
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly deformable hydrogels constructed by pH-triggered polyacid nanoparticle disassembly in aqueous dispersions†
Wenkai
Wang
a,
Dongdong
Lu
a,
Mingning
Zhu
a,
Jennifer M.
Saunders
a,
Amir H.
Milani
 a,
Steven P.
Armes
b and
Brian R.
Saunders
*a
a,
Steven P.
Armes
b and
Brian R.
Saunders
*a
aSchool of Materials, University of Manchester, MSS Tower, Manchester, M13 9PL, UK. E-mail: Brian.saunders@manchester.ac.uk
bDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK
First published on 13th April 2018
Abstract
Most hydrogels are prepared using small-molecule monomers but unfortunately this approach may not be feasible for certain biomaterial applications. Consequently, alternative gel construction strategies have been established, which include using covalent inter-linking of preformed gel particles, or microgels (MGs). For example, covalently interlinking pH-responsive MGs can produce hydrogels comprising doubly crosslinked microgels (DX MGs). We hypothesised that the deformability of such DX MGs was limited by the presence of intra-MG crosslinking. Thus, in this study we designed new nanoparticle (NP)-based gels based on pH-swellable NPs that are not internally crosslinked. Two polyacid NPs were synthesised containing methacrylic acid (MAA) and either ethyl acrylate (EA) or methyl methacrylate (MMA). The PMAA–EA and PMAA–MMA NPs were subsequently vinyl-functionalised using glycidyl methacrylate (GMA) prior to gel formation via free-radical crosslinking. The NPs mostly disassembled on raising the solution pH but some self-crosslinking was nevertheless evident. The gels constructed from the EA- and MMA-based NPs had greater breaking strains than a control DX MG. The effect of varying the solution pH during curing on the morphology and mechanical properties of gels prepared using PMAA–MMA–GMA NPs was studied and both remarkable deformability and excellent recovery were observed. The gels were strongly pH-responsive and had tensile breaking strains of up to 420% with a compressive strain-at-break of more than 93%. An optimised formulation produced the most deformable and stretchable gel yet constructed using NPs or MGs as the only building block.
Introduction
Hydrogels are water-rich polymer networks that continue to attract wide interest in the literature.1–3 This activity is largely driven by their potential applications which include biomaterials,4,5 water purification,6 photonics7 and electronic skin.8 Hydrogels are usually prepared from the bottom up via statistical copolymerisation of (small-molecule) monomers and crosslinkers.9 Alternatively, pre-formed soluble linear chains can be covalently linked together.10 Combined approaches have also been explored, whereby multiple networks are formed11 or hydrogels are prepared using nanoparticles (NPs) as crosslinkers.12 Recently, hydrogels have also been prepared without any covalent crosslinking.13 Of particular interest are pH-responsive polyacid hydrogels containing high levels of carboxylic acid groups. Such gels can exhibit strong pH-responsive or electrolyte-responsive swelling.14–20 They also offer potential biomaterial applications, because the pH-triggered swelling transition can be tuned to correspond to physiological conditions, especially for gels based on methacrylic acid (MAA).21–24 Whilst excellent mechanical properties have been reported for such gels,25 one common limitation in the context of biomaterials applications is that gel construction methods rely on the use of small molecule monomers.14,17–20,25–32 In this study we introduce a new generic approach that enables the rational design of highly deformable pH-responsive hydrogels using aqueous dispersions of pre-formed polyacid NPs as the only gel building blocks.A new approach to polyacid hydrogel construction utilising pH-responsive copolymer microgels (MGs) as building blocks was introduced in 2011 at Manchester.33 Such MGs are typically lightly crosslinked colloidal particles that are usually prepared with a crosslinking comonomer and significant swelling occurs when the solution pH approaches the pKa of the constituent copolymer chains.34 Macroscopic gels were prepared in the absence of any small molecule crosslinker by covalent interlinking of the pre-formed MGs to produce doubly-crosslinked microgels (DX MGs)33 (see Scheme 1(a)). As discussed in detail elsewhere35 this DX MG concept differs from gels constructed from small molecule monomers and MG particle macro-crosslinkers.19,20,25 Gel assembly using only pre-formed MGs is a potential advantage for biomaterials applications because using small molecules as building blocks may present cytotoxicity challenges in vivo. DX MGs exhibit suitable mechanical properties for potential application in the field of intervertebral disc repair,24 but there no literature examples of highly deformable polyacid gels prepared from dispersed NPs without added small molecule monomers.
