
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable emulsions of droplets in a solid edible organogel matrix
Andrew
Matheson
 *a,
Georgios
Dalkas
b,
Rudi
Mears
a,
Stephen R.
Euston
b and
Paul S.
Clegg
a
*a,
Georgios
Dalkas
b,
Rudi
Mears
a,
Stephen R.
Euston
b and
Paul S.
Clegg
a
aSchool of Physics and Astronomy, University of Edinburgh, James Clerk Maxwell Building, Edinburgh EH9 3FD, UK. E-mail: Andrew.Matheson@ed.ac.uk
bInstitute of Mechanical, Process and Energy Engineering, Heriot-Watt University, Edinburgh EH14 4AS, UK
First published on 20th February 2018
Abstract
Sitosterol and oryzanol self-assemble to form very firm gels in a range of organic solvents. However, due to the formation of sitosterol hydrate crystals, these gels are unstable in the presence of water, prohibiting the dispersal of water droplets throughout the gel matrix. We demonstrate that by using glycerol as the polar phase rather than water, droplets may be dispersed throughout the oil phase without disrupting the self-assembly of the gel. As increasing volumes of water are added to the glycerol, the G′ values decrease. This can be correlated to both a drop in water activity, and also the stability of the fibrils in the presence of glycerol compared to water, as elucidated by molecular dynamics simulations. We explore how changing the total volume of polar droplets, and changing the water content of these droplets alters the strength of 15% w/w sterol gels. We find that gels exhibit G′ values of ∼1 × 107 Pa even with ∼30% w/w glycerol dispersed throughout the matrix. At higher glycerol loadings, complex multiple emulsion morphologies can form.
Introduction
Organogels are formed when an organic solvent has been structured by a micro-scale network which arrests fluid flow, resulting in a transition from liquid-like to viscoelastic flow properties. The plant sterols sitosterol and oryzanol (structures shown in Fig. 1) have been shown to self-assemble into a fibril network which may gel a wide range of oils.1 This system is of particular interest as it consists entirely of food safe ingredients, and plant sterols have been shown to lower blood cholesterol,2 making it a promising candidate for saturated fat replacement, an application where it has been shown to be acceptable to consumers.3 Additionally, the extremely solid-like nature of these gels4,5 means that they could be suited to forming lozenges for the delivery of bio-active ingredients. | ||
| Fig. 1 Chemical structures of sitosterol (top) and oryzanol (bottom). | ||
Nutraceutical applications place several restrictions and requirements upon the system. It obviously must consist only of food safe ingredients, it must be robust enough to maintain its integrity such that the bio-actives are not released prematurely, and must be stable in ambient conditions such that it does not break down when in storage prior to use. Organogels have been shown to be useful for these applications previously.6–8 However, to the best of our knowledge, sitosterol–oryzanol organogels have not been used for this application.
This is largely because when water is dispersed throughout the gel, sitosterol begins to form hydrate crystals, resulting in the breakdown of the gel structure.1,9 It has been shown that this can be mitigated against to some extent through the reduction of the water activity, typically by dissolving large concentrations of salt or sugar in the water phase,1 but the addition of large volumes of salt to a pharmaceutical or nutritional supplement is clearly sub-optimal. With this in mind, we wished to explore whether water could be entirely replaced by an alternative polar-liquid, which would allow for hydrophilic active ingredients to be disperse through the organogel matrix without including large volumes of salt. Glycerol is a polar liquid which is cheap, food safe, and non-volatile, which made it a promising candidate for dispersing through the gel. Additionally, it is commonly used as a sweetener, which may make organogel emulsions of glycerol well suited to low-fat confectionary applications. The aim of this work was to determine whether glycerol droplets could be dispersed through a sitosterol–oryzanol organogel network, and if so to then explore how this affected the structure and behavior of the gel.
Experimental
Sitosterol and oryzanol were weighed out at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, and then dissolved at ∼90 °C in sunflower oil. Whilst still molten, the required volume of glycerol was added to each gel, and then mixed using an ultra-turrax IKA T10 rotor-stator high shear mixer at ∼10000 rpm, whilst sat on a hot-plate at a temperature above the gel melting point. This allowed for glycerol droplets to be dispersed throughout the oily phase without triggering shear-induced gelation.
:1 molar ratio, and then dissolved at ∼90 °C in sunflower oil. Whilst still molten, the required volume of glycerol was added to each gel, and then mixed using an ultra-turrax IKA T10 rotor-stator high shear mixer at ∼10000 rpm, whilst sat on a hot-plate at a temperature above the gel melting point. This allowed for glycerol droplets to be dispersed throughout the oily phase without triggering shear-induced gelation.
