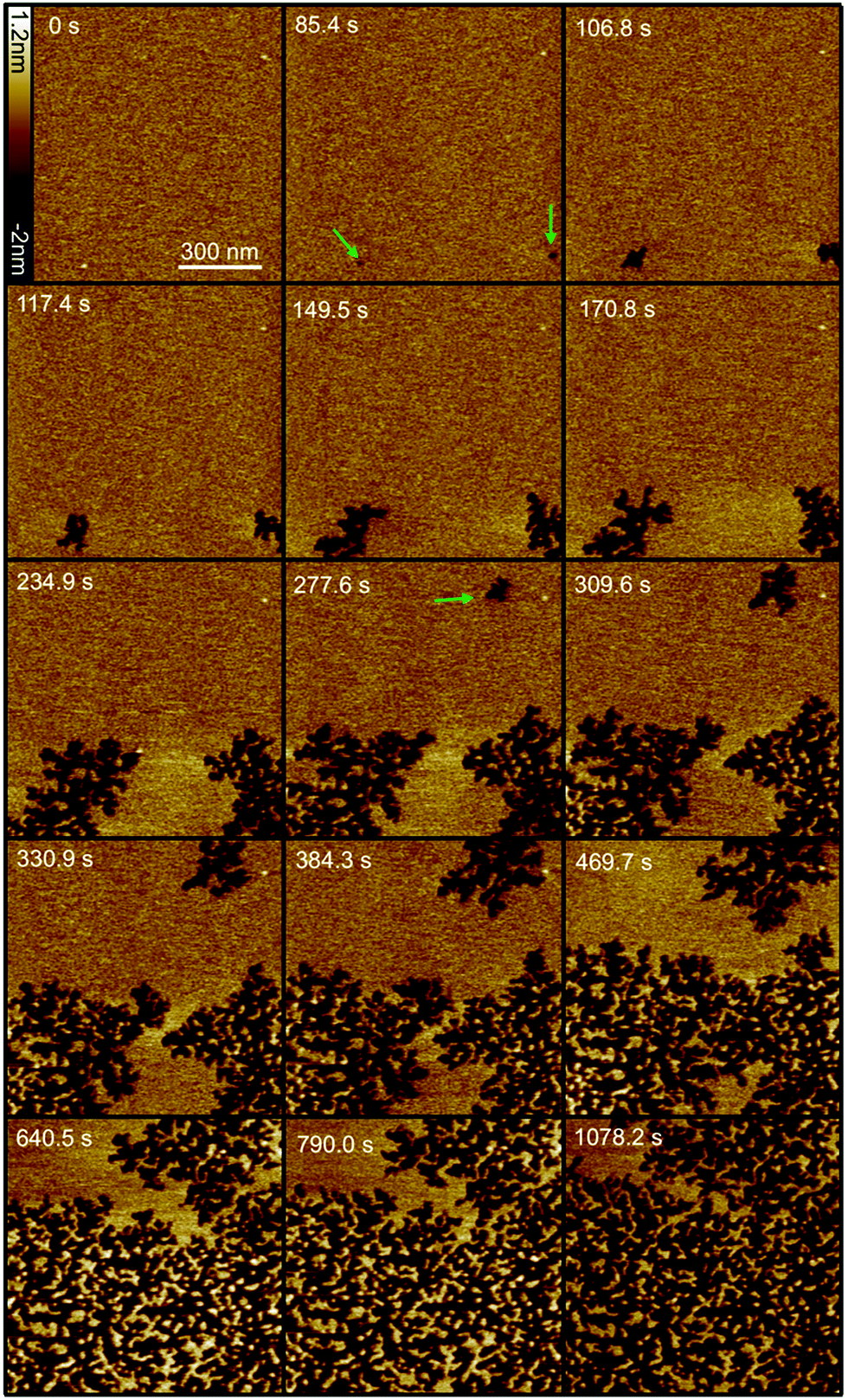 | ||
| Fig. 1 Selection of fast-scanning AFM images of a DOPC:DOPG (1:1) supported lipid bilayer in the presence of 1 μM Smp43 (P/L = 0.42). Images were taken from a series of 111 images (ESI,† Movie S1) obtained at an imaging rate of 10.7 seconds per frame. 1 μM Smp43 was added at time t = 0 s, scan size is 1 × 1 μm, z scale = 3.2 nm. Green arrows indicate the first appearance of defects. | ||
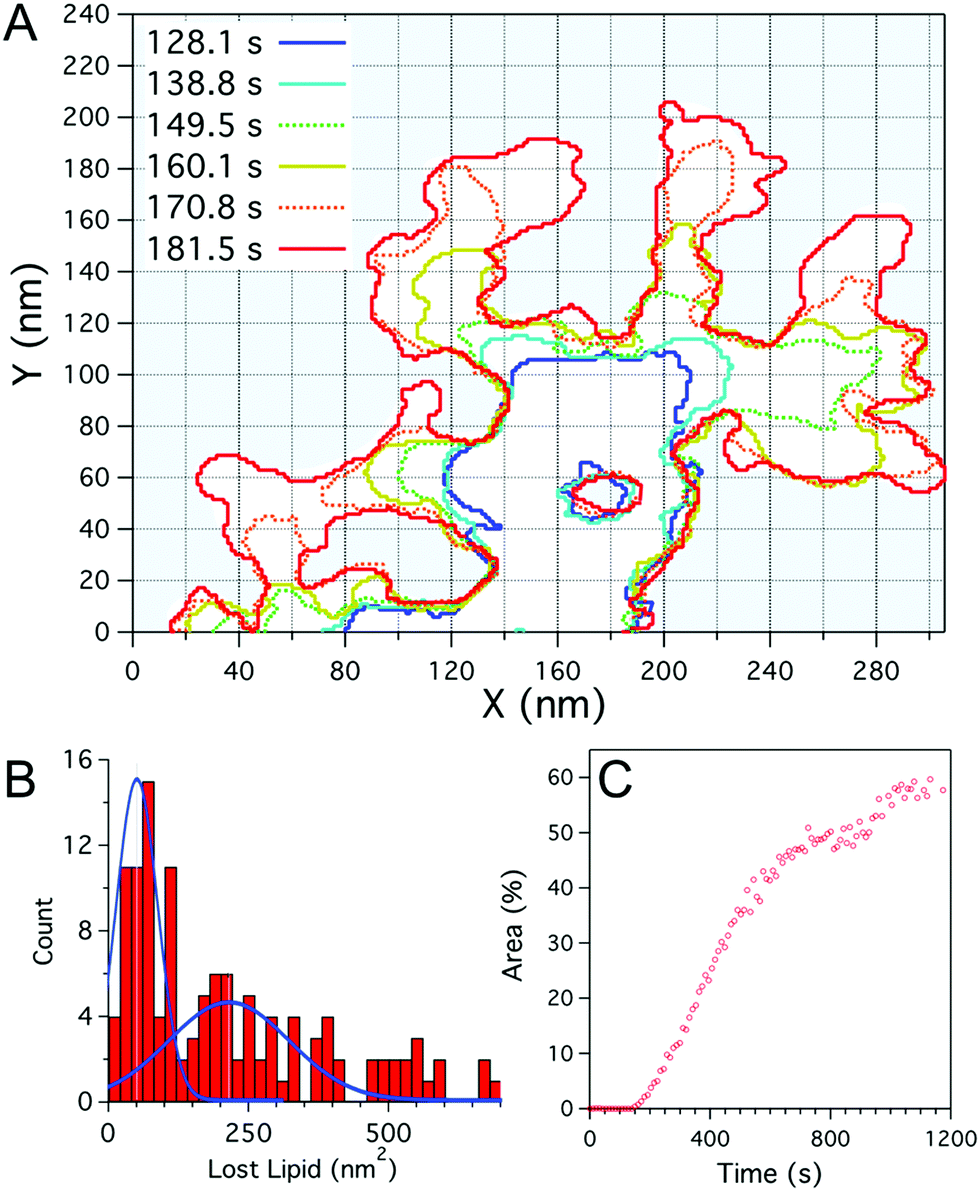 | ||
| Fig. 2 (A) Plot outlining the defect boundary over time obtained from fast-scanning AFM imaging of a DOPC:DOPG (1:1) supported lipid bilayer after incubation with Smp43(1 μM). The boundaries at given times after peptide addition are indicated by different coloured lines. (B) Histogram of the local defect area increase of individual branches over the imaging period (10.7 s), blue lines represent double Gaussian fits to the data. (C) Percentage of the total area (1 μm2) of the bilayer consumed by defects, as a function of time as shown in Fig. 1. | ||
To examine the growth of the branching defects caused by Smp43, fast-scanning AFM images were studied frame-by-frame, mapping the boundaries of the defects every 10.7 seconds. As can be seen in Fig. 2A, some regions of the defects remain stable over time whilst others branch out at various rates but with characteristic branch thicknesses. The merging of two branches results in the apparent trapping of membrane patches, as seen at 181.6 s, x = 100 nm, y = 30 nm and throughout at x = 170 nm, y = 50 nm (Fig. 2A), which remain relatively stable over time. These intact membrane “islands”, suggest that a peptide monomer cannot insert directly into the membrane and remove lipid; the data support a hypothesis whereby membrane disruption is triggered by a threshold concentration of Smp43, driven in turn by lateral diffusion of the peptide across the surface of the phospholipid membrane. This idea is also supported by the observed lag time before nucleation sites appear. It should be noted that we do not directly observe the peptides on the membrane surface as they are diffusing quickly.
