
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase separation in mixed polymer brushes on nanoparticle surfaces enables the generation of anisotropic nanoarchitectures†
Christian
Rossner
 *a,
Qiyun
Tang
*b,
Marcus
Müller
b and
Gerald
Kothleitner
a
*a,
Qiyun
Tang
*b,
Marcus
Müller
b and
Gerald
Kothleitner
a
aInstitut für Elektronenmikroskopie und Nanoanalytik, Technische Universität Graz, Steyrergasse 17, A-8010 Graz, Austria. E-mail: christian.rossner@felmi-zfe.at
bInstitut für Theoretische Physik, Universität Göttingen, Friedrich-Hund-Platz 1, D-37077 Göttingen, Germany. E-mail: qiyun.tang@theorie.physik.uni-goettingen.de
First published on 8th May 2018
Abstract
The preparation of nanoparticles and their targeted connection with other functional units is one key challenge in developing nanoscale devices. Herein, we report an experimental strategy toward the development of anisotropic nanoparticle architectures. Our approach is based on phase separation of binary mixed polymer brushes on gold nanoparticle surfaces leading to Janus-type structures, as revealed by scanning transmission electron microscopy and electron energy-loss spectroscopy and, additionally, corroborated by computer simulation. We show that such structures can be used for the site-selective functionalization with additional nanosized entities.
1. Introduction
Because of their specific properties, inorganic particles with nanoscale dimensions hold potential for device fabrication in diverse fields, such as electronics, photonics, and sensing. However, in order to use nanometer-sized building blocks in technological applications that take advantage of the specific nanomaterial properties, the ability to precisely arrange such building units in a controlled fashion is critical. These considerations have stimulated abiding research effort dedicated to devising efficient strategies for high-precision hierarchical nanoparticle arrangement.1–7Hierarchically ordered nanoparticle arrangements can take a generic structure in which specific components are directly attached to a central scaffold particle. This general approach is very versatile, as has been demonstrated impressively by Caruso and co-workers,8 and it enables access to nanoparticle clusters with e.g. unique surface-enhanced Raman and plasmonic properties.9–15 One key parameter of nanoarchitectures of this type is the distance between the central scaffold particle and the attached functional units. This parameter can be controlled (on the respective length scales) by varying the length of short-chain alkyl linkers (∼100 nm),16 and also by using rigid DNA origami scaffolds (up to ∼102 nm).17 We had recently introduced RAFT polymers with special architecture as linker and spacer to control nanoparticle distances (up to ∼101 nm) in planet–satellite nanostructures,18,19 and we have also demonstrated the flexibility of this strategy by joining different metal nanoparticles, i.e. gold and silver, into this type of arrangement structure.20 Besides particle spacing, there is another crucial structural feature in these nanoarchitectures, namely the distribution of functional units around the scaffold. Approaches toward controlling this distribution were made, which relied on non-centrosymmetric scaffold particles (i.e. gold nanorods, which can be site-selectively functionalized).21 However, such strategy does not solve the problem from a more fundamental perspective. In order to do so, the challenge of reducing the symmetry of centrosymmetric scaffolds has to be tackled. This can be achieved following a protection strategy that uses substrates, such as latex particles,22,23 polymers,24–26 or glass slides,27 onto which the scaffold particles can be assembled and then to attach functional units on the vacant site of the scaffold. A more elegant and scalable strategy to desymmetrize the scaffolds is to exploit specific interactions in ligand-modified nanoparticles in order to induce the formation of surface patterns.28–31
The ability to control surface pattern formation on polymer-coated nanoparticles recently received much focus in the polymer science community.32,33 For example, constrained de-wetting of homopolymer ligands under bad solvent conditions had been demonstrated as a versatile paradigm in this regard.34–36 Using such approach, distinct surface patterns may emerge, depending on the polymer's degree of polymerization, grafting density, and the nanoparticle's dimension and shape.34,35 Other approaches toward nanoparticle surface patterning with polymer ligands include the phase-separation of mixed polymer brushes.37–47 Here, mixed brushes formed by successive surface-initiated atom-transfer radical polymerization of both types of polymer from a common surface-immobilized asymmetric initiator were intensively studied.37–43 However, because relatively large silica nanoparticles were used in these studies and the polymer grafting points were immobile, the resulting surface morphologies were restricted to “rippled”38–42 or “wedge-shaped”43 structures. Janus-type surface patterns were concluded from contrast variation in small-angle neutron scattering experiments performed by Kim et al. on mixed poly(methyl methacrylate)/polystyrene (PMMA/PS) brushes grafted at very small (3 nm) gold nanoparticles,44 as also predicted by simulations.44,45 The group of Liz-Marzán reported Janus-type particles with larger gold cores and mixed brushes of low molecular weight polymers (1–5 kDa).46 Recently, a combination of self-consistent field theory and fluctuating dynamic mean-field theory has been introduced to study the brush structure on a particle surface.48 Besides these advances, however, the direct real-space visualization of local chemistries within mixed brushes and also the targeted connection of isolated surface polymer-patches with additional functional units remain challenging.