For rheology measurements, these mixtures were poured onto the bottom plate of a TA Instruments AR2000 rheometer. A cross-hatched plate–plate geometry with a gap of 1 mm was used for all rheology measurements, with an oscillatory shear stress of 175 Pa applied at 10 Hz. These parameters are the same as we used previously when analyzing gelation in sitosterol–oryzanol gels,5 and are informed by the work of Bot et al. into how shearing affects gel formation.4
For confocal microscopy, fluorescein sodium salt was dissolved in the glycerol phase to give contrast between the phases. The glycerol was dispersed through the gel using a high-shear mixer as described above, before the sample was removed from the hot plate, vortex mixed to encourage gelation then allowed to cool before a portion was placed onto microscope slides for imaging. A Zeiss LSM T-PMT/LSM700 confocal laser scanning inverted microscope was used, samples were excited at 488 nm and fluorescence gathered using 10×, 20× and 40× objective lenses to image the sample.
For water activity measurements, samples were mixed, poured into a Petri-dish and loaded into the Rotronic AwTHERM water activity meter.
Docking and molecular dynamic (MD) simulations were used to define an initial tubule structure. Initially, fragment and knowledge-based docking approaches were used so as to obtain the initial layer of the tubular structure. First, a dimer of β-sitosterol + γ-oryzanol is constructed using molecular docking software; this is then used to build a tubule through consecutive stacking of dimers. The 3D models of β-sitosterol, γ-oryzanol and glycerol were generated from SMILES representation using the program OMEGA 2.510 and the conformation of the triglyceride was constructed using the Antechamber package.11 Gasteiger charges were applied using AutoDockTools 1.5.6.12 The search space was defined by a grid box centered on oryzanol with 80 grid points of 0.375 Å spacing in each dimension. 100 docking rounds were calculated with AutoDock 4.212 using the Lamarckian genetic algorithm with the default parameters of AutoDock4. The maximum number of energy evaluations was set to 5 × 106, and the solvent dielectric constant was set to 3.0. The value of the solvent dielectric constant reflects the triglyceride of the solvent in the organogelating systems. The resulting docked conformations were clustered using a tolerance of 2.0 Å and the most populated cluster with the highest free energy of binding ΔG was selected. In such a conformation, β-sitosterol displays its sterane group stacked with the γ-oryzanol's sterane group and, at the same time, a hydrogen bond between the hydroxyl group of the β-sitosterol and the carbonyl group of the γ-oryzanol is formed. Step by step, 38 sterols and sterol esters were docked radially on a plane with the alkyl chains pointing inward and the ferulic acid moieties of the oryzanol molecules were outward, forming a helix-like structure. The adopted tubule structure is based on available neutron scattering and molecular modelling data available in the literature.13,14 The starting diameter of the structure was 85–95 Å. Larger diameter structures were not considered for consistency with experimental studies15–17 in which mixtures of γ-oryzanol and β-sitosterol were found to self-assemble into hollow tubules with an average diameter of ∼10 nm. Subsequently, the simulated structure was subjected to energy minimization using the AMBER v16 suite of programs.18 The initial structure was first minimized using 5000 steps of steepest descent minimization followed by 5000 steps of conjugate gradient minimization.
The second layer was taken to be identical to the first layer, with the distance between the last compound of the first layer and the first compound of the second layer to be less than 5 Å, and with the distance between layers taken to be around 5 Å. Subsequently, triglyceride molecules were inserted using the Packmol19 software package in order to satisfy all of the geometric constraints and to generate molecular packing with no overlaps. The final structure was then minimized. Two different MD simulations were carried out in order to explore how glycerol and water affect the structure of the system sitosterol–oryzanol–triglyceride. The systems were solvated in two separate boxes, containing glycerol and TIP3P water, respectively, with a minimum extension of 10 Å from the solute, using the XLEaP module of AMBER16. The total charge of the system was neutral and periodic boundary conditions were used in the simulations.
Force-field parameters for all the molecules were prepared using the Antechamber package11 and the AM1-BCC atomic charges. The force-field parameters were applied to the molecules using the XLEaP module of AMBER16. A three-stage equilibration protocol, consisting of energy minimization and molecular dynamics (MD), was used to eliminate any unfavorable contacts which may have occurred as a result of model building process and to gently adjust the initial structure to the molecular mechanics force field prior to production of MD simulations. The energy of the initial structure was first minimized using 5000 steps of steepest descent minimization followed by 5000 steps of conjugate gradient minimization. Next, the system was slowly heated to the production temperature of 300 K in two stages. All atoms in the system were restrained to their initial coordinates during the heating in order to prevent large structural deviations, using a harmonic restraint force constant of 10 kcal mol−1 Å−2. In the first heating stage the system target temperature was slowly raised from 0 to 100 K for 10 ps with a constant volume simulation using the Langevin thermostat with a collision frequency of 1.0 ps−1.20 The second phase of heating slowly increased the temperature to the production temperature of 300 K over 90 ps with constant pressure periodic boundary conditions.