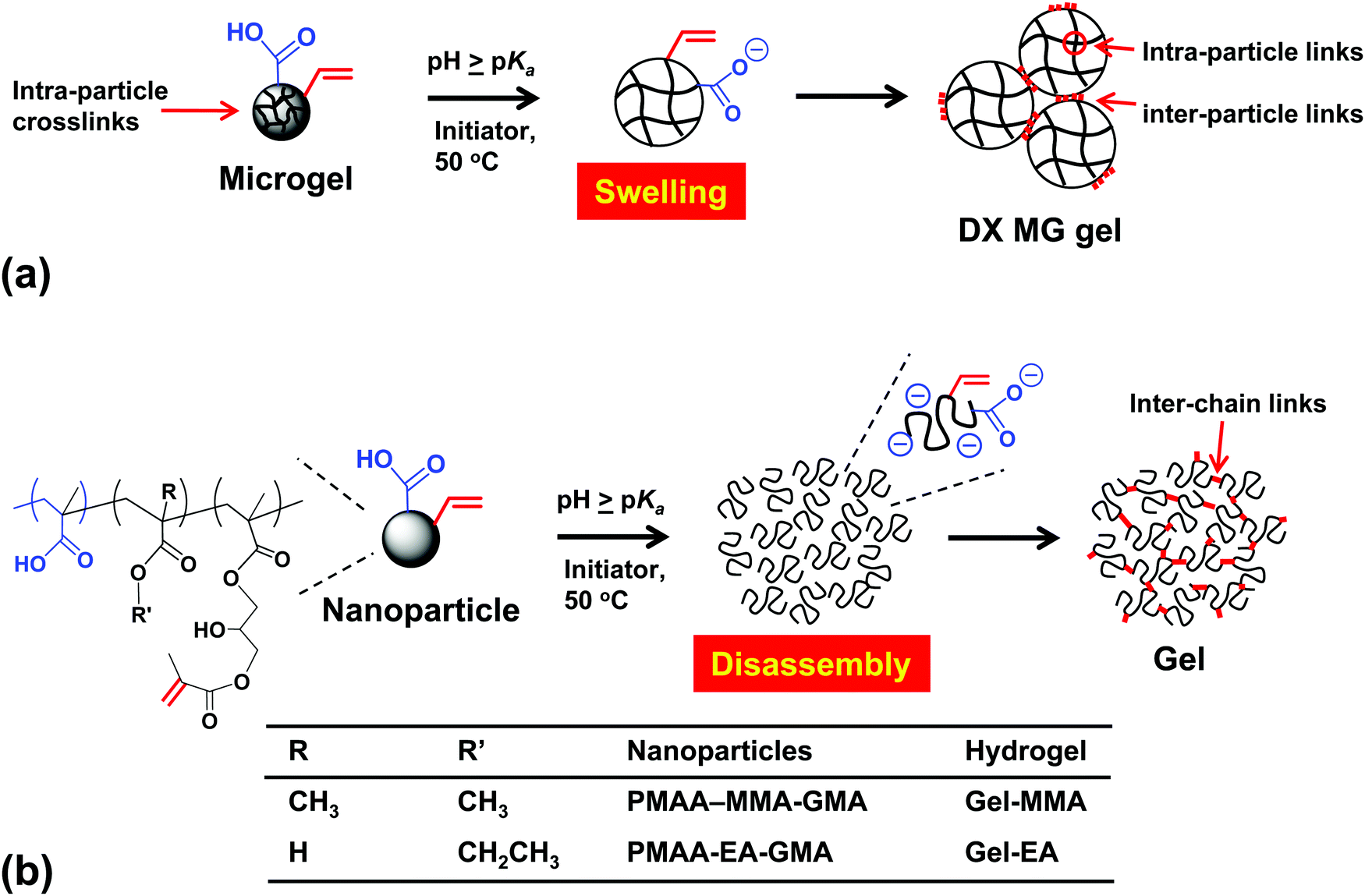 | ||
| Scheme 1 (a) Schematic representation of vinyl-functionalised microgels (MGs) that swell on raising the solution pH and are then linked together to form a double-crosslinked MG (DX MG) gel. (b) pH-Triggered disassembly of vinyl-functionalised nanoparticles (NPs) and subsequent hydrogel formation. This study focuses on gels formed via (b) but the results are compared to DX MGs prepared via (a). Note that no free monomer is present in either (a) or (b). | ||
A major driver for development of new gels is enhanced mechanical properties. Unfortunately, heterogeneity in the elastically-effective chain length is common for hydrogels. This results in gel embrittlement because stress is concentrated at the shortest chains.11 In principle, highly stretchable tough gels can be obtained by making all the elastically-effective chains of similar length.36 Alternatively, tough gels can be prepared by introducing sacrificial3 or reversible37 crosslinks to dissipate energy. Our previous DX MGs consisted of interlinked MGs that comprise two populations of elastically-effective chains; intra-MG chains as well as inter-MG chains (Scheme 1(a)). Here, we sought to substantially reduce the former population by preparing pH-swellable nanoparticles (NPs) without added crosslinker (Scheme 1(b)). We hypothesised that such gels should contain only one major population of elastically-effective chains which was expected to improve gel deformability.
MGs are usually prepared using a divinyl crosslinker.38 However, seminal studies by the Lyon group have shown that N-isopropylacrylamide (NIPAM) MGs can be prepared in the absence of such comonomers.39,40 Moreover, such self-crosslinked MGs can be ultrasoft and highly deformable.41 Although the precise mechanism responsible for MG self-crosslinking has not been established, it seems likely that chain transfer to polymer is responsible.39,40 In the present study, we use two different formulations comprising either ethyl acrylate (EA) or methyl methacrylate (MMA) to prepare the first examples of self-crosslinked polyacid NPs (Scheme 1(b)).
Our approach to gel construction is summarised in Scheme 1(b). We seek to enhance pH-triggered particle swelling by preparing MAA-rich NPs with no intra-particle crosslinker. Thus, these NPs simply contain mainly MAA plus a minor proportion of either MMA or EA comonomer. They were subsequently vinyl-functionalised by reaction of a minor fraction of the MAA units with glycidyl methacrylate (GMA; see Scheme S1, ESI†). On raising the solution pH the vinyl-functionalised NPs initially swell and eventually undergo disassembly. The interpenetrating copolymer chains were then crosslinked to afford deformable pH-responsive gels. This new approach employs preformed NPs to deliver vinyl-functionalised polyacid chains on demand using pH as the trigger. The present gels differ substantially from vinyl-functionalised micellar-based gels that contained –COOH groups and required added small monomers for their formation19,20 and also from gels constructed using a combination of vinyl-functionalised nanogels as crosslinkers and small molecule monomers such as NIPAM.25
Herein, we first characterise the two NP systems and study their pH-triggered disassembly. The effect of varying the solution pH on the dilute dispersion properties is compared to that for a control pH-responsive MG. We then compare the mechanical properties of gels constructed from such NPs at pH 7.4 to data previously reported for a related DX MG. In order to understand and optimise the influence of NP disassembly on gel mechanical properties the effect of the preparation pH is then investigated. These gels are shown to be the most deformable (and toughest) that have been constructed entirely from polyacid NP building blocks to date. This study is an important step in the development of injectable NP-based injectable gels for high toughness applications such as cartilage repair.