The growth of an individual defect branch away from the main defect was analysed by measuring the increase in area from image to image at the leading edge of each branch (Fig. 2A). As shown in Fig. 2B the distribution for the amount of lipid lost locally in the time between images (10.7 s) has a broad distribution with a maximum at 60–80 nm2. Assuming that the darkened areas reflect an absence of phospholipids from the mica surface, this value is likely to represent the preferred lipid extraction size. The lipid removed for this area is not enough to form a vesicular structure but closely corresponds to a spherical lipid micelle with a radius of 2.2–2.5 nm (assuming a fixed area per lipid head group and neglecting peptides within the micelle). The material ejected from the membrane likely also includes a fraction of peptide to stabilize the micelle assembly. The distribution in lost lipid area has a secondary peak at 200 nm2, which may correspond to two micelles being lost during one scan. Whilst in the DOPC:DOPG membranes lipid removal occurs in relatively discrete steps, the defect expansion in the DOPC:DOPE membranes appears slow and continuous. Analysis of image to image area growth (ESI,† Fig. S3), as performed for Fig. 2A and B, showed changes over the 11 second time frame on the same scale as the boundary fluctuations of the defect.
The effects of 1 μM Smp43 on DOPC:DOPE (1:1) supported lipid bilayers as mammalian model membranes was also investigated using the same fast-scanning AFM technique (Fig. 3, ESI,† Movie S2). A similar mechanism to DOPC:DOPG membranes was observed against the DOPC:DOPE bilayer but on a much slower timescale and with significantly less total membrane disruption. The lag phase before the first appearance of membrane disruption (evidenced by two independent nucleation sites) was six times longer with DOPC:DOPE membranes (519 ± 11 vs. 85 ± 11 s). After the initial nucleation, the area consumed by defects grew at a linear rate of 0.24% per min (Fig. 3B), much slower than the 7.2% per min observed for the DOPC:DOPG membrane system (Fig. 2C). Maximum defect coverage for DOPC:DOPE membranes of 5.5 ± 0.1% was only obtained after 40 min (cf. 58% after 20 min for DOPC:DOPG membranes). High resolution imaging of the DOPC:DOPE defects (Fig. 4A) shows a fractal structure similar to that of the DOPC:DOPG system (Fig. 4B) but with much smaller and more densely packed branch features. The highly convoluted boundary of the defects suggests that Smp43 is able to stabilize the defective membrane edges. In a lipid-only system we would expect defects to close or become more circular to reduce the energetic cost associated with exposed hydrophobic membrane edges in a surrounding aqueous milieu.