After heating the system, a production run of 50 ns of molecular dynamics was simulated at constant temperature and pressure using the Langevin thermostat at 300 K with a collision frequency of 1.0 ps−1. The pressure was regulated at 1 bar using the Berendsen weak-coupling algorithm with a relaxation time of 2.0 ps.21 The SHAKE algorithm22 was used to constrain bonds involving hydrogen with a relative tolerance of 1 × 10−7. The Particle Mesh Ewald (PME) method was used for long-range electrostatic and van der Waals interactions,23 with a real-space cut-off of 8 Å. A time step of 1 fs was used for equilibration and increased to 2 fs for the production of MD.
Analysis of the trajectories was performed using the CPPTRAJ module24 of AmberTools v16, and VMD 1.925 was used to visualize the trajectories and prepare the figures. CPPTRAJ was used to find and track hydrogen bonds over the course of the trajectories by extracting their simple geometric features as a function of simulation time: the donor to acceptor heavy atom distance and the donor–hydrogen–acceptor angle. Both distance and angle cut-offs were equal to the default values for hydrogen bonds in CPPTRAJ.
Results and discussion
In Fig. 2 we see an image of three samples; a sterol in sunflower oil organogel, a sample prepared by mixing glycerol in a molten gel, and a sample prepared by mixing water in a molten gel. Neither of the gels prepared with an additional polar phase are as transparent as the pure gel, due to scattering from droplets. However, the gel prepared with glycerol droplets has not taken on quite the degree of opacity as that prepared with water, due in part to sunflower oil having a refractive index closer to that of glycerol than water. | ||
| Fig. 2 A sterol in sunflower oil organogel (left), a gel from the same batch with glycerol droplets added (centre), and a gel from the same batch with water added (right). | ||
We may quantify how water and glycerol affect the properties of the gel by adding glycerol and water to gels at varying ratios. We define the ratio as w = Vwater/(Vwater + Vglycerol) and add a fixed volume (∼0.5 ml) of polar liquid to ∼2 g of sterol gel. We then track the evolution of the gel's rheological properties.
In Fig. 3(a) we see the evolution of the storage modulus, G′, over time for samples with a range of w values, as well as a pure gel prepared without the addition of any polar phase. The G′ values for each of the samples prepared with polar droplets is lower than that of the pure gel, indicating that the addition of the polar phase is reducing the solidity of the system. We also see that for the pure gel and those samples with w < 0.4 gelation occurs on a timescale of <1000 s whereas the G′ values of those with 0.4 < w < 1 take longer to approach their plateau values.
 | ||
| Fig. 3 (a) Oscillatory rheology of samples prepared by mixing polar droplets into the molten organogel during the gelation process. The composition of the polar droplets is defined by w = Vwater/(Vwater + Vglycerol), and is varied throughout. A pure gel sample prepared without polar droplets is shown for comparison. (b) The plateau G′ (black squares) and G′′ (green triangles) values for each of these samples versus w, and (in red) the corresponding (1 − Aw) value of the polar phase. | ||
In Fig. 3(b) we plot the plateau G′ values as a function of w for the samples prepared with a polar phase. If we look at the system with pure glycerol (w = 0) droplets dispersed, we see that although weaker than the pure gel, this composite system is several orders of magnitude stronger than the gel with pure water (w = 1) droplets dispersed. Our observations are consistent with previous reports on the inability for the sterols to self-assemble in the presence of water.6,7 If we look at the samples with mixed water and glycerol droplets (0 < w < 1) we see a fairly consistent trend that increasing w, the water content, results in a reduction of G′. For all samples, G′ > G′′, demonstrating that although significantly weakened, even gels with w = 1 remain solid-like in nature. In Fig. 3(b) we also plot (1 − Aw), with Aw the water activity for the polar phase, alongside the plateau G′ values. We clearly see that increased G′ values tracks very closely with the difference between Aw and that of pure water values (i.e. increasing (1 − Aw)). By reducing w the effect on Aw is two-fold – (a) water is being removed and replaced from the system and (b) the Aw of the remaining water in the system is lower than the equivalent volume of water with no glycerol. Previously it has been shown that by reducing Aw, the interactions between sitosterol and water may be minimized somewhat protecting the gel structure.1 Our results are consistent with this, but by using glycerol rather than dissolving a preservative molecule in the water phase we are able to have a larger effect on Aw whilst retaining the same volume of polar droplets.