Experimental
Materials
MMA (99%), MAA, (99%), GMA (97%) and NaOH (97%), ammonium persulfate (APS, 99%), N,N,N′,N′-tetramethylethylenediamine (TEMED, 99%) and sodium dodecyl sulphate (SDS, ≥ 99%) were purchased from Sigma-Aldrich and used as received. The control MGs considered in this study contained EA, MAA, 1–4 butanediol diacrylate (BDDA) and GMA and were reported earlier.42 They are denoted here as PEA–MAA–GMA MG. The latter MGs were prepared by emulsion polymerisation and vinyl functionalised with GMA.42 Ultrahigh purity (distilled and deionised) water was used in all experiments.Synthesis of vinyl-functionalised polyacid nanoparticles
The synthesis of the precursor NPs was conducted using a modified “seed-feed” emulsion polymerisation method to minimise self-crosslinking. For preparation of PMAA–MMA NPs a comonomer mixture (35.0 g) comprising MMA (14.0 g, 0.14 mol) and MAA (21.0 g, 0.25 mol) was used. Seed formation was achieved using 5.0 g of this comonomer stock solution, which was added to water (480 g) containing SDS (2.4 g, 8.0 mmol) and then TEMED solution (1.0 ml of 11.0 wt% solution) and APS (5.0 g of a 11.0 wt% solution) were added. The seed was prepared at 50 °C with continuous mechanical stirring for 15 min. A total of 20.0 g of the comonomer solution was then added at a uniform rate of 0.50 ml min−1 over 40 min and the temperature was raised to 60 °C. APS (2.75 g of a 11.0 wt% solution) was added and the remaining comonomer solution was fed into the mixture at the same addition rate. Particle growth continued for a further 1 h at 60 °C. The crude product was purified by extensive dialysis using water. GMA (1.25 g, 9.0 mmol) was added to the purified precursor NP dispersion (100 g, 3.0 wt%) and the ring-opening reaction (Scheme S1, ESI†) was allowed to proceed for 8.0 h at pH ∼ 5.0. Unreacted GMA was removed by extensive dialysis using water. An identical protocol was used for the preparation of PMAA–EA–GMA NPs.Nanoparticle disassembly studies
NaOH solution was added to NP dispersions to achieve the pH required. Dynamic light scattering (DLS) measurements were obtained after 24 h equilibration at 25 °C.Hydrogel preparation
Aqueous NaOH solution (4.0 M) was added to the NPs (4.0 g of a 13.3 wt% aqueous dispersion) and the pH adjusted to the desired pH (5.8, 7.4 or 10.0) to form a slightly viscous dispersion. Gels were formed by mixing of an aqueous APS solution (50 μl, 5.0 wt%) with the MG dispersion via stirring and subsequently heating the dispersions (10.6 wt%) in sealed molds at 50 °C for 24 h.Physical measurements
DLS measurements were conducted using a 50 mW He–Ne laser operating at 633 nm with a standard avalanche photodiode (APD) and detection optics connected to a Malvern Zetasizer Nano ZS90 autocorrelator. This instrument set-up provided the z-average diameter (dz) as well as the electrophoretic mobility. Potentiometric titration was conducted using a Mettler Toledo DL15 titrator. Titrations were performed in the presence of aqueous NaCl (0.010 M) as background electrolyte and the titration solution was NaOH (0.10 M). TEM images were obtained using an FEI Tecnai Bio Twin instrument (100 kV) and samples were prepared using 300 mesh copper TEM grids. The dispersion concentration used was 0.0010 wt% and uranyl acetate staining (0.25 wt% solution) was used. SEM measurements were obtained using a Philips XL30 FEG SEM instrument. Grids were coated with a thin layer of either platinum or carbon to prevent sample charging.Uniaxial compression measurements were performed using an Instron series 5569 load frame equipped with a 100 N compression testing head. The modulus was calculated using the gradient of the stress–strain data measured over the first 10% strain. Cyclic compression experiments were conducted at 10 mm s−1. Engineering stress (σ) and strain values (ε) are reported. Uniaxial tensile stress–strain measurements were conducted using an Instron series 5569 load frame equipped with a 10 N compression testing head at a rate of 10 mm s−1 using dog-bone shaped molds with a central rectangular region of length 20 mm. The width and thickness were both 4.0 mm, while paddle regions had a width of 17.5 mm. Gel samples for the qualitative tensile tests involving knot formation were prepared using a cylindrical mold.
Gel swelling measurements were conducted by immersing gel samples in buffer for 24 h. The volume swelling ratios (Q) were calculated using gravimetry according to the following equation.
 | (1) |
Results and discussion
Nanoparticle characterisation and pH-triggered disassembly
Two types of MAA-rich, crosslinker-free NPs were prepared to demonstrate the versatility of our new approach for the construction of deformable gels using NP building blocks: PMAA–EA–GMA and PMAA–MMA–GMA. Potentiometric titration data (Fig. S1, ESI† and Table 1) revealed that these NPs contained 57.0–75.9 mol% MAA and 8.6–13.2 mol% GMA and exhibited pKa values of 5.8–5.9. The EA-based system contained a lower MAA content and higher GMA content. These differences are attributed to the greater hydrophobicity of the EA-based NPs. The water contact angles for PEA and PMMA homopolymer films are ∼100° and 68°, respectively.43,44 Attempts to prepare control samples of MAA-rich MGs via addition of a divinyl crosslinker resulted in severe coagulation. Instead, we compare NP data to that obtained for an earlier PEA–MAA–GMA MG prepared using EA, MAA and BDDA.42 PEA–MAA–GMA MG contained 32 mol% MAA and had a pKa of 6.2 (see Table 1).| Copolymer | Particle type | MAAa/mol% | MMAa/mol% | EAa/mol% | GMA/mol% | pKab | d TEM /nm [CV] | d z /nm [PDI] |
|---|---|---|---|---|---|---|---|---|
| a Calculated from potentiometric titration data shown in Fig. S1 (ESI). b Apparent pKa values were obtained from Fig. S1 (ESI). c Number-average diameters determined from TEM images. CV is the coefficient of variation. d Mean (z-average) diameter determined by DLS at pH 4.0. e These particles were internally crosslinked with BDDA. | ||||||||
| PMAA–EA–GMA | NP | 57.0 | — | 29.8 | 13.2 | 5.9 | 81 [21] | 94 [0.08] |
| PMAA–MMA–GMA | NP | 75.9 | 15.5 | — | 8.6 | 5.8 | 66 [25] | 74 [0.26] |
| PEA–MAA–GMAe | MG | 32.0 | — | 61.0 | 7.0 | 6.2 | 62 [17] | 67 [0.04] |
TEM images confirmed that the as-made PMAA–EA–GMA (Fig. 1a) and PMAA–MMA–GMA (Fig. 1b) NPs were spherical and had number-average diameters, dTEM, of 81 and 66 nm, respectively. (Larger area TEM images are shown in Fig. S2(a) and (b), ESI†.) There was some minor debris present for PMAA–MMA–GMA (Fig. 1(b)). The control PEA–MAA–GMA MGs were spherical with a dTEM value of 62 nm. DLS data for PMAA–EA–GMA and PMAA–MMA–GMA NPs indicated dz values of 94 and 74 nm, respectively, at pH 4.0. The dz value for PEA–MAA–GMA MG was 67 nm at pH 4.0. The dz values were greater than the dTEM values for each of the three systems because DLS is biased towards larger particles for size distributions of finite width.