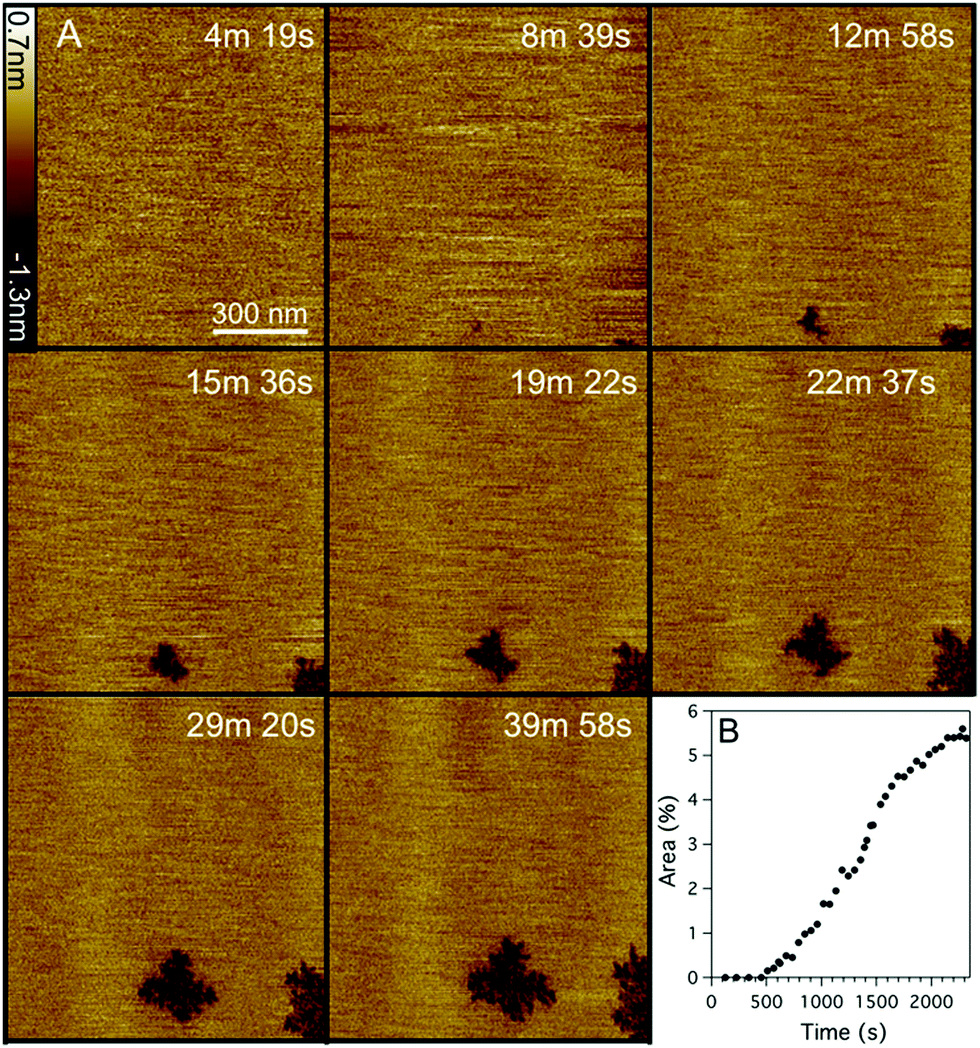 | ||
| Fig. 3 (A) Selection of fast-scanning AFM images of a DOPC:DOPE (1:1) supported lipid bilayer in the presence of Smp43 (1 μM, P/L = 0.42). Images were taken from a series of 210 images (ESI,† Movie S2) obtained at an imaging rate of 11.3 seconds per frame. Peptide was added at time t = 0 s, scan size is 1 × 1 μm, z scale = 2.0 nm. (B) Percentage of the total area (1 μm2) of the bilayer consumed by defects, as a function of time. | ||
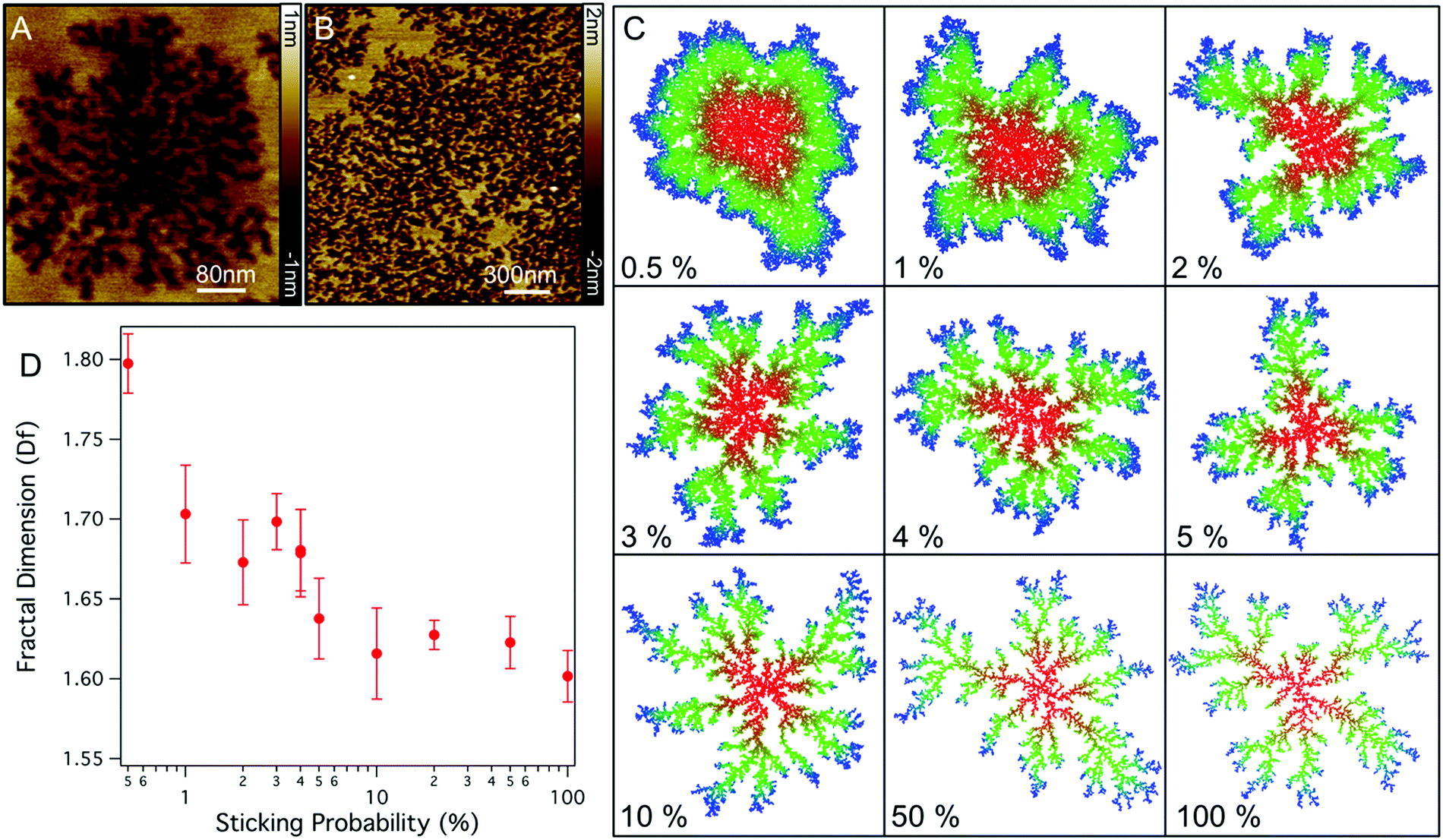 | ||
| Fig. 4 (A) High resolution AFM images of the structure of Smp43 pores in DOPC:DOPE lipid bilayers. (B) Zoom out of Smp43 disruption across a larger section of DOPC:DOPG membrane (C) Images showing the morphology of aggregates in 2D diffusion limited aggregation simulations for varying sticking probabilities from 0.5% to 100% (each simulation was run until the aggregate reached 10000 particles). (D) Fractal dimension of simulated aggregates for varying sticking probability. (Bars represent standard deviation.) | ||
The fractal nature of these defects has a similar appearance to many structures found in nature which can be described by two-dimensional diffusion limited aggregation (DLA),25 a model whereby particles moving with Brownian motion coalesce upon coming into contact with an aggregate.26 The branched structure in DLA arises as particles are captured by the extremities of the aggregate before being able to diffuse to the centre.27,28 Physical DLA models generally assume that a particle immediately attaches to a cluster, however, for many kinetic processes in biology binding occurs with a certain probability due to required binding orientation and the need to overcome an energy barrier. To model the effect of binding probability we ran 2D DLA simulations and varied the probability of a particle binding (P) to the aggregate (see methods). As shown in Fig. 4C, lowering the sticking probability results in aggregates with thicker and fewer branches, eventually resulting in rounded aggregates with no branching at P = 1–0.5% in agreement with previous models.29 The changes in morphologies arise as particles with lower P are able to diffuse further into the centre of the aggregate before they are captured. For the dynamics of lipid removal by Smp43 the sticking probability can be related to the probability of Smp43 association at an aggregate boundary and the subsequent lipid removal. Qualitatively, models with sticking probabilities in the region of 4% reproduce the morphology of the defects observed in DOPC:DOPE bilayers. Defects in DOPC:DOPG membranes are slightly different however, with branches which have a preference to connect to each other.