In this contribution, we built on the above considerations in order to maneuver the structure of complex nanoparticle architectures. We prepared PMMA and PS homopolymers that were intended to affect pattern formation on gold nanoparticles. In addition, by implementing an α,ω-bifunctional (telechelic) polymeric architecture, the PS homopolymer was designed such as to allow additional functionalization reactions on the surface-bound polymer by attaching functional units to separated PS domains. We show that such approach can lead to anisotropic nanoparticle arrangement structures.
2. Materials and methods
Materials
RAFT agent 1, sodium borohydride, sodium citrate tribasic dihydrate, (Aldrich) and tetra-N-octylammonium bromide (ABCR), were purchased at the highest purity available and used as received. Hydrogen tetrachloroaurate trihydrate (ABCR, 99.9%) was used as received. Methyl methacrylate and styrene (Aldrich) were passed through a short column of activated basic alumina prior to use. AIBN (Akzo Nobel) was recrystallized from methanol and stored at 4 °C prior to use. Dichloromethane, N,N-diemthylformamide, hexane, methanol, tetrahydrofurane, and toluene (p.a. grade, Fisher) were used as received. The preparation of RAFT agents 249 and 350 had already been described earlier. Nanopure (type I) water was obtained using a Millipore filtration system.Size-exclusion chromatography (SEC)
SEC characterization was performed on a SEC 3010 (WGE Dr Bures, Germany) with a precolumn, two separation columns (8 × 300 mm, MZ-Gel SD plus, MZ-Analysentechnik, Germany), and a refractive index detector. A temperature of 30 °C was maintained in the column compartment and THF was used as the eluent. The system was calibrated using linear polystyrene standards with narrow size distribution.Electron microscopy
Electron microscopic characterization of the nanostructures was carried out on a FEI Tecnai F20 microscope operated at an acceleration voltage of 200 kV. The experiments were conducted by cooling the specimen (77 K) in order to minimize electron beam induced damage.51 All samples were applied onto PELCO® graphene films supported by a lacey carbon film on 300 mesh copper grids. Mapping of gold was performed by HAADF detection. Mapping of the different polymer species was performed in STEM mode, by analysing the low-loss region of the EEL spectrum. Maps were extracted by MLLS fitting52 of reference spectra for PMMA and PS (see Fig. 1) to the acquired spectrum images, in a spectral range from 6 eV to 25 eV, after careful calibration of the zero-loss peak. Mapping of the polymeric species at lateral positions with gold present was not possible with this approach. | ||
| Fig. 1 Illustration of the structure of PMMA and PS polymer used in this work (a) and EEL spectra for these two polymers, which were used as reference data in this work (b). The peak between 6 and 7 eV in the polystyrene data can be attributed to π–π* transitions of the aromatic ring. | ||
Polymerizations
See also Scheme S1 (ESI†). The respective RAFT agent, AIBN, monomer and (in the case of PMMA) toluene (the amounts for the different polymerization reactions are given in Table 1) were weighed into polymerization flasks and the mixtures were purged with nitrogen for 10 minutes. The polymerizations were then initiated by heating to the respective temperatures as indicated in Table 1. After predetermined times, polymerizations were stopped by cooling on ice and exposing the mixtures to air. The polymeric material was isolated by threefold precipitation into hexane (PMMA sample) or methanol (PS samples).| Polymer | RAFT agenta | [Monomer]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[RAFT]0:[AIBN]0 :[RAFT]0:[AIBN]0 |
t/h | θ/°C | M n /(103 g mol−1) | Đ |
|---|---|---|---|---|---|---|
| a See ESI (Scheme S1) for chemical formulas of these RAFT agents. b In 50 wt% toluene. c Determined by SEC (calibrated against PS standards) in THF at 30 °C, as monitored by refractive index detection. d Correction applied using the Mark–Houwink parameters KPS = 0.0128 cm3 g−1, aPS = 0.712 and KPMMA = 0.0128 cm3 g−1, aPMMA = 0.690. | ||||||
| PMMA | 1 | 200:1:0.1b |
16 | 65 | 14.9d | 1.08 |
| Telechelic PS | 2 | 600:1:0.1 |
17 | 65 | 14.4 | 1.09 |
| Monofunctional PS | 3 | 66:1:1 |
2.67 | 60 | 2.0 | 1.07 |
Grafting of polymers to the gold nanoparticles
Gold nanoparticles had been prepared by the citrate-reduction method53 as already described elsewhere.54 PMMA and telechelic polystyrene were dissolved in DMF (40 mL, 0.25 mg mL−1 for both polymers, i.e. a molar ratio of approx. 1:1 due to the almost identical number average molar masses). A solution of gold nanoparticles from citrate-reduction (2 mL, see above) was added dropwise over a period of 2 minutes under sonication. The formed core–shell particles were purified by three centrifugation/redispersion cycles: 6 h, 15300 g, −5 °C.