Our results show that despite having three hydroxyl groups, the glycerol does not interact with sitosterol in a manner which undermines self-assembly. To better understand how the tubules which make up the gel structure interact with the glycerol, we performed molecular dynamic simulations, which we have previously found to be highly instructive in the understanding of organogel systems.26 Tubule structures were prepared in line with the structures previously determined from spectroscopy, scattering and AFM imaging.5,13 The cross-section of the tubule is shown in Fig. 4(a), along with the resulting cross-section after immersion in glycerol (top) and water (bottom) for 50 ns. We see that the tubule in glycerol retains its initial structure, whilst that in water has begun to alter.
 | ||
| Fig. 4 (a) Snapshots of the sitosterol–oryzanol organogel with glycerol and water at the beginning and at the end of the course of the MD simulation. Sitosterol is shown with cyan carbons and oryzanol with orange carbons. (b) Time-dependent root mean square deviation (RMSD) from the starting conformation, (c) mass-weighted radius of gyration and (d) number of sitosterol–oryzanol hydrogen bonds measured for the 50 ns MD simulations of the organogel system in water and in glycerol (colored black and red, respectively). Alignments and measurements were performed for the carbon atoms. | ||
To quantify this, the RMSD was calculated by comparing the structure of the model during the simulation time with the starting conformation. This can be computed using the equation  , where rfinal(i) is the final coordinates of an atom i, and rinitial(i) is the initial coordinate of the atom i, and N is the number of atoms. Fig. 4(b) shows the changes of RMSD values of each carbon atom of the two systems during the course of the MDs. The RMSD values of the system with glycerol remained low within the first ns, and then shot up as the system relaxed within the solvent. After that, the RMSD is relatively stable during the whole simulation time with no large oscillations, indicating that glycerol results in an overall stabilization of the organogel system. The system with water produced higher RMSD values with relatively larger fluctuations during the simulation time, suggesting significant changes in the structure as it begins to interact with water. The radius of gyration (Rg) of the model quantifies the distribution of the atoms in space relative to their centre of mass. We calculated the Rg in order to understand the changes in the size of the model under the influence of glycerol and water. The Rg can be calculated using the equation
, where rfinal(i) is the final coordinates of an atom i, and rinitial(i) is the initial coordinate of the atom i, and N is the number of atoms. Fig. 4(b) shows the changes of RMSD values of each carbon atom of the two systems during the course of the MDs. The RMSD values of the system with glycerol remained low within the first ns, and then shot up as the system relaxed within the solvent. After that, the RMSD is relatively stable during the whole simulation time with no large oscillations, indicating that glycerol results in an overall stabilization of the organogel system. The system with water produced higher RMSD values with relatively larger fluctuations during the simulation time, suggesting significant changes in the structure as it begins to interact with water. The radius of gyration (Rg) of the model quantifies the distribution of the atoms in space relative to their centre of mass. We calculated the Rg in order to understand the changes in the size of the model under the influence of glycerol and water. The Rg can be calculated using the equation 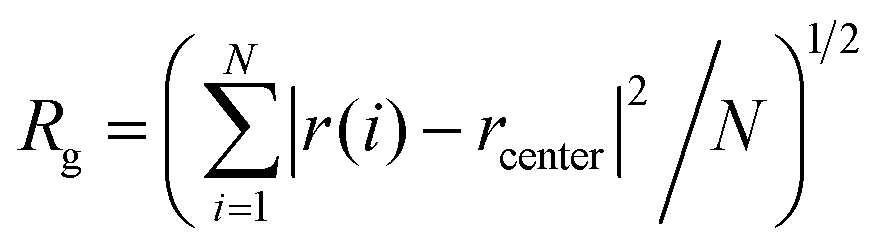 , where r(i) is the final coordinates of an atom i, rcenter is the coordinates of the organogel's center of mass, and N is the number of atoms. Fig. 4(c) shows the Rg plot from the two MD simulations. The Rg values for the model under the influence of glycerol did not vary much with respect to the initial conformation, because no significant change to the structure was observed. Conversely, the fluctuations of the Rg values of the model with the influence of water were higher compared to the model with glycerol, indicating that the structure of the sitosterol–oryzanol organogel was changed over the simulation timescale. Importantly, the calculated maximum Rg, which is the maximum distance of a selected atom from the center, was found to be 46 Å for the model with glycerol. Thus, according to the Rg computation, the sitosterol–oryzanol organogel has a diameter of about 9.2 nm which is consistent with the experimental results.5,13 It has previously been shown that a hydrogen bond between the hydroxyl group of the sitosterol and the carbonyl group of the oryzanol is key to the formation of the tubular structure.15,27 Importantly, we see the population of sitosterol–oryzanol hydrogen bonds in the system with glycerol was much higher and more stable than the system with water, as shown in Fig. 4(d).