To further investigate if the branched structures are likely to be formed by a DLA process we can measure the fractal dimension (Df) of the defects in the AFM images. Df is a ratio which describes how the complexity of a geometric pattern changes with the scale at which it is measured; in two dimensions this ratio lies between 1 and 2. Using the box counting method25 we find values for Df of 1.60 ± 0.03 and 1.72 ± 0.04 for the fully formed DOPC:DOPE and DOPC:DOPG defects respectively. These values are in good agreement with previous measures of the Df for DLA structures where values have been reported between 1.68–1.73,27 implying that diffusion-limited aggregation is determining the structures formed. Measuring the fractal dimension of the model DLA system shows that as sticking probability decreases Df increases (Fig. 4D). This trend, which has been previously observed in similar models,29 implies that the DOPC:DOPG system has a lower sticking (lipid extraction) probability.
Applying DLA to model the growth of membrane defects created by Smp43 fits well with current principles of peptide binding, peptide insertion and membrane removal. After initial peptide binding to a membrane, diffusing peptides reach a critical concentration and insert (partly) in to the membrane, stochastically nucleating a defect. Additional Smp43 molecules reach the edges of the defect via two-dimensional diffusion, at which point the peptide concentration is high enough to remove lipid in the form of a lipid-Smp43 micelle.
Whilst AFM provides a tool to resolve the nanoscale changes of the membrane it is unable to resolve the highly mobile peptide. We therefore used QCM-D to follow peptide binding to the two different lipid compositions (Fig. 5A and B). After the formation of a DOPC:DOPG bilayer, creating a frequency change of approximately −25 Hz,30,31 Smp43 binding can be observed by a decrease in frequency which occurs via a two-step process; there is an initial rapid decrease (−24.8 Hz to −25.7 Hz) over the first 200 s, followed by a more gradual decline to −27.4 Hz over 24 min. There is a similar two-step process observed within the dissipation with an initial increase from 0.2 to 0.28 over the first 300 s and then a more gradual increase to 0.4 over 30 min. This two-step process is indicative of the classical AMP concentration threshold model, in which peptides accumulate on to the membrane bilayer and reach a certain threshold before inserting into the membrane and causing disruption. The gradual decline in frequency observed in the second step (Fig. 5A) is most likely due to a combination of peptide still binding to the membranes whilst lipid-peptide micelles are simultaneously ejected. As the surface saturates with peptide, the removal becomes the dominant process 24 min after peptide injection where the frequency begins to rise. Against the DOPC:DOPE (Fig. 5B) bilayer the frequency change shows a more prolonged two-stage decrease from −24.7 Hz to −27.1 Hz over 21 min with a further small decrease down to −28.1 Hz after an additional 17 min. This suggests that the critical threshold surface concentration required for insertion of Smp43 into DOPC:DOPE membranes is much greater than for DOPC:DOPG membranes. As observed by AFM, loss of mass from the DOPC:DOPE membrane was very little. These QCM-D measurements support the differences seen by AFM and provide complementary evidence for membrane binding followed by insertion at different critical threshold surface concentrations.