 | ||
| Fig. 1 TEM images obtained for PMAA–EA–GMA NPs (a and d), PMAA–MMA–GMA NPs (b and e) and PEA–MAA–GMA MGs (c and f) deposited from pH ∼ 4.0 (top row) and pH 7.4 (bottom row) solutions. The arrows highlight some NPs that have not undergone full disassembly. Scale bars: 100 nm. | ||
Increasing the aqueous dispersion pH had a dramatic effect on the NP morphology. TEM images for PMAA–EA–GMA and PMAA–MMA–GMA NPs deposited from aqueous solution at pH 7.4 (Fig. 1(d) and (e), respectively) are consistent with the expected NP disassembly (see also lower magnification TEM images in Fig. S2(d) and (e), ESI†). Nevertheless, some of the original NPs were still present (red arrows). This suggests that some (albeit relatively low) degree of self-crosslinking has occurred. Self-crosslinking has been reported for PNIPAM MGs by Lyon et al.39 However, it has not been previously observed for polyacid MGs. Self-crosslinking most likely involves chain transfer to polymer. Following the approach adopted by Lyon et al., we used TEMED at low temperature to minimise this side reaction during particle formation and growth. However, it was not possible to completely eliminate self-crosslinking. TEM images obtained for these NPs differ markedly from those obtained for the control MG particles at pH 7.4 (Fig. 1(f) and Fig. S2(f), ESI†) where only fully intact spheres were present. These TEM observations demonstrate that omitting divinyl crosslinker leads to substantial loss of the original NP morphology on raising the solution pH above the NP pKa.
DLS was employed to study the pH-triggered disassembly of dispersed PMAA–EA–GMA and PMAA–MMA–GMA NPs in Fig. 2(a) and (b), respectively. As the solution pH is gradually increased, the NP size distributions shift to larger diameters and then become bimodal at pH 6.8 before shifting back to smaller diameters at pH 10. (Unfortunately, reliable DLS data could not be measured for the PMAA–EA–GMA NPs at pH 10.) These data indicate initial NP swelling at pH 6.8–7.4, followed by almost complete NP disassembly at pH 10. In striking contrast, the control MG DLS size distributions (Fig. 2(c)) remain monomodal and show only an increase in mean diameter at pH 6.8, which remained constant at higher pH.
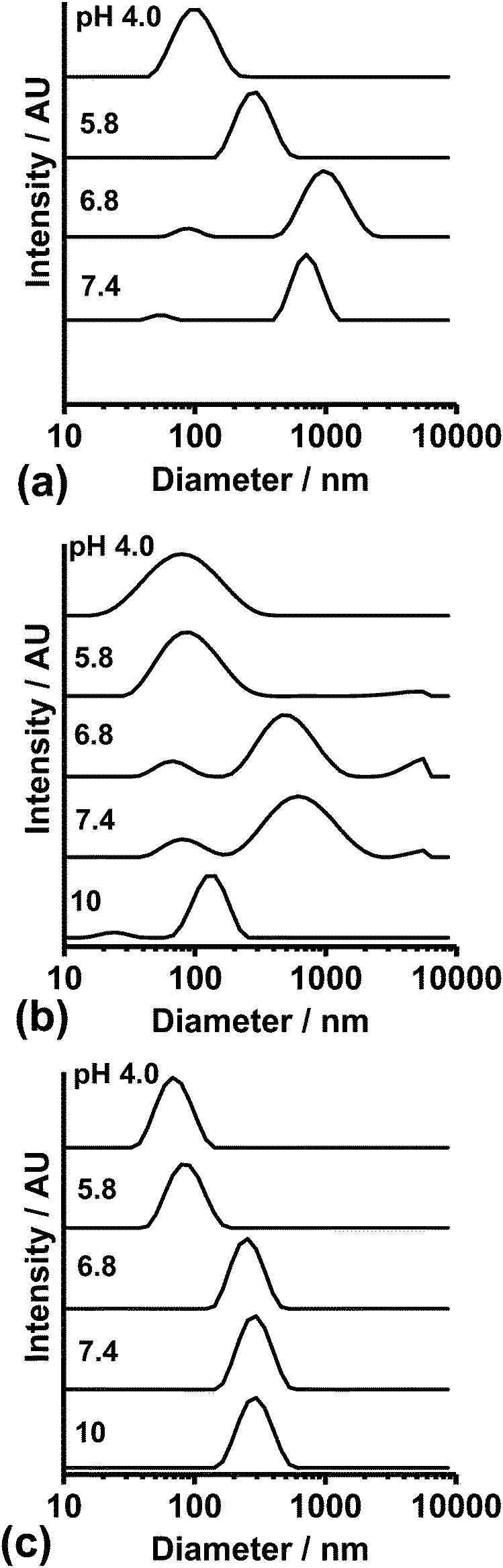 | ||
| Fig. 2 DLS size distributions recorded at selected pH values for (a) PMAA–EA–GMA NPs, (b) PMAA–MMA–GMA NPs and (c) PEA–MAA–GMA MGs. | ||
The pH-dependence for the dz and polydispersity index (PDI) of the NPs is shown in Fig. S3(a) and (b) (ESI†) for completeness. These data show that the onset of NP swelling occurs at a critical solution pH that lies close to their pKa values (5.8–5.9). The NP dz values are less reliable at higher pH values because the PDI values were very high (i.e., greater than 0.30). The dz data obtained for the pH-responsive MGs indicate a significant increase in size near the particle pKa (of 6.2) followed by a plateau (Fig. S3(c), ESI†). To gain further insight into the NP swelling process, pH-dependent electrophoretic mobilities (μ) were measured for PMAA–MMA–GMA NPs via aqueous electrophoresis (Fig. S4, ESI†). The μ magnitude increased strongly when the pH exceeded 6.5, which is attributed to the formation of anionic carboxylate groups. This critical pH coincided with a dramatic increase in dz (Fig. S3(b), ESI†), which suggests that NP disassembly occurs as a result of a pH-triggered increase in electrostatic repulsion between anionic PMAA–MMA–GMA chains.