Model and simulation technique
Here we use the coarse-grained model to describe the adsorption of PMMA and PS on the gold nanoparticle surface. The repeat units of PMMA and PS are labeled as A and B monomers correspondingly. We ignore the small differences between the molar masses and the volumes of the repeat units of PMMA and PS molecules, and set the length of coarse-grained chains to be N = 41 for both PMMA and PS molecules. The bonded interactions between neighboring monomers along polymers are modeled by a FENE potential:55where the parameters lmax = 1.5σ, k = 30.0ε/σ2, and kBT = 1.0ε are chosen to prevent chain crossings.

The nonbonded interactions between all monomers are treated via a truncated and shifted Lennard-Jones potential:

The cut-off length between A and B monomers is chosen as 21/6σ, which gives rise to a purely repulsive interaction. For A–A and B–B monomers, the cut-off is selected as 2·21/6σ and 21/6σ, corresponding to a bad and good solvent case. Here we choose three different cases, summarized in Table S1 (ESI†). The interaction strengths are selected as εAA = 1.0, εBB = 1.0, εAB = 3.0, corresponding to a large χ between PS and PMMA. This model has been demonstrated to be good for studying the phase behaviour of AB diblock copolymers.56
The strong interaction between the end groups and gold nanoparticle surface are chosen as
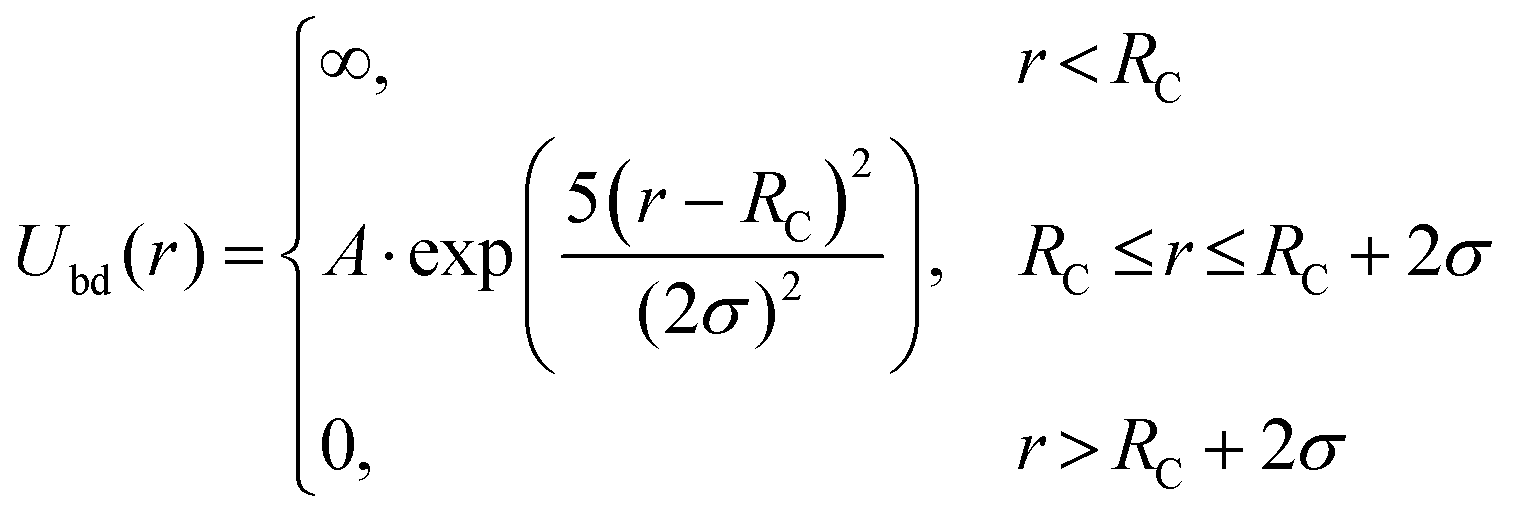
The coefficient A is set to be A = −14.52kBT, corresponding to the adsorption free enthalpy of −36 kJ mol−1 measured from experiments at 21 °C.19 Here the Rc = 6.8σ for the big gold particle and 1.9σ for the small ones. In the simulations, the total grafting density of PMMA and PS is set to be 0.41 chains nm−2, according to previous experiments.19 The density of adsorbed PS macromolecules varies from 0.17 chains nm−2, 0.20 chains nm−2, to 0.24 chains nm−2.