, where r(i) is the final coordinates of an atom i, rcenter is the coordinates of the organogel's center of mass, and N is the number of atoms. Fig. 4(c) shows the Rg plot from the two MD simulations. The Rg values for the model under the influence of glycerol did not vary much with respect to the initial conformation, because no significant change to the structure was observed. Conversely, the fluctuations of the Rg values of the model with the influence of water were higher compared to the model with glycerol, indicating that the structure of the sitosterol–oryzanol organogel was changed over the simulation timescale. Importantly, the calculated maximum Rg, which is the maximum distance of a selected atom from the center, was found to be 46 Å for the model with glycerol. Thus, according to the Rg computation, the sitosterol–oryzanol organogel has a diameter of about 9.2 nm which is consistent with the experimental results.5,13 It has previously been shown that a hydrogen bond between the hydroxyl group of the sitosterol and the carbonyl group of the oryzanol is key to the formation of the tubular structure.15,27 Importantly, we see the population of sitosterol–oryzanol hydrogen bonds in the system with glycerol was much higher and more stable than the system with water, as shown in Fig. 4(d).
A snapshot of the hydrogen-bonding network that is generated by the sitosterol and oryzanol in glycerol is shown in Fig. 5. We find that glycerol forms hydrogen bonds with the methoxy, phenol and carbonyl groups of the oryzanol and the hydroxyl group of the sitosterol in a manner that enhances the stability of the cylindrical fibre structure relative to a system prepared in a pure triglyceride environment. It may seem contradictory that the addition of glycerol increases the stability of the fibril structure, yet we see in Fig. 3 that samples prepared with pure glycerol distributed throughout (i.e. w = 0) have lower G′ values than those without. However, it should be noted that the exceptional strength of the sitosterol–oryzanol gel is not purely due to the stability of individual fibrils, but also due to the dense network formed due to inter-fibril interactions,4,5 which is disrupted by the insertion of glycerol droplets. Therefore, any increase in strength of fibrils at the edge of glycerol droplet is likely cancelled out by a lack of fibrils in the glycerol itself.
 | ||
| Fig. 5 (a) Top view of the sitosterol–oryzanol organogel in glycerol after 50 ns of simulation time. (b) Hydrogen bond network between the hydroxyl group of sitosterols and the carbonyl group oryzanols. (c) Hydrogen bond network between sitosterol, oryzanol and glycerol. Sitosterol is shown with cyan carbons, oryzanol with orange carbons, glycerol with white carbons and the hydrogen bonds are in circles. | ||
In the system with water, the number of hydrogen bonds between sitosterol and oryzanol decreases during the course of the MD simulation. This is a consequence of the hydroxyl group of sitosterol and the carbonyl group of oryzanol beginning to form hydrogen bonds with water (see Fig. 6), which decreases the stability of the structure. It is also worth noting that although we observe hydrogen bonds forming between sitosterol and water, these are not entirely stable, and may form, break and then reform several times during the simulations. This is normal in MD simulations, and the violation of tight distance and angle criteria for hydrogen bonding does not necessarily mean a hydrogen bond has broken; it can be a consequence of vibrational motion of the two atoms which still remain connected by a hydrogen bond even as they move away from each other. Given longer MD simulations of the order of milliseconds, we would expect stable hydrogen bonds to form as the system equilibrates.
 | ||
| Fig. 6 (a) Top view of the sitosterol–oryzanol organogel in water after 50 ns of simulation time. (b) Hydrogen bond network between the hydroxyl group of sitosterols and water molecules. (c) Hydrogen bond between sitosterols. Sitosterol is shown with cyan carbons and the hydrogen bonds are in circles. | ||
Having demonstrated that glycerol does not have the same effect on gel stability as water, we wished to see what the maximum volume of pure glycerol we could disperse throughout the gel whilst still retaining structural integrity.