Effect of comonomer type on hydrogel mechanical properties
Hydrogels were constructed from concentrated EA- or MMA-based NP dispersions at pH 7.4 using free-radical crosslinking (Scheme 1(b)). [Note: hydrogels constructed from the PMAA–EA–GMA and PMAA–MMA–GMA NPs at pH 7.4 are denoted as Gel-EA-7.4 and Gel-MMA-7.4, respectively.] The as-made gels were transparent and easily supported their own weight (see inset of Fig. 3(a)). Compressive stress–strain data for these gels are shown in Fig. 3(a) and Table 2. Gel-EA-7.4 and Gel-MMA-7.4 had breaking strain values of 45% and 72%, respectively (Fig. 3(b)). The latter gel was softer and more deformable than the former. This implies that the elastically-effective chains are relatively long for Gel-MMA-7.4, which is reasonable given the compositions of the parent NPs used to construct these gels. PMAA–MMA–GMA NPs had a lower GMA content (8.6 mol%) compared to the PMMA–EA–GMA NPs (13.2 mol%).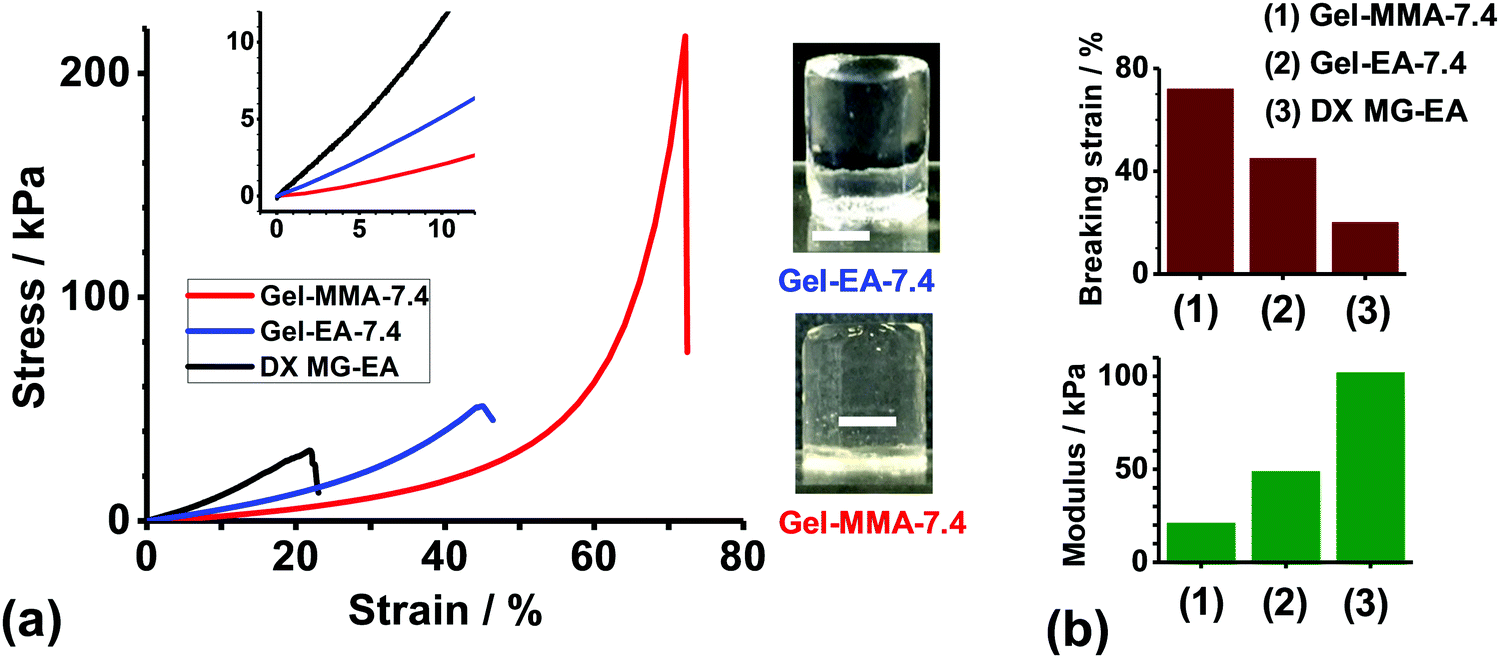 | ||
| Fig. 3 (a) Uniaxial compressive stress–strain curves for Gel-MMA-7.4, Gel-EA-7.4 and a comparable DX MG–EA system reported earlier.45 The insets show and expanded view that includes the strain region used to calculate the modulus values (0–10%) as well as representative images of each gel. Scale bars: 5 mm. (b) Breaking strain and modulus values for the three gels from (a). | ||
The compression stress–strain data obtained for Gel-EA-7.4 and Gel-MMA-7.4 are compared to those previously reported45 for a DX MG gel prepared from PEA–MAA–GMA MGs. The MG used to prepare that gel (denoted as DX MG–EA) had an almost identical composition and particle size as those shown in Fig. 1(c) and Table 1. The GMA content of the PEA–MAA–GMA MG precursor particles used to prepare the DX MG–EA gel was 3.5 mol%. If GMA-crosslinking controlled the mechanical properties, a more deformable gel would be expected compared to the present gels. In contrast, Gel-EA-7.4 and Gel-MMA-7.4 had higher breaking strains and hence were more deformable gels (Fig. 3). Gel-EA-7.4 and Gel-MMA-7.4 exhibited lower moduli compared to DX MG–EA (Fig. 3(b)). Accordingly, Gel-EA-7.4 and Gel-MMA-7.4 had lower number densities of elastically-effective chains compared to DX MG–EA, which accounts for the enhanced gel properties. We attribute the superior deformability of Gel-MMA-7.4 and Gel-EA-7.4 to the absence of any divinyl crosslinker within the NP building blocks.