3. Results and discussion
PMMA and telechelic PS (see illustration in Fig. 1, and Table 1 for polymer characteristics) were prepared by RAFT polymerization57 and attached to gold nanoparticles (AuNPs) via their trithiocarbonate end groups58–60 following a “grafting-to” approach.54,61 This resulted in binary mixed polymer brushes. PMMA and PS are immiscible polymers and hence have a tendency to phase separate; the phase separation of surface-grafted PMMA/PS mixed brushes has already been experimentally observed.44,62,63 For investigations of the phase behavior of the systems studied within this work, we used scanning transmission electron microscopy (STEM) and electron energy-loss spectroscopy (EELS) to spatially map the different polymer species in the prepared nanostructures60 (see experimental details). Although selective staining procedures are often applied to produce contrast between distinct polymer phases (as exemplarily shown in Fig. S1 (ESI†) for one of the samples studied in this work), such staining may result in artifacts such as overstaining,47 especially if the investigated materials are interface-rich and if separated domains are comparably small. Such staining artifacts are avoided using direct mapping by means of STEM-EELS. To obtain insights into the mechanism of phase separation, we also performed Monte Carlo simulations.19,55,56Reconstruction of the three-dimensional structure of core–shell scaffold structures by means of STEM-EELS tomography was impeded here, due to the beam sensitivity of the investigated polymer.51 Therefore, instead of performing tomography, we analyzed several different core–shell particles, in order to capture different particle orientations that may result from the deposition on the graphene film support. The analysis of 18 different core–shell structures reveals two principal results: Firstly, the PS appears to be located always in a bottom layer, whereas the PMMA extends farther from the gold core (see middle column of Fig. 2). Our simulations revealed that this can be attributed to polymer–solvent interactions which are weakly unfavorable for PS but favor PMMA during the preparation of the hybrid nanostructures (see also ESI,† Fig. S2 for comparative simulations performed for different solvent qualities), leading to segregated PS domains. The polymers were grafted to gold nanoparticles in mixed solvent DMF/water 20:1 and the solvent selectivity is related to the Flory–Huggins interaction parameters: χPMMA–DMF = 0.22;64χPS–DMF = 0.46;65χPMMA–water = 3.5;66χPS–water = 6.0.66 These parameters indeed reflect the solvent qualities predicted by simulation results. Secondly, the distribution of the PS is found to be laterally isotropic in 5 out of 18 investigated cases (as exemplarily shown in Fig. 2a), whereas in 4 out of 18 cases PS is found only on one side of the particles and fully depleted on the opposite side, i.e. a Janus-type structure (as exemplarily shown in Fig. 2c). (The remaining 9 cases are intermediate between these two cases, as exemplarily shown in Fig. 2b.) These different lateral polymer distributions in spectrum image maps, which are 2-dimensional projections of the investigated 3-dimensional structures, correspond to different side views from a single simulated core–shell structure (right column in Fig. 2). Our simulations are hence supportive of the claim that analyzing different core–shell particles can provide a sampling of different particle orientations resulting from casting on the graphene support prior to the microscopic experiments. Our observations point to a non-uniform distribution of PS around the gold cores in the form of a Janus-like structure.
 | ||
| Fig. 2 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of core–shell particles (left column) and corresponding spectrum image maps of these particles (middle column). The images in the middle column show maps of gold (red), PS (green), and PMMA (blue). [Note that mapping of the polymeric species at the gold positions was not possible with the employed approach.] Particles with isotropic (a) and anisotropic (b and c) lateral distribution of PS were found. The right column displays different side views of a single simulation core–shell particle with mixed PMMA/PS RAFT polymers attached to the gold core. | ||
We also analyzed the robustness of the formation of Janus-like structures, by varying the amount of grafted PS in simulations. These simulations were performed by keeping an overall grafting density of 0.41 chains nm−2,19 and varying the amount of adsorbed PS from 0.17 chains nm−2, 0.20 chains nm−2, to 0.24 chains nm−2. Such analysis reveals that a fluctuating density of grafted PS chains does not significantly affect the formation of the segregated PS domains, which form during the entire range of PS grafting density investigated (Fig. 3a–c).
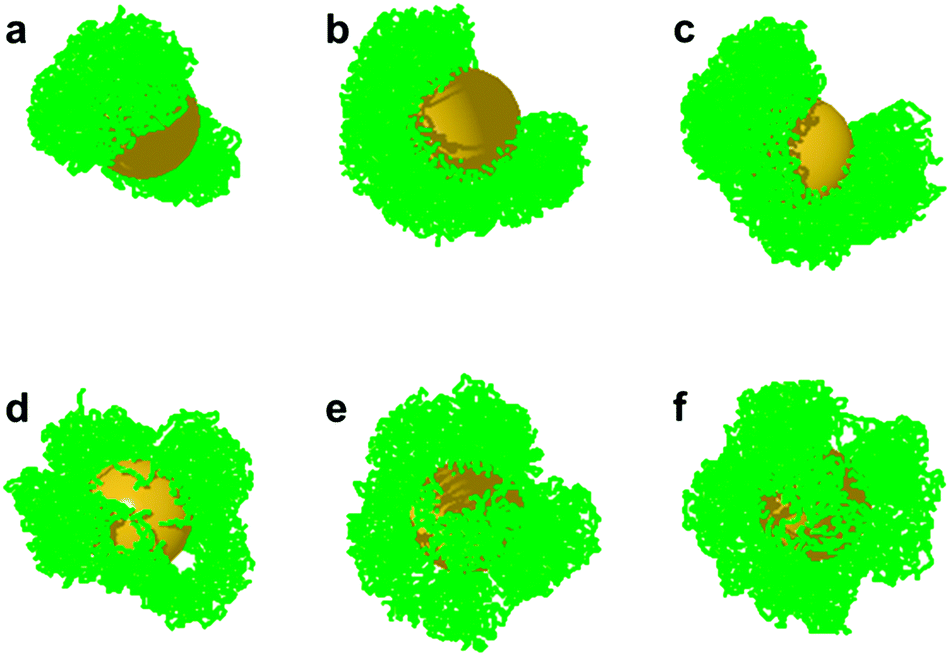 | ||
| Fig. 3 Snapshots of the segregated PS domain at the AuNP surface for the case of laterally mobile grafting points (a–c) and fixed grafting points (d–f). The side views are selected for clear display. PS grafting densities are: (a and d), 0.17 chains nm−2; (b and e), 0.20 chains nm−2; (c and f), 0.24 chains nm−2. | ||
To investigate whether the lateral mobility of the grafted polymer ligands provides another degree of freedom that influences the phase separation, we performed control simulations with immobile surface grafting points (see also Fig. S3, ESI†). In such situation, the PS domains are more jagged compared with the situations involving mobile grafting points, and no well phase-separated PS domains emerge (Fig. 3d–f). From this comparison and from the segregated PS domains experimentally observed, we can therefore conclude that the adsorbed polymers are laterally mobile, as had already been observed for self-assembled monolayers on flat gold surfaces.67 It should be emphasized that a coarse-grained simulation with Martini model was previously employed to show Janus-like configurations of the PS and PEO brushes on gold nanoparticle surface.45 However, our simulations are distinct in the following aspects: First, the grafting points in the cited simulations are immobile, and the Janus-like structure is set in prior. In our simulation, the Janus-like configurations are obtained by the phase separation of PS and PMMA on the AuNP surface. Second, the parameters used in our simulation, such as the chain length, nanoparticle size, grafting densities of PS and PMMA, and the solvent qualities are directly extracted from the experimental systems (see the previous sections). These two aspects ensure the direct comparison of our simulations to the experimental findings.
We were particularly intrigued if these Janus-like core–shell nanostructures can be employed as anisotropic scaffolds for the site-specific attachment of additional functional units. We reasoned that the fraction of telechelic PS, which does not attach to the AuNP core with both chain ends, should provide free trithiocarbonate moieties onto which smaller AuNPs could be attached.19 As a proof-of-concept, we followed an experimental strategy described earlier by us18,19 and treated the core–shell particles from Fig. 2 with smaller satellite AuNPs from two-phase Brust–Schiffrin synthesis. This was followed by addition of monofunctional PS (Table 1) to shield the outer surfaces of the satellite particles60 in order to provide colloidal stabilization. This resulted in the formation of planet–satellite nanostructures, as evidenced from HAADF images (Fig. 4, left column). Mapping of the PMMA and PS polymeric species in these planet–satellite structures from both experiments and simulations shows that the satellite particles are found in the outer region of the separated PS bottom layer (Fig. 4, middle and right columns). This reveals that, (i) satellite AuNPs can penetrate the outer PMMA layer to become attached to the planet particle via association with telechelic PS; and (ii) AuNPs with phase-separated mixed polymer brushes can be site-selectively functionalized, if one of the polymeric species provides binding sites for the attachment of additional nanoparticles.
 | ||
| Fig. 4 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of planet–satellite structures particles (left column) and corresponding spectrum image maps of these particles (middle column). The images in the middle column show maps of gold (red), PS (green), and PMMA (blue). The satellite particles are located at the PS outlayer with isotropic (a) and anisotropic (b) lateral distributions (see white arrows). The right column shows the simulation snapshots with PS (green) and AuNPs (yellow), which confirm the adsorption of satellite AuNPs on the PS surface, and also indicate that the isotropic lateral distribution in (a) is the side-view of an anisotropic PS distribution. | ||
4. Conclusions
In conclusion, we have developed a modular synthetic approach toward anisotropic nanoparticle arrangements with hierarchical structure. By exploiting the phase separation of immiscible polymers on AuNP surfaces directionality can be imparted to hybrid core–shell structures, and anisotropic nanoparticle arrangements are thusly accessible from there.Whereas staining procedures are typically used for the microscopic visualization of phase separation phenomena on nanoparticle surfaces, we herein revealed the morphologies of mixed polymer brush systems by direct mapping of the polymeric species using scanning transmission electron microscopy and electron energy-loss spectroscopy. Employing Monte Carlo simulations, we could also show that the phase separation into Janus-type structures is a result of laterally mobile surface grafting points. By equipping one of the constituent polymers in these separating mixed brushes with functional groups, site-selective incorporation of additional functional units can be performed. Our concept was demonstrated by preparing gold-planet–gold-satellite nanoparticle arrangements with Janus-like structures, but it may in principle enable the fabrication of a variety of different anisotropic nanostructures as well. The approach may therefore find applications in the preparation of well-defined nanostructures capable of undergoing directed self-assembly or performing directed motion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Professor Gregor Trimmel (TU Graz) for providing access to his laboratory and the Zentrum für Elektronenmikroskopie Graz (ZFE) for access to their microscopes. C. R. acknowledges support from the Leopoldina Fellowship Programme, German National Academy of Sciences Leopoldina (Project No. LPDS 2017-02). Q. T. and M. M. acknowledge the financial support from Deutsche Forschungsgemeinschaft-CONICET under grant Mu1674/15-1. The simulations have been performed at the GWDG Göttingen, the HLRN Hannover/Berlin, and the von-Neumann Institute for Computing, Jülich, Germany.References
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- K. Liu, Z. Nie, N. Zhao, W. Li, M. Rubinstein and E. Kumacheva, Science, 2010, 329, 197–200 CrossRef PubMed.
- A. Guerrero-Martínez, B. Auguié, J. L. Alonso-Gómez, Z. Džolič, S. Gómez-Grańa, M. Žinić, M. M. Cid and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2011, 50, 5499–5503 CrossRef PubMed.
- J. Wang, H. Xia, Y. Zhang, H. Lu, R. Kamat, A. V. Dobrynin, J. Cheng and Y. Lin, J. Am. Chem. Soc., 2013, 135, 11417–11420 CrossRef PubMed.
- C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. Hao, E. Auyeung, G. Liu, X. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741–746 CrossRef PubMed.
- T. G. W. Edwardson, K. L. Lau, D. Bousmail, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2016, 8, 162–170 CrossRef PubMed.
- S. Mei, H. Qi, T. Zhou and C. Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13645–13649 CrossRef PubMed.
- J. Guo, B. L. Tardy, A. J. Christofferson, Y. Dai, J. J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R. R. Dagastine, I. Yarovsky and F. Caruso, Nat. Nanotechnol., 2016, 11, 1105–1111 CrossRef PubMed.
- N. Gandra and S. Singamaneni, Chem. Commun., 2012, 48, 11540–11542 RSC.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645–2651 CrossRef PubMed.
- R. P. M. Höller, M. Dulle, S. Thomä, M. Mayer, A. M. Steiner, S. Förster, A. Fery, C. Kuttner and M. Chanana, ACS Nano, 2016, 10, 5740–5750 CrossRef PubMed.
- S. Borsley, S. Flook and E. R. Kay, Chem. Commun., 2015, 51, 7812–7815 RSC.
- P. Dey, S. Zhu, K. J. Thurecht, P. M. Fredericks and I. Blakey, J. Mater. Chem. B, 2014, 2, 2827–2837 RSC.
- Z. Fan, M. Tebbe, A. Fery, S. Agarwal and A. Greiner, Part. Part. Syst. Charact., 2016, 33, 698–702 CrossRef.
- L. Wu, U. Glebe and A. Böker, Adv. Mater. Interfaces, 2017, 4, 1700092 CrossRef.
- J. H. Yoon and S. Yoon, Langmuir, 2013, 29, 14772–14778 CrossRef PubMed.
- R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schüller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2014, 9, 74–78 CrossRef PubMed.
- C. Rossner and P. Vana, Angew. Chem., Int. Ed., 2014, 53, 12639–12642 Search PubMed.
- C. Rossner, Q. Tang, O. Glatter, M. Müller and P. Vana, Langmuir, 2017, 33, 2017–2026 CrossRef PubMed.
- W. Peng, C. Rossner, V. Roddatis and P. Vana, ACS Macro Lett., 2016, 5, 1227–1231 CrossRef.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699–1709 CrossRef PubMed.
- Q. Yang, M. H. de Vries, F. Picchioni and K. Loos, Nanoscale, 2013, 5, 10420–10427 RSC.
- Q. Yang and K. Loos, Macromol. Chem. Phys., 2017, 218, 1600451 CrossRef.
- M. D. McConnell, M. J. Kraeutler, S. Yang and R. J. Composto, Nano Lett., 2010, 10, 603–609 CrossRef PubMed.
- C. C. Lin, C. W. Liao, Y. C. Chao and C. Kuo, ACS Appl. Mater. Interfaces, 2010, 2, 3185–3191 Search PubMed.
- B. Wang, B. Li, B. Zhao and C. Y. Li, J. Am. Chem. Soc., 2008, 11594–11595 CrossRef PubMed.
- J. H. Yoon, J. Lim and S. Yoon, ACS Nano, 2012, 6, 7199–7208 CrossRef PubMed.
- A. M. Jackson, J. W. Myerson and F. Stellacci, Nat. Mater., 2004, 3, 330–336 CrossRef PubMed.
- C. Singh, P. K. Ghorai, M. A. Horsch, A. M. Jackson, R. G. Larson, F. Stellacci and S. C. Glotzer, Phys. Rev. Lett., 2007, 99, 226106 CrossRef PubMed.
- R. P. Carney, G. A. DeVries, C. Dubois, H. Kim, Y. K. Jin, C. Singh, P. K. Ghorai, J. B. Tracy, R. L. Stiles, R. W. Murray, S. C. Glotzer and F. Stellacci, J. Am. Chem. Soc., 2008, 130, 798–799 CrossRef PubMed.
- S. K. Meena, C. Goldmann, D. Nassoko, M. Seydou, T. Marchandier, S. Moldovan, O. Ersen, F. Ribot, C. Chanéac, C. Sanchez, D. Portehault, F. Tielens and M. Sulpizi, ACS Nano, 2017, 11, 7371–7381 CrossRef PubMed.
- L. Chen and H.-A. Klok, Soft Matter, 2013, 9, 10678–10688 RSC.
- Y. Wang, G. Yang, P. Tang, F. Qiu, Y. Yang and L. Zhu, J. Chem. Phys., 2011, 134, 134903 CrossRef PubMed.
- R. M. Choueiri, E. Galati, H. Thérien-Aubin, A. Klinkova, E. M. Larin, A. Querejeta-Fernández, L. Han, H. L. Xin, O. Gang, E. B. Zhulina, M. Rubinstein and E. Kumacheva, Nature, 2016, 538, 79–83 CrossRef PubMed.
- E. Galati, M. Tebbe, A. Querejeta-Fernández, H. L. Xin, O. Gang, E. B. Zhulina and E. Kumacheva, ACS Nano, 2017, 11, 4995–5002 CrossRef PubMed.