Fig. 7(a) shows the evolution of the G′ values of the samples over time, whilst Fig. 7(b) shows the plateau values achieved after 6000 s. For samples with ∼30% glycerol added there is a significant weakening of the gel, from G′ ∼ 9 MPa to G′ ∼ 6 MPa. However, this is still of the same order of magnitude as the pure gel, and an extremely high G′ value for an organogel. As concentrations reach ∼40% there is a stark decrease in G′, as it falls to ∼0.2 MPa. Thus we can infer that the glycerol droplets we add to the gel are acting as a passive rather than active filler, i.e. they do not anchor to the network instead they dilute it.28
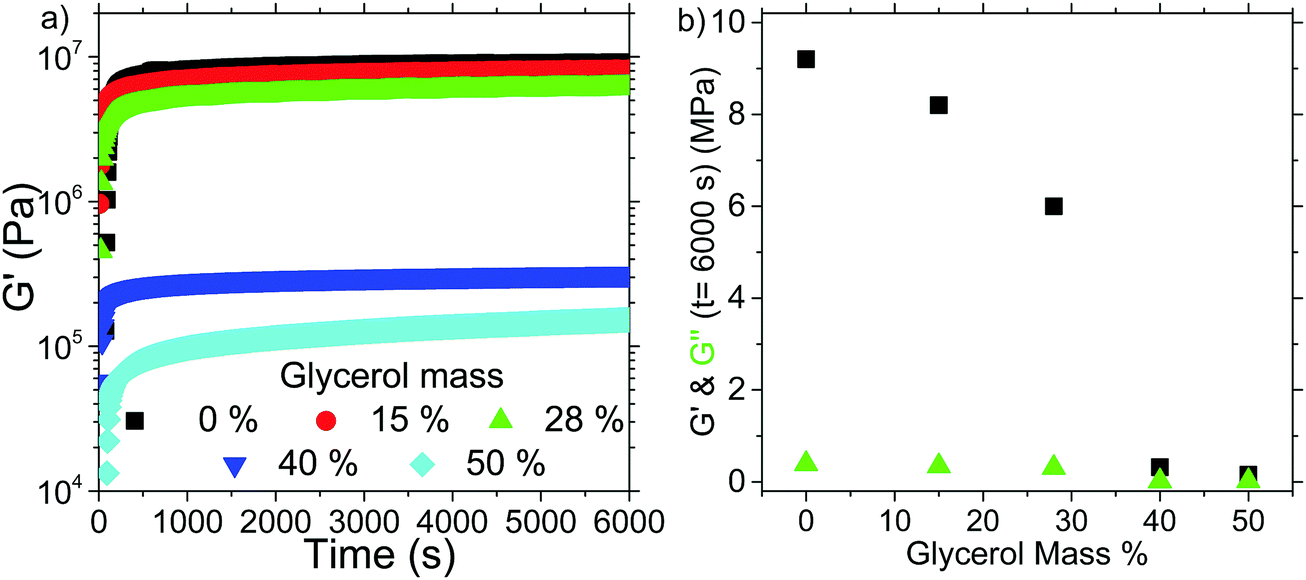 | ||
| Fig. 7 (a) Oscillatory rheology of samples prepared by mixing differing masses of pure glycerol (i.e. w = 0) into the molten organogel and then pouring onto rheometer plate, (b) plot of G′ (black squares) and G′′ (green triangles) at t = 6000 s as a function of glycerol mass. | ||
It is clear from the rheology that for the 15% w/w gels we are using, ∼30% glycerol is the maximum droplet content which can be sustained without significantly compromising the gel strength. However, we cannot tell from rheology alone if the stark reduction in G′ is due to the weakening of the gel structure due to large volumes of unstructured fluid acting as passive filler, or due to a profound change in the morphology of the gel. To investigate this we use confocal microscopy. In Fig. 8(a) we see a confocal image of a gel prepared via the dispersion of ∼20% by mass of glycerol through the gel. The droplets (dyed green with fluorescein sodium salt) are ∼10–40 μm in diameter and dispersed evenly throughout the organogel matrix. Fig. 8(b and c) shows images of a gel prepared with the addition of ∼40% glycerol (by mass) into the network. The continuous phase of the system is still organogel, although the glycerol droplets are larger (∼100 μm in diameter) and the percolation pathways of the gel around the droplets are much thinner, which may go some way to explaining why this gel exhibits a much lower value of G′. Interestingly, we see that the glycerol droplets now have organic phase dispersed within them, forming an organogel/glycerol/organogel (o/g/o) multiple emulsion. We look at these multiple emulsion droplets in more detail in Fig. 8(c), and see that the internal organogel droplets show a broad distribution from ∼2–20 μm in diameter. As glycerol content is increased to ∼50% in Fig. 8(d) we do see phase inversion, with glycerol now making up the continuous phase, forming a triple emulsion (o/g/o/g).
 | ||
| Fig. 8 Confocal microscopy images for an organogel with (a) ∼20% glycerol by mass, (b and c) ∼40% glycerol by mass, (d) ∼50% glycerol by mass dispersed through the system. The glycerol has been dyed green for contrast, dark regions are organogel. | ||
These images demonstrate the wide range of possible gel morphologies obtainable with these materials. Considering these images in tandem with the rheology results shown in Fig. 7 would suggest that significant reductions in gel strength occur before the full inversion of the system, but may be related to the development of the complex multiple emulsions observed in Fig. 8(b–d), as this results in the formation of gel domains which are no longer percolated with the overall gel network. Such complex multiple emulsions are more commonly formed in a sequence of steps or using microfluidic techniques,29,30 therefore, although not optimal for the application we initially envisaged, the morphologies observed at high loading structures are highly interesting as an example of multiple emulsions formed with no interfacial stabilization.