Effect of preparation pH on nanoparticle-based hydrogel properties
A key aim of this study was to examine our hypothesis that the absence of intra-particle crosslinking would produce stronger, more resilient hydrogels compared to the previously studied DX MGs.45,46 Gel-MMA-7.4 was selected for further study because it had a much higher breaking strain than Gel-EA-7.4 (Table 2). Moreover, this MMA-based gel is solely constructed from components that have either already been used in bone cement (MMA) or a hydrolysis product (MAA) of the key component of bone cement.47 Arguably, the most important question concerning these gels is what role the preparation pH has on the final gel mechanical properties. To address this question, two further gels were prepared at pH 5.8 (denoted Gel-MMA-5.8) and pH 10.0 (denoted Gel-MMA-10). According to the DLS studies (Fig. 2(b)), these three pH values of 5.8, 7.4 and 10 correspond to NPs that are slightly swollen, partially dissociated and mostly dissociated, respectively.Concentrated dispersions of conventional MGs prepared with a crosslinker (e.g., BDDA) form strong physical gels when the solution pH exceeds the particle pKa because of MG particle swelling.33 In contrast, the present PMAA–MMA–GMA NPs exhibited only a modest increase in viscosity at pH 5.8, 7.4 and 10 and did not form strong physical gels (see Fig. S5, ESI†). These results demonstrate that the NPs did not restrict each other's motion significantly during macrogel network formation. The NPs were not swollen at pH 5.8, whereas they became swollen, and underwent disassembly at higher pH, with interpenetration of the dissolved copolymer chains (Fig. 2(b)). Transparent gels were obtained after curing at each pH (see insets in Fig. 4(a)–(c)). Thus, the building blocks for gel formation are fundamentally different from those of the previously studied DX MGs.33
 | ||
| Fig. 4 (left) SEM images obtained for freeze-dried samples of (a) Gel-MMA-5.8, (b) Gel-MMA-7.4 and (c) Gel-MMA-10.0. Scale bars: SEM images, 10 μm; gel images, 10 mm. (right) FTIR spectra recorded for freeze-dried samples of (d) Gel-MMA-5.8, (e) Gel-MMA-7.4 and (f) Gel-MMA-10.0. The highlighted IR bands denote those assigned to hydrogen-bonded carboxylic acid dimers (yellow) and anionic carboxylate groups (pink), respectively. | ||
SEM images were obtained for freeze-dried Gel-MMA-X (X = 5.8, 7.4 and 10.0) samples and showed highly porous morphologies (see Fig. 4(a)–(c)). Digital image analysis indicated mean pore diameters of 4.0 ± 0.9, 7.2 ± 1.5 and 15.4 ± 4.5 μm, respectively. Such pores originate from ice formation.48 Control of pore size is relevant for membrane or tissue scaffold application.48,49 This parameter also provides useful qualitative information concerning the mechanical properties of the gels. In our previous work on DX MGs, it was shown that the pore size increased as the gel modulus is reduced.50 Here, larger pores were obtained at higher pH, suggesting that the gels became less stiff (lower moduli). These images suggest that increasing the preparation pH leads to more deformable gels because gel stiffness is often inversely related to deformability.
FTIR spectroscopy was used to probe the extent of ionisation of the carboxylic acid groups and spectra were recorded for freeze-dried gels prepared at pH 5.8, 7.4 and 10 (Fig. 4(d)–(f)). Bands at 1710 and 1544 cm−1 are assigned to carboxylic acid and anionic carboxylate groups, respectively.51,52 As the gel preparation pH increased the relative –COO−/–COOH absorbance ratio for these bands increased from 0.3 (pH 5.8) to 1.5 (pH 7.4) and to 4.0 (at pH 10.0). Hence, the MAA residues gradually became neutralised, as expected. Notably, neutralisation was not complete at pH 7.4. Such “hindered” ionisation is well-documented for polyacid hydrogels.53
The three Gel-MMA-X (X = 5.8, 7.4 and 10) samples were pH-responsive (Fig. S6, ESI†) and exhibited their lowest volume swelling ratios (Q) at pH 5.0, which is well below the particle pKa. These gels swelled strongly and attained Q values of 40–47 when equilibrated in buffer with a pH of 7.4 or pH 10. The swelling is driven by the formation of anionic carboxylate groups, which leads to inter-chain electrostatic repulsion as discussed above in relation to Fig. S4 (ESI†). Increasing the solution pH further to pH 10 did not significantly change the Q value for Gel-MMA-10 which is presumably because the –COOH groups were fully neutralised. However, the Q values for the Gel-MMA-5.8 and Gel-MMA-7.4 gels were significantly lower. The corresponding FTIR spectra (Fig. 4(d) and (e)) provided evidence for incomplete neutralisation of the latter gels. We propose that a higher intra-gel ionic strength (and hence electrostatic screening) occurred for those gels as a result of the higher proportions of carboxylic acid groups that required neutralisation when subsequently swollen in pH 10 buffer.
The preparation pH had a major effect on the mechanical properties of the gels. Fig. 5(a) shows compression stress–strain data for Gel-MMA-5.8, Gel-MMA-7.4 and Gel-MMA-10, which exhibited breaking strains (and gel moduli) of 66% (35.6 kPa), 72% (21.1 kPa) and >93% (3.09 kPa), respectively (Table 3). Hence, these three gels exhibited significantly greater deformability as they became less stiff. These data enabled calculation of the number-density of elastically-effective chains (νe). The modulus (E) is related to νe by
| E = 3νekTϕ21/3 | (2) |
 | (3) |
 | ||
| Fig. 5 (a) Compressive stress–strain curves for Gel-MMA-5.8, Gel-MMA-7.4 and Gel-MMA-10. Note that Gel-MMA-10 did not break even at the maximum strain studied. (b) Calculated number density of elastically-effective chains for these three gels (see text). (c) A monolithic Gel-MMA-10 sample withstands substantial compressive deformation. (Scale bar: 10 mm.) | ||
| Gel | Modulusa/kPa | Stress-at-break/kPa | Breaking-strain/% | ν e /× 10−23 m−3 | x c |
|---|---|---|---|---|---|
| a The polymer volume fraction used for these experiments was 9.0 vol%. b Number-density of elastically-effective chains (see text). c Average number of segments between crosslinks (see text). d Sample did not fail even at the maximum strain recorded by our instrument. | |||||
| Gel-MMA-5.8 | 35.6 | 133 | 66.6 | 64.3 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210 210 |
| Gel-MMA-7.4 | 21.1 | 215 | 72.0 | 38.1 | 2040 |
| Gel-MMA-10 | 3.09 | >1170d | >93.0d | 5.6 | 13920 |
We probed the mechanism responsible for the excellent deformability of Gel-MMA-5.8 and Gel-MMA-10 using cyclic compression measurements (Fig. S7, ESI†). The area calculated from the difference in the loading and unloading curves is the hysteresis. These area values represent only 13 and 17% of the respective areas under the loading curves for Gel-MMA-5.8 and Gel-MMA-10. These data reveal modest hysteresis and hence relatively low energy dissipation. There was also no residual strain, which confirms excellent recovery for these hydrogels. If either reversible or dynamic crosslinks made a major contribution, then much higher hysteresis and significant residual strain would be expected.58 Consequently, covalent crosslinking is primarily responsible for the observed mechanical properties. Covalent crosslinks are mainly formed via free-radical coupling of the vinyl groups on the chains (plus a minor proportion of self-crosslinks remaining within the NPs, as discussed above).
Not only did the three Gel-MMA-X samples possess good to excellent mechanical properties when subjected to compression, they also proved to be remarkably stretchable. This property enabled tensile stress–strain data to be determined. We emphasise that this is the first time that such data has been reported for MAA-rich hydrogels constructed from NP (or MG) building blocks without using added small molecule monofunctional or difunctional comonomers. Tensile stress–strain data (Fig. 6(a)) showed approximately linear behaviour with only moderate strain-hardening. (The tensile data are summarised in Table 4.) Interestingly, such behaviour is qualitatively similar to that reported for cartilage.59 The tensile breaking strain and Young's modulus values are shown in Fig. 6(b) and (c), respectively. The stretchability (as determined by the breaking strain) proved to be very sensitive to the gel preparation pH. Thus, Gel-MMA-5.8, Gel-MMA-7.4 and Gel-MMA-10 could be stretched to 200, 340 and 520% of their original lengths, respectively. The gel moduli also showed the same trend indicated by the compression data (Table 3). The high breaking strain obtained for the hydrogel prepared at pH 7.4 (Gel-MMA-7.4) together with its remarkable toughness (49.4 kJ m−3, Table 4) are very encouraging for future biomaterials applications. The breaking strain obtained for Gel-MMA-10 compares favourably to data recently reported for alternative next-generation hydrogels.60,61 Indeed, Gel-MMA-10 strings could be readily tied in knots and stretched to more than 250% of its original length without damage (see Fig. 6(d) and video, ESI†), which is unprecedented for this class of gel.
 | ||
| Fig. 6 Summary of hydrogel mechanical properties: (a) tensile stress–strain data, (b) tensile breaking strains and (c) Young's moduli for the three new gels reported herein. (d) Gel-MMA-10.0 was also prepared in the form of strings that could be tied in knots and were able to withstand substantial stretching without breaking. A video showing this manipulation is available in the ESI.† | ||
What is the origin of the high deformability of these gels? There are two key differences between the NPs used in the present study and the MGs reported previously:33,34 (i) the omission of a crosslinking comonomer and (ii) the higher MAA contents of the NPs. The absence of any crosslinker should enable faster diffusion of the GMA reagent throughout the precursor NPs during the coupling reaction between GMA and a minor fraction of the carboxylic acid groups on the monomer repeat units (Scheme S1, ESI†). In principle, this should result in copolymer chains containing a uniform distribution of vinyl groups. Furthermore, their relatively high MAA content means that the NPs should swell strongly and undergo almost complete disassembly when the solution pH is raised beyond the NP pKa, which has the effect of further distributing the crosslinking points and increasing xc. Combined with the substantial interpenetration of the copolymer chains and greater extent of NP disassembly in alkaline solution, a relatively uniform distribution of elastically-effective chains can be expected for these hydrogels (as depicted in Scheme 1(b)). The excellent deformability obtained for the optimised formulation (Gel-MMA-10) arises because more complete disassembly can be achieved at this higher pH. This interpretation is supported by a representative TEM image recorded for the parent PMAA–MMA–GMA dispersion deposited from a mildly alkaline (pH 10) solution (Fig. S8, ESI†) which confirmed that very few NPs remained intact under such conditions.
Conclusions
In summary, a versatile new NP-based approach that does not require small molecule comonomers is reported for the preparation of highly deformable hydrogels with excellent recovery. These MMA- or EA-based NPs contain very high MAA contents and can be vinyl-functionalised using GMA. Triggered NP disassembly occurs on increasing the pH and subsequent crosslinking leads to the formation of highly deformable and strongly pH-responsive polyacid hydrogels. The MMA-based gels proved to be more deformable than the EA-based gels. The mechanical properties of the MMA-based gels could be tuned by adjusting the preparation pH. The most deformable gel (Gel-MMA-10) had a strain-at-break of 420%. Importantly, the deliberate omission of any intra-particle crosslinker enabled a substantial improvement in both gel deformability and toughness compared to previously reported DX MGs. These new NP-based hydrogels are comparable to next-generation gels constructed using small molecule monomers in terms of their mechanical performance. Future work will investigate the use of lower synthesis temperatures (e.g., 37 °C) in order to target injectable highly deformable gels for load-bearing tissue and possibly cartilage repair applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a five-year EPSRC Established Career Fellowship awarded to BRS (M002020/1) as well as EP/K030949/1.References
- M. A. Haque, T. Kurokawa and J. P. Gong, Polymer, 2012, 53, 1805–1822 CrossRef CAS.
- I. Jeon, J. Cui, W. R. K. Illeperuma, J. Aizenberg and J. J. Vlassak, Adv. Mater., 2016, 28, 4678–4683 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, D. R. King, T. Kurokawa, Y. Zhao, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Macromolecules, 2016, 49, 2750–2760 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS.
- T. E. Brown and K. S. Anseth, Chem. Soc. Rev., 2017, 46, 6532–6552 RSC.
- Y. Chen, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 1992–2001 CAS.
- L. Dong, A. K. Agarwal, D. J. Beebe and H. Jiang, Nature, 2006, 442, 551 CrossRef CAS.
- S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger and R. Schwödiauer, Adv. Mater., 2014, 26, 149–162 CrossRef CAS PubMed.
- J. G. H. Joosten, J. L. McCarthy and P. N. Pusey, Macromolecules, 1991, 24, 6690–6699 CrossRef CAS.
- K. Yue, G. Trujillo-deSantiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- K. Haraguchi, Macromol. Symp., 2007, 256, 120–130 CrossRef CAS.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1604951 CrossRef PubMed.
- T. V. Terziyan, A. P. Safronov and Y. G. Belous, Polym. Sci., Ser. A, 2015, 57, 200–208 CrossRef CAS.
- U. Gulyuz and O. Okay, Soft Matter, 2013, 9, 10287–10293 RSC.
- S. Chauhan, Chem. Sin., 2015, 6, 61–72 CAS.
- T. Baskan, D. C. Tuncaboylu and O. Okay, Polymer, 2013, 54, 2979–2987 CrossRef CAS.
- A. R. Khare and N. A. Peppas, Biomaterials, 1995, 16, 559–567 CrossRef CAS PubMed.
- L. Xiao, C. Liu, J. Zhu, D. J. Pochan and X. Jia, Soft Matter, 2010, 6, 5293–5297 RSC.
- L. Xiao, J. Zhu, J. D. Londono, D. J. Pochan and X. Jia, Soft Matter, 2012, 8, 10233–10237 RSC.
- V. Albright, I. Zhuk, Y. Wang, V. Selin, B. van de Belt-Gritter, H. J. Busscher, H. C. van der Mei and S. A. Sukhishvili, Acta Biomater., 2017, 61, 66–74 CrossRef CAS PubMed.
- K. Pafiti, Z. Cui, D. Adlam, J. Hoyland, A. J. Freemont and B. R. Saunders, Biomacromolecules, 2016, 17, 2448–2458 CrossRef CAS PubMed.
- C. P. McCoy, R. A. Craig, S. M. McGlinchey, L. Carson, D. S. Jones and S. P. Gorman, Biomaterials, 2012, 33, 7952–7958 CrossRef CAS PubMed.
- A. H. Milani, A. J. Freemont, J. A. Hoyland, D. J. Adlam and B. R. Saunders, Biomacromolecules, 2012, 13, 2793–2801 CrossRef CAS PubMed.
- L.-W. Xia, R. Xie, X.-J. Ju, W. Wang, Q. Chen and L.-Y. Chu, Nat. Commun., 2013, 4, 2226 Search PubMed.
- L. Toman, M. Janata, J. Spěváček, J. Brus, A. Sikora, J. Horský, P. Vlček and P. Látalová, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1284–1291 CrossRef CAS.
- D.-G. Gao, J.-Z. Ma and H.-Q. Guo, New J. Chem., 2010, 34, 2034–2039 RSC.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- J. Jovanovic, B. Potkonjak, T. Adnadjevic and B. Adnadjevic, Polym. Eng. Sci., 2016, 56, 87–96 CAS.
- H. M. N. El-Din, E.-S. A. Hegazy and D. M. Ibraheim, Polym. Compos., 2009, 30, 569–575 CrossRef CAS.
- S. Noppakundilograt, S. Choopromkaw and S. Kiatkamjornwong, J. Appl. Polym. Sci., 2018, 135, 45654 CrossRef.
- T. Sandu, A. Sârbu, F. Constantin, S. Vulpe and H. Iovu, J. Appl. Polym. Sci., 2013, 127, 4061–4071 CrossRef CAS.
- R. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696–4704 RSC.
- A. H. Milani, J. M. Saunders, N. T. Truong, L. P. D. Ratcliffe, D. J. Adlam, A. J. Freemont, J. A. Hoyland, S. P. Armes and B. R. Saunders, Soft Matter, 2017, 1554–1560 RSC.
- W. Richtering and B. R. Saunders, Soft Matter, 2014, 10, 3695–3702 RSC.
- D. E. Apostolides, T. Sakai and C. S. Patrickios, Macromolecules, 2017, 50, 2155–2164 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147–148, 251–262 CrossRef CAS PubMed.
- X. Hu, Z. Tong and L. A. Lyon, Langmuir, 2011, 27, 4142–4148 CrossRef CAS.
- J. Gao and B. J. Frisken, Langmuir, 2003, 19, 5212–5216 CrossRef CAS.
- H. Bachman, A. C. Brown, K. C. Clarke, K. S. Dhada, A. Douglas, C. E. Hansen, E. Herman, J. S. Hyatt, P. Kodlekere, Z. Meng, S. Saxena, M. W. Spears Jr., N. Welsch and L. A. Lyon, Soft Matter, 2015, 11, 2018–2028 RSC.
- W. Wang, A. H. Milani, Z. Cui, M. Zhu and B. R. Saunders, Langmuir, 2017, 33, 8192–8200 CrossRef CAS PubMed.
- Y. Ma, X. Cao, X. Feng, Y. Ma and H. Zou, Polymer, 2007, 48, 7455–7460 CrossRef CAS.
- H. Mnatsakanyan, P. Rico, E. Grigoriou, A. M. Candelas, A. Rodrigo-Navarro, M. Salmeron-Sanchez and R. Sabater i Serra, ACS Appl. Mater. Interfaces, 2015, 7, 18125–18135 CAS.
- T. Lane, J. L. Holloway, A. H. Milani, J. M. Saunders, A. J. Freemont and B. R. Saunders, Soft Matter, 2013, 9, 7934–7941 RSC.
- Z. Cui, A. H. Milani, P. J. Greensmith, J. Yan, D. J. Adlam, J. A. Hoyland, I. A. Kinloch, A. J. Freemont and B. R. Saunders, Langmuir, 2014, 30, 13384–13393 CrossRef CAS PubMed.
- A. S. Gladman, A.-D. N. Celestine, N. R. Sottos and S. R. White, Adv. Healthcare Mater., 2015, 4, 202–207 CrossRef CAS PubMed.
- X. Yao, H. Yao and Y. Li, J. Mater. Chem., 2009, 19, 6516–6520 RSC.
- M. Chau, K. J. De France, B. Kopera, V. R. Machado, S. Rosenfeldt, L. Reyes, K. J. W. Chan, S. Förster, E. D. Cranston, T. Hoare and E. Kumacheva, Chem. Mater., 2016, 28, 3406–3415 CrossRef CAS.
- Z. Cui, W. Wang, M. Obeng, M. Chen, S. Wu, I. Kinloch and B. R. Saunders, Soft Matter, 2016, 12, 6985–6994 RSC.
- B. A. Brozoski, M. M. Coleman and P. C. Painter, Macromolecules, 1984, 17, 230–234 CrossRef CAS.
- U. K. Mandal, Polym. Int., 2000, 49, 1653–1657 CrossRef CAS.
- O. E. Philippova, D. Hourdet, R. Audebert and A. R. Khokhlov, Macromolecules, 1997, 30, 8278–8285 CrossRef CAS.
- L. R. G. Treloar, The Physics of Rubber Elasticity, Clarendon Press, Oxford, 1975 Search PubMed.
- A. N. Gent, Rubber Chem. Technol., 1996, 69, 59–61 CrossRef CAS.
- A. H. Milani, J. Bramhill, A. J. Freemont and B. R. Saunders, Soft Matter, 2015, 11, 2586–2595 RSC.
- A. D. Drozdov and J. d. Christiansen, Macromolecules, 2018, 51, 1462–1473 CrossRef CAS.
- H. Jia, Z. Huang, Z. Fei, P. J. Dyson, Z. Zheng and X. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 31339–31347 CAS.
- A. J. S. Fox, A. Bedi and S. A. Rodeo, Orthop., 2009, 1, 461–468 Search PubMed.
- C. Bilici, S. Ide and O. Okay, Macromolecules, 2017, 50, 3647–3654 CrossRef CAS.
- J. Du, S. Xu, S. Feng, L. Yu, J. Wang and Y. Liu, Soft Matter, 2016, 12, 1649–1654 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00325d |
| This journal is © The Royal Society of Chemistry 2018 |