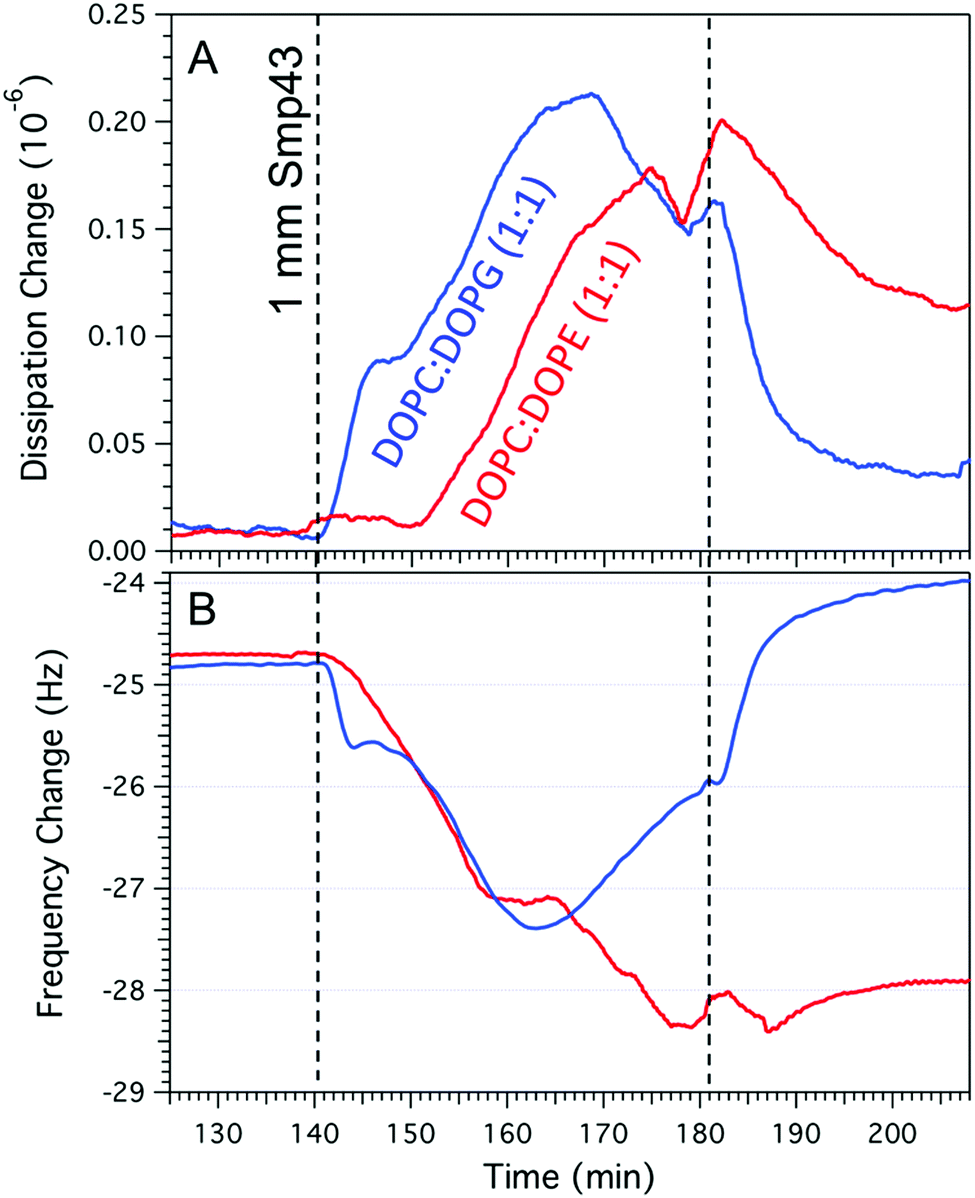 | ||
| Fig. 5 QCM-D data of DOPC:DOPG (A) and DOPC:DOPE (B) supported lipid bilayers after the addition of 1 μM Smp43 (dashed line at 140 min) followed by rinsing with deionized water (dashed line at 180 min). QCM-D experiments were performed at a flow rate of 70 μl min−1 at a constant temperature of 22 °C. | ||
Cross sectional analysis of the defect topography (Fig. 6A) shows depths of 2.1 + 0.2 nm and 1.0 + 0.2 nm for DOPC:DOPG and DOPC:DOPE membranes respectively. The depth of the defects in the DOPC:DOPG bilayer does not correspond to the full depth of a bilayer (∼5 nm) and suggests that the top monolayer is removed with the peptide possibly covering the exposed hydrophobic tails. In the case of the DOPC:DOPE membranes the depth is less and may correspond to a membrane thinning effect (Fig. 6B) Both these assemblies have been previously described by Mecke et al. for interactions of the antimicrobial peptide MSI-78 with DMPC membranes which have a similar lateral structure to the defects observed in our Smp43 studies.13 However, it should be noted that the tip geometry may not allow a full probing of defect depths which have lateral dimensions smaller than the tip diameter (approximately 10–20 nm) as is the case for much of the DOPC:DOPE defect structure.
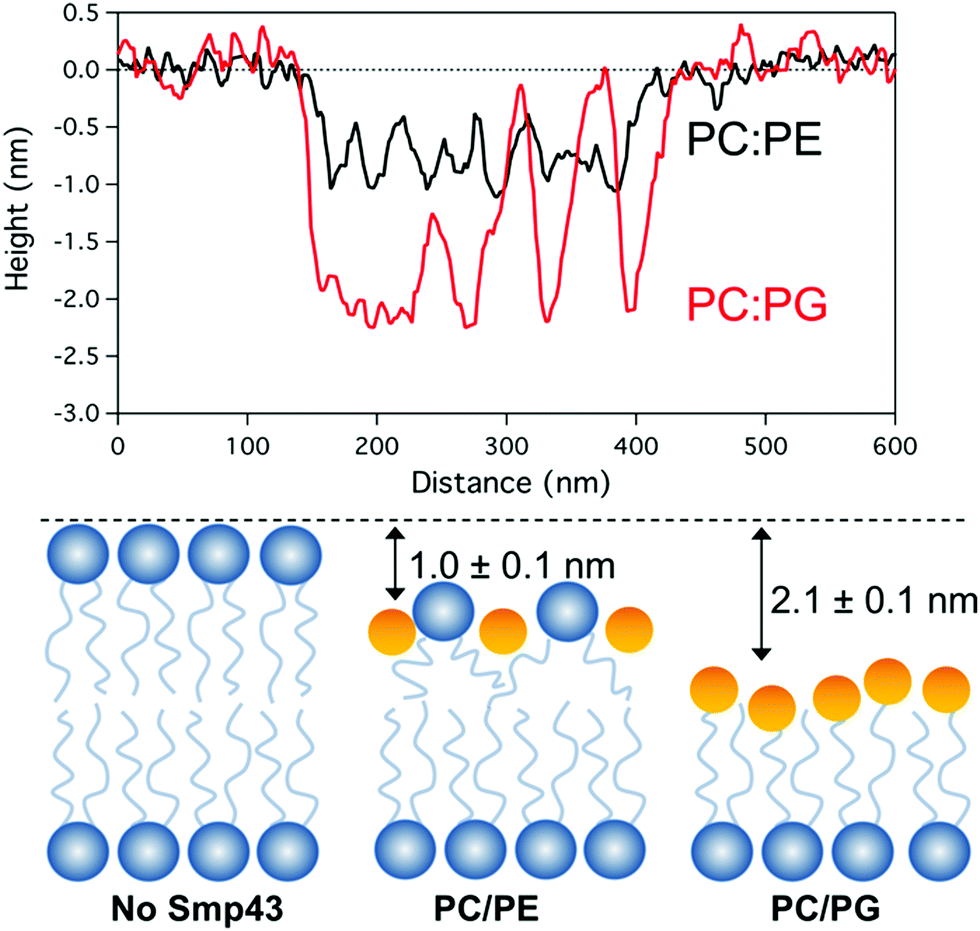 | ||
| Fig. 6 (A) Cross sectional analysis of the defect topography of both DOPC:DOPG & DOPC:DOPE bilayers after Smp43 peptide interaction. (B) Illustration showing possible differences of Smp43 pepitde interaction with the top layer of the bilayers. | ||
Discussion
In this study, we have demonstrated the propensity of Smp43 to rapidly and extensively disrupt negatively charged phospholipid bilayers over zwitterionic ones. A comparison of the effects of AMPs on charged and uncharged bilayers provides a convenient model system to investigate the selectivity of AMPs for prokaryotes, as a negatively charged membrane is a prominent feature of prokaryotic membranes, distinguishing them from eukaryotic membranes which are more zwitterionic in nature.32 Our results suggest that complete membrane collapse takes place on the negatively charged bilayer whilst only discrete and limited disruption occurs on the zwitterionic bilayer. This correlates well with our previous biological characterization of Smp43, demonstrating good antimicrobial activity and low toxicity in haemolytic assays.22In vivo mechanistic studies with membrane integrity assays on both Gram classes along with a reporter gene assay of the stress response of Bacillus subtilis cells also suggested that no intracellular targets were present, where only stress responses at the level of the cell envelope were recorded.22CD spectral analysis of Smp43 has revealed two regions of approximately 70% N-terminal and 25% C-terminal helical content, suggesting it adopts a di-helical structure, linked by a random coil region.22 This places Smp43 among many scorpion venom di-helical long chain AMPs which have favorable biological activity that make them ideal candidates for future drug development.33 Another member of this family is Pandinin-1 (Pin-1) which shares 86% sequence homology with Smp43, including three conserved tryptophan residues at position 4, 6 and 15 (Fig. 7).
 | ||
| Fig. 7 Smp43 amino acid comparison with Pandinin-1. | ||
NMR studies of the interaction of Pin-1 with PC liposomes revealed a detergent-like effect due to the formation of cubic phase non-lamellar structures.34 In the Pin-1 mechanism, the peptide is thought to sit at the interface between the hydrophobic core and the phospholipid polar head groups of the membrane, with the N-terminal helices tilted at a 30° angle with respect to the plane of the horizontal bilayer, which contains the C-terminal helices. NMR data suggests interactions between the peptide and the membrane occur via the π-electron cloud of the conserved tryptophan indole groups and the positively charged group of the phosphatidylcholine head groups. Membrane disruption is thought to be a consequence of the rotation of the N-terminal helices of the peptide around the average helical axis, causing membrane tearing and cubic phase structure formation.34
Although we have not observed the formation of cubic phases after the addition of Smp43, the high degree of homology between Smp43 and Pin-1 should be taken into account when considering the mechanism of action of the former. Both peptides have a long chain helix–hinge–helix topology with all the conserved tryptophan residues on the N-terminal helix. Taking into account its helical structure, the chain length of Smp43 results in an approximately ∼6.5 nm long peptide which is too large to span the bilayer membrane (4.8 ± 0.4 nm30). From our AFM data we can conclude the following of Smp43-membrane interactions: after initial binding and nucleation, defects grow by the two-dimensional diffusion of surface bound peptides to existing defects sites. Additionally, the convoluted geometry of the defects implies that Smp43 resides at the defect boundary where it stabilizes the high edge energies associated by exposing the hydrophobic tails of the membrane.
These observations taken together with Smp43's homology to Pin-1, points to a mechanism whereby, after initial electrostatic attraction to the membrane surface, peptide–lipid bonds are stabilized by the π-electron cloud of the conserved tryptophan indole groups. Upon a set concentration being reached insertion can occur, a stable pore is created with disruption then increasing as additional peptide diffuses to the defect resulting in removal of lipid micelles. Our observations show this process proceeds rapidly, potentially limited by the arrival of new peptide to defect boundaries and the probability of peptide binding/lipid removal. Whilst it is often reported that insertion occurs at a critical peptide surface concentration and is then followed by membrane disruption, from our results we cannot distinguish if peptide insertion occurs before or after diffusion to the observed membrane defects.
Although diffusion limited aggregation models are typically used to describe the growth of non-biological aggregates, here we consider the model to explain the growth of defects as a consequence of the peptide diffusion and accumulation; hence we have called this model ‘diffusion limited disruption’. A comparison between the DOPC:DOPG and DOPC:DOPE model membranes highlights the difference lipid composition has on the speed and scope of this process, with defect growth being observed at a 30-fold faster rate on DOPC:DOPG membranes. The rapid disruption of the DOPC:DOPG bilayer is likely amplified by the favorable electrostatic interaction between the negatively charged PG head group and the cationic amphipathic peptide. This effect can be seen by the QCM-D data, where the rate of absorption is initially much greater for the DOPC:DOPG membrane system, up until a critical surface concentration is reached, whereupon it is likely that we observe a combination of peptide binding and membrane removal. In the case of the zwitterionic/neutral DOPC:DOPE model membranes, binding still occurs but more slowly, via van der Waals interactions between the hydrophobic tails of the phospholipids and hydrophobic residues of Smp43.35 This weaker interaction can be observed in the QCM-D data which suggests that the critical surface concentration of Smp43 is three times higher for DOPC:DOPE bilayers than for DOPC:DOPG bilayers. The weaker interaction with neutrally changed membranes is also observed in the liposomal leakage experiments (Fig. S1, ESI†). In addition to the weaker driving force for membrane binding/insertion, the reduced damage of the mammalian model membranes can be explained by PE lipids preferring negative spontaneous curvature (a splayed tail, small headgroup configuration) and therefore having a reduced preference to form pores and/or lipid micelles.35
As we have observed, Smp43 has very little effect upon neutrally charged bilayers, which correlates well with previous haemolytic assays. Since, however it is perhaps fanciful to suggest that even AMPs with favorable therapeutic indices will not have any effect on host cell membranes, unintended synergies between AMPs and other circulatory proteins/peptides should be considered. Biochemical techniques such as liposome leakage assays cannot be relied upon to distinguish these subtle mechanistic changes and therefore more high resolution and real time experiments are required to move AMPs a step closer to the clinical setting.
Conclusions
Using fast-scanning AFM to capture an AMP attack in real time on planar lipid bilayers allows us to build on the expanding pore model proposed by Rakowska et al., 2013.11 The data presented here helps us to refine our understanding of this process and to propose a model of AMP-membrane interactions ‘diffusion limited disruption’. Further investigations using fluorescently tagged peptides to monitor membrane-associated peptide aggregation and FRET labelled peptides and phospholipids to monitor peptide–lipid interaction during the expansion of pores are required to further refine the model. In future studies, it would also be useful to see the effect of a more diverse range of AMPs, including tryptophan substituted variants, on a range of lipid compositions containing cholesterol and various other lipid components creating more complex mixtures which mimic closer eukaryotic membranes.Taking into account the favorable therapeutic index of AMPs with a helical-hinge-helical topology not only from scorpion venom but from other diverse sources such as the silk moth Hyalophora cecropia36 and Dermaseptin B237 from the Amazonian tree Phyllomedusa bicolour, this study offers important insights of how these peptides may exert their effects, which will ultimately allow us develop the therapeutic potential of this scaffold by minimizing undesirable membrane interactions.
Author contributions
G. R. H., P. L. H., P. N. S., K. M. and S. D. E. conceived and designed the experiments. G. R. H. and P. L. H. performed the experiments and analysed the data. G. R. H., P. L. H., P. N. S., K. M. and S. D. E. wrote the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
SDE would like to thank the EPSRC (EP/P023266/1 and EP/P00122X/1) for financial support.References
- Y. J. Gordon, E. G. Romanowski and A. M. McDermott, A Review of antimicrobial peptides and their therapeutic potential as anti-infective drugs, Curr. Eye Res., 2009, 30, 505–515 CrossRef PubMed.
- K. V. R. Reddy, R. D. Yedery and C. Aranha, Antimicrobial peptides: premises and promises, Int. J. Antimicrob. Agents, 2004, 24, 536–547 CrossRef PubMed.
- N. B. Leite, et al., PE and PS lipids synergistically enhance membrane poration by a peptide with anticancer properties, Biophys. J., 2015, 109, 936–947 CrossRef PubMed.
- D. W. Hoskin and A. Ramamoorthy, Studies on anticancer activities of antimicrobial peptides, Biochim. Biophys. Acta, 1778, 357–375, 2008 Search PubMed.
- G. Wang, L. Xia and Z. Wang, APD3: the antimicrobial peptide database as a tool for research and education, Nucleic Acids Res., 2016, 4(44), D1087–D1093 CrossRef PubMed.
- K. A. Brogden, Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef PubMed.
- V. Teixeira, M. J. Feio and M. Bastos, Role of lipids in the interaction of antimicrobial peptides with membranes, Prog. Lipid Res., 2012, 51, 149–177 CrossRef PubMed.
- H. W. Huang, Molecular mechanism of antimicrobial peptides: The origin of cooperativity, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1292–1302 CrossRef PubMed.
- R. Mani, et al., Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16242–16247 CrossRef PubMed.
- H. W. Huang, Action of Antimicrobial Peptides: Two-State Model, Biochemistry, 2000, 39, 8347–8352 CrossRef PubMed.
- P. D. Rakowska, et al., Nanoscale imaging reveals laterally expanding antimicrobial pores in lipid bilayers, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8918–8923 CrossRef PubMed.
- K. Hall, T.-H. Lee, A. I. Mechler, M. J. Swann and M.-I. Aguilar, Real-time measurement of membrane conformational states induced by antimicrobial peptides: balance between recovery and lysis, Sci. Rep., 2014, 4, 5479 CrossRef PubMed.
- A. Mecke, D.-K. Lee, A. Ramamoorthy, B. G. Orr and M. M. Banaszak Holl, Membrane thinning due to antimicrobial peptide binding: an atomic force microscopy study of MSI-78 in lipid bilayers, Biophys. J., 2005, 89, 4043–4050 CrossRef PubMed.
- J. M. Henderson, A. J. Waring, F. Separovic and K. Y. C. Lee, Antimicrobial peptides share a common interaction driven by membrane line tension reduction, Biophys. J., 2016, 111(10), 2176–2189 CrossRef PubMed.
- P. L. Harrison, et al., Phospholipid dependent mechanism of smp24, an α-helical antimicrobial peptide from scorpion venom, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2737–2744 CrossRef PubMed.
- T. Ando, High-speed atomic force microscopy coming of age, Nanotechnology, 2012, 23, 62001 CrossRef PubMed.
- G. R. Heath, J. Roth, S. D. Connell and S. D. Evans, Diffusion in low-dimensional lipid membranes, Nano Lett., 2014, 14, 5984–5988 CrossRef PubMed.
- I. Casuso, et al., Characterization of the motion of membrane proteins using high-speed atomic force microscopy, Nat. Nanotechnol., 2012, 7, 525–529 CrossRef PubMed.
- N. Kodera, D. Yamamoto, R. Ishikawa and T. Ando, Video imaging of walking myosin V by high-speed atomic force microscopy, Nature, 2010, 468, 72–76 CrossRef PubMed.
- G. E. Fantner, R. J. Barbero, D. S. Gray and A. M. Belcher, Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy, Nat. Nanotechnol., 2010, 5, 280–285 CrossRef PubMed.
- M. A. Abdel-Rahman, V. Quintero-Hernandez and L. D. Possani, Venom proteomic and venomous glands transcriptomic analysis of the Egyptian scorpion Scorpio maurus palmatus (Arachnida: Scorpionidae), Toxicon, 2013, 74, 193–207 CrossRef PubMed.
- P. L. Harrison, M. A. Abdel-Rahman, P. N. Strong, M. M. Tawfik and K. Miller, Characterisation of three alpha-helical antimicrobial peptides from the venom of Scorpio maurus palmatus, Toxicon, 2016, 117, 30–36 CrossRef PubMed.
- M. R. Yeaman and N. Y. Yount, Mechanisms of antimicrobial peptide action and resistance, Pharmacol. Rev., 2003, 55, 27 CrossRef PubMed.
- L. Engelhardt, The diffusion limited aggregation in continuous 2D space model. Version 1.0, Java 1.5, 2011.
- F. Caserta, et al., Physical mechanisms underlying neurite outgrowth: A quantitative analysis of neuronal shape, Phys. Rev. Lett., 1990, 64, 95–98 CrossRef PubMed.
- Y.-B. Huang and P. Somasundaran, Effects of random-walk size on the structure of diffusion-limited aggregates, Phys. Rev. A: At., Mol., Opt. Phys., 1987, 36, 4518–4521 CrossRef.
- P. Meakin, Diffusion-controlled cluster formation in 2–6-dimensional space, Phys. Rev. A: At., Mol., Opt. Phys., 1983, 27, 1495–1507 CrossRef.
- L. M. Sander, Diffusion-limited aggregation: A kinetic critical phenomenon?, Contemp. Phys., 2000, 41, 203–218 CrossRef.
- B. Ranguelov, D. Goranova, V. Tonchev and R. Yakimova, Diffusion limited aggregation with modified local rules, C. R. Acad. Bulg. Sci., 2011, 65, 913–918 Search PubMed.
- G. R. Heath, et al., Layer-by-Layer assembly of supported lipid bilayer poly- l-lysine multilayers, Biomacromolecules, 2016, 17, 324–335 CrossRef PubMed.
- G. A. McCubbin, S. Praporski, S. Piantavigna, D. Knappe, R. Hoffmann, J. H. Bowie and L. L. Martin, QCM-D fingerprinting of membrane-active peptides, Eur. Biophys. J., 2011, 40(4), 437–446 CrossRef PubMed.
- M. Zasloff, Antimicrobial peptides of multicellular organisms, Nature, 2002, 415(6870), 389–395 CrossRef PubMed.
- P. L. Harrison, M. A. Abdel-Rahman, K. Miller and P. N. Strong, Antimicrobial peptides from scorpion venoms, Toxicon, 2014, 88, 115–137 CrossRef PubMed.
- K. Nomura, G. Ferrat, T. Nakajima, H. Darbon and T. Iwashita, Induction of morphological changes in model lipid membranes and the mechanism of membrane disruption by a large scorpion-derived pore-forming peptide, Biophys. J., 2005, 89, 4067–4080 CrossRef PubMed.
- D. Koller and K. Lohner, The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2250–2259 CrossRef PubMed.
- T. A. Holak, et al., The solution conformation of the antibacterial peptide cecropin A: a nuclear magnetic resonance and dynamical simulated annealing study, Biochemistry, 1988, 27, 7620–7629 CrossRef PubMed.
- C. Galanth, et al., Mechanism of antibacterial action of dermaseptin B2: interplay between helix−hinge−helix structure and membrane curvature strain, Biochemistry, 2009, 48, 313–327 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00707a |
| This journal is © The Royal Society of Chemistry 2018 |