- R. M. Choueiri, A. Klinkova, S. Pearce, I. Manners and E. Kumacheva, Macromol. Rapid Commun., 2017, 17000554 Search PubMed.
- D. Li, X. Sheng and B. Zhao, J. Am. Chem. Soc., 2005, 127, 6248–6256 CrossRef PubMed.
- B. Zhao and L. Zhu, J. Am. Chem. Soc., 2006, 128, 4574–4575 CrossRef PubMed.
- L. Zhu and B. Zhao, J. Phys. Chem. B, 2008, 112, 11529–11536 CrossRef PubMed.
- X. Jiang, G. Zhong, J. M. Horton, N. Jin, L. Zhu and B. Zhao, Macromolecules, 2010, 43, 5387–5395 CrossRef.
- C. Bao, S. Tang, R. A. E. Wright, P. Tang, F. Qiu, L. Zhu and B. Zhao, Macromolecules, 2014, 47, 6824–6835 CrossRef.
- C. Bao, S. Tang, J. M. Horton, X. Jiang, P. Tang, F. Qiu, L. Zhu and B. Zhao, Macromolecules, 2012, 45, 8027–8036 CrossRef.
- J. M. Horton, S. Tang, C. Bao, P. Tang, F. Qiu, L. Zhu and B. Zhao, ACS Macro Lett., 2012, 1, 1061–1065 CrossRef.
- S. Kim, T. H. Kim, J. Huh, J. Bang and S. H. Choi, ACS Macro Lett., 2015, 4, 417–421 CrossRef.
- J. Dong and J. Zhou, Macromol. Theory Simul., 2013, 22, 174–186 CrossRef.
- A. M. Percebom, J. J. Giner-Casares, N. Claes, S. Bals, W. Loh and L. M. Liz-Marzán, Chem. Commun., 2016, 52, 4278–4281 RSC.
- S. Tang, T.-Y. Lo, J. M. Horton, C. Bao, P. Tang, F. Qiu, R.-M. Ho, B. Zhao and L. Zhu, Macromolecules, 2013, 46, 6575–6584 CrossRef.
- J. P. Koski and A. L. Frischknecht, ACS Nano, 2018, 12, 1664–1672 CrossRef PubMed.
- C. Rossner, O. Glatter, O. Saldanha, S. Koester and P. Vana, Langmuir, 2015, 31, 10573–10582 CrossRef PubMed.
- C. Rossner, B. Ebeling and P. Vana, ACS Macro Lett., 2013, 2, 1073–1076 CrossRef.
- R. F. Egerton, P. Li and M. Malac, Micron, 2004, 35, 399–409 CrossRef PubMed.
- S. Yakovlev and M. Libera, Micron, 2008, 39, 734–740 CrossRef PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 55–75 RSC.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef.
- G. S. Grest and K. Kremer, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3628–3631 CrossRef.
- M. Murat, G. S. Grest and K. Kremer, Macromolecules, 1999, 32, 595–609 CrossRef.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef.
- A.-S. Duwez, P. Guillet, C. Colard, J.-F. Gohy and C.-A. Fustin, Macromolecules, 2006, 39, 2729–2731 CrossRef.
- S. Slavin, A. H. Soeriyadi, L. Voorhaar, M. R. Whittaker, C. R. Becer, C. Boyer, T. P. Davis and D. M. Haddleton, Soft Matter, 2012, 8, 118–128 RSC.
- C. Rossner, V. Roddatis, S. Lopatin and P. Vana, Macromol. Rapid Commun., 2016, 37, 1742–1747 CrossRef PubMed.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef.
- B. Zhao, R. T. Haasch and S. MacLaren, Polymer, 2004, 45, 7979–7988 CrossRef.
- S. Santer, A. Kopyshev, J. Donges, J. Rühe, X. Jiang, B. Zhao and M. Müller, Langmuir, 2007, 23, 279–285 CrossRef PubMed.
- H. Pang, Y. Bao, L. Xu, D.-X. Yan, W.-Q. Zhang, J.-H. Wang and Z.-M. Li, J. Mater. Chem. A, 2013, 1, 4177 Search PubMed.
- B. A. Wolf and G. Blaum, Makromol. Chem., 1978, 179, 2265–2277 CrossRef.
- E. M. Davis and Y. A. Elabd, J. Phys. Chem. B, 2013, 117, 10629–10640 CrossRef PubMed.
- S. J. Stranick, A. N. Parikh, Y.-T. Tao, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 7636–7646 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00545a |
| This journal is © The Royal Society of Chemistry 2018 |