In an emulsion, it is a common observation that the minority phase is more likely to become droplets within the majority phase (all else being equal). This is observed at low glycerol concentration (such as the 20% glycerol sample shown in Fig. 8(a)), where mixing disperses the glycerol droplets, and the gel sets before the droplets may coalesce. The reason for the change in phase behavior at higher glycerol loadings is intriguing. The emulsification process is heavily influenced by the time dependent properties of the oleogel; as the sample cools (either while being vortexed in the vial, or sheared on the rheometer) the oleaginous phase becomes more viscous, and eventually solid. We see in Fig. 7(a) that gelation onsets progressively more slowly as the concentration of glycerol is increased. At higher glycerol concentration any gain in sample viscosity is observed to be much slower. Initially droplets of the low viscosity, hot oil phase start to form; however, as the oil viscosity grows the glycerol becomes the less viscous phase and hence forms droplets leading to the multiple emulsion observed in Fig. 8(b and c). Finally, the 50% glycerol sample proceeds along the same trajectory but with the viscosity rising even more slowly. Once the oil-in-glycerol-in-oil droplets have formed the continuous oil phase has still not fully gelled. Hence there is time for extensive coalescence of these droplets. This gives rise to a system with a continuous phase of glycerol, containing oleogel droplets which act as a solid shell preventing further coalescence of the glycerol droplets dispersed within them, these droplets themselves contain tiny oleogel droplets formed at very early times. We have illustrated the processes described above, in Fig. 9. The top row shows evolution of the 20% glycerol sample, the middle row shows the evolution of the 40% glycerol sample, and the bottom row shows the evolution of the 50% glycerol sample.
 | ||
| Fig. 9 Diagram showing the evolution of the samples following emulsification for each of the three compositions imaged in Fig. 8. The top row shows a 20% glycerol system, where the sample quickly gels immediately after the glycerol in oil emulsion is formed. The middle row shows a 40% gel where an oil in glycerol emulsion is initially formed, with secondary droplets forming as the oil phase becomes more viscous. The bottom row shows a 50% gel, which proceeds in the same manner, but with the glycerol droplets coalescing before the oleogel phase fully arrests. | ||
We also suspected that the phase inversion may be in part facilitated by the high shear stresses applied during gelation being greater than the yield stress of the oloegel network in the high glycerol samples, but not of the low glycerol samples. To test this we prepared 40% glycerol 60% organogel samples, using a high shear mixer to disperse droplets in the molten gel and then pouring samples onto the rheometer bottom plate, in the same manner as the samples measured in Fig. 3. However, instead of applying an oscillatory stress of 175 Pa during the gelation process, we applied lower stresses (1 Pa and 20 Pa) to try and avoid yielding the gel. We found that the gel did not set quickly enough to prevent phase separation between the glycerol and organogel phases, rendering the results somewhat meaningless as the rheological response was dominated by the regions of pure organogel which offered very high G′ values. This illustrates the multifarious role of shear stress in the production of these organogel emulsions; high shear stresses encourage the formation of firmer gels, presumably through fibril aggregation4,5 and are thus necessary to set the gel before wholescale phase separation occurs, yet these high shear stresses also appear to result in the phase inversion of the system. It is interesting to note that we observe in Fig. 7(b) that G′ > G′′ when performing rheology measurements on the 50% gel, but when examining the gel via microscopy, we see that glycerol constitutes the continuous phase. It may be that there are some percolated “columns” of organogel which traverse the gap between the two rheometer plates and result in the system as a whole exhibiting solid like properties whilst undergoing oscillatory shear, or that the densely packed solid gel droplets jam to give solid-like properties.31
Taking into account all the results, it is clear that ∼10 μm glycerol droplets can be dispersed throughout the organogel matrix with only moderate effects on its structural integrity, but that the water content of the glycerol must be minimized, and there is a maximum volume fraction of glycerol which may be used before the gel structure is significantly disrupted. One possible application for this system could be drug delivery or delayed release pharmaceuticals; sitosterol, oryzanol, sunflower oil and glycerol are all food safe ingredients, it is relatively easy to cast lozenges of material, it allows water soluble active ingredients to be dispersed through a very firm water-insoluble matrix which slowly breaks up when placed in water, and as we have not observed any phase separation over ∼6 months this system is highly stable in ambient conditions. Additionally, there is possibility that water insoluble bio-actives could also be dispersed throughout the organogel phase. This may open the possibility of placing in the organic phase complementary bioactives, or a precursor chemical which reacts in situ, with the molecules dispersed through the glycerol phase.
Conclusions
We have shown that by using glycerol instead of water we can add polar droplets to sitosterol–oryzanol in sunflower oil gels, without significantly compromising on gel strength in the system. Molecular dynamics simulations show that the manner in which glycerol interacts with sitosterol and oryzanol is compatible with tubule formation, whereas hydrogen bonding between sitosterol and water triggers instability in tubule structures. Although glycerol does not prohibit the formation of fibrils, at glycerol loadings >40% the morphology shifts from being glycerol in gel to a more complex multiple emulsion, which disrupts the percolation of the gel network and results in a significant reduction in G′. However, this is a qualitatively different failure mode from that induced by water. This opens up the possibility of creating oleogel capsules loaded with bioactives dispersed in glycerol.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Arjen Bot of Unilever R&D for useful conversations and materials. This work was supported by the BBSRC DRINC project [Grant BBSRC BB/M027597/1]. This project made use of time on ARCHER granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://hecbiosim.ac.uk), supported by EPSRC (grant no. EP/L000253/1).References
- H. Sawalha, R. den Adel, P. Venema, A. Bot, E. Flöter and E. van der Linden, J. Agric. Food Chem., 2012, 60, 3462–3470 CrossRef CAS PubMed.
- M. B. Katan, S. M. Grundy, P. Jones, M. Law, T. Miettinen and R. Paoletti, Mayo Clin. Proc., 2003, 78, 965–978 CrossRef CAS PubMed.
- E. Panagiotopoulou, T. Moschakis and E. Katsanidis, LWT -- Food Sci. Technol., 2016, 73, 351–356 CrossRef CAS.
- A. Bot and W. G. M. Agterof, J. Am. Oil Chem. Soc., 2006, 83, 513–521 CrossRef CAS.
- A. B. Matheson, V. Koutsos, G. Dalkas, S. Euston and P. Clegg, Langmuir, 2017, 33, 4537–4542 CrossRef CAS PubMed.
- S. Murdan, G. Gregoriadis and A. T. Florence, J. Pharm. Sci., 1999, 88, 608–614 CrossRef CAS PubMed.
- A. Vintiloiu and J.-C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS PubMed.
- S. Murdan, Expert Opin. Drug Delivery, 2005, 2, 489–505 CrossRef CAS PubMed.
- R. den Adel, C. M. H. Patricia and B. Arjen, J. Phys.: Conf. Ser., 2010, 247, 012025 CrossRef.
- P. C. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. Kollmann and D. Case, J. Comput. Chem., 2005, 25, 1157–1174 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. Bot, E. P. Gilbert, W. G. Bouwman, H. Sawalha, R. den Adel, V. M. Garamus, P. Venema, E. van der Linden and E. Floter, Faraday Discuss., 2012, 158, 223–238 RSC.
- M. Pernetti, K. F. van Malssen, E. Flöter and A. Bot, Curr. Opin. Colloid Interface Sci., 2007, 12, 221–231 CrossRef CAS.
- A. Bot and W. G. M. Agterof, J. Am. Oil Chem. Soc., 2006, 83, 513–521 CrossRef CAS.
- A. Bot, R. den Adel and E. C. Roijers, J. Am. Oil Chem. Soc., 2008, 85, 1127–1134 CrossRef CAS.
- A. Bot, E. P. Gilbert, W. G. Bouwman, H. Sawalha, R. den Adel, V. M. Garamus, P. Venema, E. van der Linden and E. Flöter, Faraday Discuss., 2012, 158, 223 RSC.
- D. S. C. D. A. Case, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, D. Greene, N. Homeyer, S. Izadi, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, Amber 14, University of California, San Francisco, 2017 Search PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- R. W. Pastor, B. R. Brooks and A. Szabo, Mol. Phys., 1988, 65, 1409–1419 CrossRef.
- H. J. C. Berendsen, J. P. M. Postma, W. F. V. Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33-38), 27–38 Search PubMed.
- A. B. Matheson, G. Dalkas, A. Gromov, S. R. Euston and P. S. Clegg, Food Funct., 2017, 8, 4547–4554 CAS.
- R. D. Adel, P. C. M. Heussen and A. Bot, J. Phys.: Conf. Ser., 2010, 247, 012025 CrossRef.
- E. Dickinson and J. Chen, J. Dispersion Sci. Technol., 1999, 20, 197–213 CrossRef CAS.
- S.-H. Kim and D. A. Weitz, Angew. Chem., Int. Ed., 2011, 50, 8731–8734 CrossRef CAS PubMed.
- P. S. Clegg, J. W. Tavacoli and P. J. Wilde, Soft Matter, 2016, 12, 998–1008 RSC.
- T. G. Mason, M.-D. Lacasse, G. S. Grest, D. Levine, J. Bibette and D. A. Weitz, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 3150–3166 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |