
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom 2D to 3D patches on multifunctional particles: how microcontact printing creates a new dimension of functionality†
Marc
Zimmermann
 *ab,
Daniela
John
a,
Dmitry
Grigoriev
a,
Nikolay
Puretskiy
a and
Alexander
Böker
*ab
*ab,
Daniela
John
a,
Dmitry
Grigoriev
a,
Nikolay
Puretskiy
a and
Alexander
Böker
*ab
aFraunhofer Institute for Applied Polymer Research IAP, D-14476 Potsdam-Golm, Germany. E-mail: marc.zimmemann@iap.fraunhofer.de; alexander.boeker@iap.fraunhofer.de
bChair of Polymer Materials and Polymer Technologies, University Potsdam, D-14476 Potsdam-Golm, Germany
First published on 16th February 2018
Abstract
A straightforward approach for the precise multifunctional surface modification of particles with three-dimensional patches using microcontact printing is presented. By comparison to previous works it was possible to not only control the diameter, but also to finely tune the thickness of the deposited layer, opening up the way for three-dimensional structures and orthogonal multifunctionality. The use of PEI as polymeric ink, PDMS stamps for microcontact printing on silica particles and the influence of different solvents during particle release on the creation of functional particles with three-dimensional patches are described. Finally, by introducing fluorescent properties by incorporation of quantum dots into patches and by particle self-assembly via avidin–biotin coupling, the versatility of this novel modification method is demonstrated.
Introduction
Over the years the research and synthesis of patchy particles have developed a huge diversity of methods giving us a broad selection of possible solutions tailored for various applications and challenges.The main ambition for the preparation of patchy particles is the possibility of guided self-assembly of these building blocks into new materials.1–3 In general; this ability is driven by anisotropy which is induced by the structural or chemical composition of these particles. Without this property, controlled and directed self-assembly would not be possible, but random aggregation or assemblies would rather occur.
Glancing angle deposition of metals can be used to apply thin inorganic films on polymer particles.4,5 Organic films on the other hand can be deposited onto silica particles using etching and casting or by vapour-assisted deposition.6,7 Other methods involve the controlled agglomeration of particles into clusters with subsequent encapsulation, the guided co-assembly of soft nanoparticles or seed mediated nucleation and growth synthesis of different metals to obtain patchy particles.8–11 Related approaches use a roughness gradient across particle clusters for directed self-assembly.12 To adjust the geometry of patchy particles, solid templates for shielding or the interface between two immiscible liquids can be used.13–16 The production of small patches on larger particles was shown using a dip-coating method and by precise absorption of polyelectrolytes.17–19 Furthermore, external forces like magnetic or electric fields were also used as common tools to improve alignment of anisotropic particles.20–24 Compared to all these methods, the microcontact printing of particles is a very flexible technique due to its robust and facile procedure.
Microcontact printing for the preparation of single- or double-patched particles has been shown for different ink systems using triggers like temperature, light or a suitable catalyst, but only limited research has been performed on the direct printing of silica particles.25–31 However, all of these printing techniques are restricted to surface functionalization of said particles without the possibility to govern the deposited surface structures in 3D.
Inspired by Tigges et al. and their use of sophisticated polymeric inks, we introduce an easy and fast approach to prepare particles with three-dimensional patches: printing high molecular polyethylenimine (PEI), directly onto blank silica microparticles using electrostatic interactions.28 We further discuss the possibility of switching between two-dimensional surface modification (2D) and three-dimensional structure printing (3D) by simple alteration of the solvent during particle release. We successfully used the generated volume of the patches for the leakage-free embedding of small nanoparticles in the 3D structures. In the following, the modification of patches using avidin and biotin functionalities leads to the self-assembly of particles induced by the interaction of these patches.
Although there have been concepts for increasing the yield of patchy particles we believe that our approach, due to its simplicity and relatively cheap materials used, could be an optimum tool for up-scale production of multifunctional patchy particles at high yield and with advanced control of patch geometry.32–34
Results and discussion
At first the reasons for the chosen approach and chemicals will be explained using the example of a conventional microcontact printing (μCP) procedure which is schematically represented in Fig. 1a. | ||
| Fig. 1 (a) Visualization of the microcontact printing approach. From left to right: Production of PDMS stamps loaded with different concentrated PEI solutions; drop casting of silica particle dispersion for monolayer creation; printing process and particle release from stamp using ultrasonic treatment in a suitable solvent. (b) Solvent influence on the patch structure during particle release. | ||
Initially, flat PDMS substrates are coated with different concentrated PEI solutions to produce loaded stamps.35,36 Silica particle monolayers on glass substrates are generated by drop casting from ethanol suspension. The loaded PDMS stamps are then pressed onto the silica monolayer, lifting off particles during substrate separation. Finally, the patchy particles are released from the stamp using ultrasonic treatment in a suitable solvent.
PDMS substrates are common materials for μCP, relatively cheap and easy to use.37–41 A great selection of different surface functionalization, patterning techniques or the simple variation of composition makes them ideal materials for our approach and future applications.31,42–45 However, to preserve the simplicity of our approach presented here, no further variations of PDMS were needed. A commonly mentioned peculiarity of these stamps is the transfer of low molecular weight fragments of PDMS during the printing process, which can contaminate the substrate. Investigations concerning this transfer have shown its almost complete elimination using oxygen plasma treatment to seal the surface of the stamp by oxidizing PDMS.46 We used the same process with comparable parameters prior to printing and additional experiments regarding this problem were conducted (see Fig. S1, ESI†).
The ink used for this process should possess two important properties: a high adhesion to the silica surface and easily accessible active chemical groups for further functionalization. PEI fulfils both preconditions. This polyelectrolyte possesses a significant density of amino groups for potential chemical modification. The high density of positive charge yields the polycationic character, which is an excellent counterpart for negatively charged surfaces like the surface of silica particles.47–51 This opens up the way for a strong electrostatic binding between the two materials, even stable after excessive cleaning and dispersing in an ultrasonic bath. Additionally, it is an economic, well-studied compound with a wide range of applications and interesting buffering and structuring properties.52–58
Monolayer preparation of 5 μm silica particles was conducted using drop casting onto glass substrates from ethanol dispersions. Silica particles also possess a wide variety of possible surface functionalizations and particles in a size range starting from 50 nm up to 5 μm are commercially available.59–65 Compared to polymer particles, they have a significant resistance against various solvents, increased temperature and possess a good colloidal stability.66,67 The last property is important for enabling a controlled assembly of functionalized particles. Self-assembly should only be induced in a specific and controlled manner.
Earlier studies on microcontact printing of particles reported different printing pressures to control patch diameter.29 This is, in particular, possible by changing the mechanical properties of the stamp via altering the monomer:crosslinker composition. Although changing the mechanical properties of the stamp material is an elegant way, the method proposed here can even go beyond this.
To achieve a broader diversity of patch geometry, we desisted of using a delicate pressure control during printing and kept the mechanical properties of the stamp constant, but changed the thickness of the polymer ink layer on the PDMS stamp. This strategy would not be possible using molecular inks. Furthermore no additional washing step or temperature dependent reaction is necessary to maintain high patch precision after particle release.29,68 Due to the electrostatic interactions between the negative PDMS surface and the PEI, the insufficient solubility of latter and the interactions of long polymer chains, the ink remains on the stamp during particle release.
The last but essential step of our approach is the particle release procedure from the PDMS stamp. Ultrasonic treatment is a common technique to remove particles from a sticky surface or a loaded stamp, but the correct choice of solvent affects the final structure of the patch (see Fig. 1b).69
Even high molecular weight PEI has a sufficient solubility in ethanol which causes the detachment of the silica particles from the stamp, leaving a 2D polymeric layer on them. Hereby the solvent is able to overcome the cohesive forces inside the PEI film, dividing it into two parts. In contrast to this acetone will not dissolve the ink film and due to strong cohesion forces, the internal film structure persists. The higher negative surface charge of the particle compared to the PDMS stamp surface finally leads to the higher adhesion of ink film to the former. This peculiarity together with the low solubility of PEI in this solvent causes a complete release of the PEI film adhered to the particle, yielding 3D structures on the particle surface. These two cases are considered below in detail.
Patches in 2D
Releasing the particles after μCP in ethanol is a fast and easy procedure. Due to the solubility of PEI in ethanol the particles detach from the PDMS stamp while a thin PEI layer remains on the particle surface.Fig. 3 displays fluorescence microscopy images and corresponding SEM pictures of patchy particles which were printed with four increasing PEI concentrations, 1, 2, 3 and 4 wt%, and then released using ethanol. A patch diameter distribution for these experiments was calculated on the basis of numerical processing of fluorescent images taken for at least 60 particle patches. The corresponding statistics for the 3 wt% sample and for other ink concentrations are shown in Fig. 3 and Table 1, respectively. Additionally, we calculated the area fraction as the ratio of patch area and particle surface. This value is an important factor for possible future assembly experiments and should give an impression of the area occupied by the PEI patch on the particle.
| PEI [wt%] | d e-LPEI [μm] | f e-LPEI [%] | d a-LPEI [μm] | f a-LPEI [%] | d a-BPEI [μm] | f a-BPEI [%] |
|---|---|---|---|---|---|---|
| 1 | 1.8 ± 0.2 | 7.2 | 1.8 ± 0.2 | 7.2 | 1.8 ± 0.3 | 7.2 |
| 2 | 2.0 ± 0.2 | 8.0 | 2.2 ± 0.4 | 8.8 | 2.1 ± 0.3 | 8.4 |
| 3 | 2.2 ± 0.3 | 8.8 | 2.4 ± 0.3 | 9.6 | 2.3 ± 0.3 | 9.2 |
| 4 | 2.4 ± 0.3 | 9.6 | 2.6 ± 0.3 | 10.4 | 2.4 ± 0.4 | 9.6 |
The average patch diameter increases from 1.8 μm to 2.4 μm upon increasing the PEI film thickness. All samples exhibit rough patch surfaces, allowing the assumption that the polymer in the patch was slightly dissolved or at least swollen during the process with some chains protruding outwards the patch surface. Only the 1 wt% sample features a smooth patch, enabling the suggestion that the detachment of the entire PEI film took place. The data for the film thickness measurements (Fig. 2) show that, the PEI film on the stamp for this ink concentration is 25 nm thick. For higher PEI concentrations, the stratification of the ink film on the stamp can occur resulting in the separation of the initial film into two parts – one on the particle surface and other residual one – on the stamp. Thus, the thickness of the layer in the case of 1 wt% PEI could be considered as a lower threshold for a complete detachment of the PEI film. This indicates further that the overall thickness of 2D patches for other ink concentrations should also be around 25 nm.
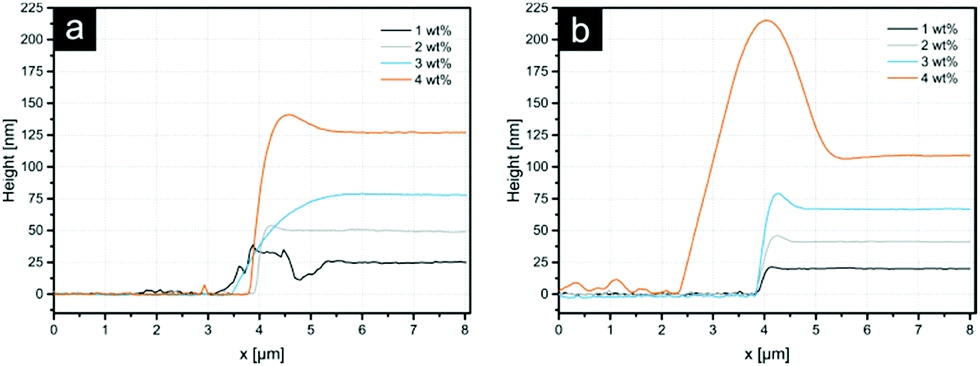 | ||
| Fig. 2 Diagrams show the film thicknesses resulting from differently concentrated linear (a) and branched (b) PEI solutions. The measurements were conducted by spin coating aqueous PEI solutions on silica wafer. Next the film was scratched using a cannula and the thickness was measured using SFM. | ||
In spite of an apparent similarity of the described printing procedure with techniques reported earlier, the amount of accessible groups is increased compared to molecular inks, which is of great importance for further developments. Additionally, this method can be considered as another proof of patch composition (PEI), evidencing the reaction of the fluorescence dye FITC with the amino groups of the polyelectrolyte.
Patches in 3D
The next step is the fabrication of new 3D structures on silica particles by microcontact printing. The corresponding results for the patchy particles released from stamps in acetone are shown in Fig. 4. Again, the patch diameter distribution was obtained by numerical measurements of the fluorescence zone dimension on the images collected for over 100 patchy particles. These results for the samples printed with 3 wt% LPEI and BPEI are shown in Fig. 4 (c′ and g′ parts, respectively). The comprehensive data for PEI types, concentrations used and area fraction are displayed in Table 1. Because PEI is practically insoluble in acetone, the release time in the ultrasonic bath had to be increased to attain the complete release of the patchy particles.The sample printed with a stamp coated by 1 wt% LPEI has a strong resemblance with the sample released in ethanol (compare Fig. 3). Patch diameter, thickness and roughness observed via SEM are alike; showing that for both samples, released in ethanol and acetone, a full detachment of the PEI film takes place.
 | ||
| Fig. 3 Fluorescence microscopy and SEM images of patchy particles with 2D patches released in ethanol printed with different LPEI concentrations (a–d). Calculated patch diameter distribution of the 3 wt% LPEI sample (c′). | ||
But when the thickness of LPEI films grows the difference between 2D and 3D patches becomes significant. The patch thickness increases with simultaneous changes in the patch morphology: from a flat ‘coin-like’ morphology for patches from 2 and 3 wt% LPEI to a ‘cap-like’ one for the sample printed from 4 wt% LPEI. This morphological alteration is also clearly observable with fluorescence microscopy, giving an even better contrast between very distinct patch forms. Additionally, the patch diameter increases stronger compared to the particles released in ethanol, reaching a value of 2.6 μm for the 4 wt% sample (see Table 1).
For further quantitative investigation of 3D PEI patches, dried PDMS stamps after particle release were examined using SFM. During the release process the particles tear off PEI patches, leaving a surface with indentations. The surface furthermore exhibits cracks in the polymer film, which are most likely a consequence of the swelling of PDMS in acetone. During this process, the PEI film could become ruptured. The corresponding SFM images can be found in the ESI† (see Fig. S2 and S3 for LPEI and BPEI, respectively). It is to say that comparable investigations for the thickness of 2D patches released in ethanol could not be conducted. The PEI film on the PDMS stamps, after release in ethanol, did not show sufficient indentations for this kind of measurement. This is another good indication for the predicted release mechanism in ethanol: the PEI film is slightly dissolved and due to this compensating for any dislodged layer of PEI.
Average depths of indentations were compared with the thicknesses of PEI films obtained by spin coating on silicon wafers (see Fig. 2). These results are summarized in Table 2.
| PEI [wt%] | t LPEI [nm] | DepthLPEI [nm] | t BPEI [nm] | DepthBPEI [nm] |
|---|---|---|---|---|
| 1 | 25 ± 1 | 23 ± 5 | 20 ± 2 | 16 ± 3 |
| 2 | 50 ± 2 | 48 ± 4 | 42 ± 3 | 34 ± 3 |
| 3 | 85 ± 2 | 82 ± 2 | 68 ± 2 | 62 ± 2 |
| 4 | 127 ± 3 | 124 ± 6 | 107 ± 4 | 107 ± 5 |
As one can see, there is a good agreement between the two sets of data confirming the complete removal of PEI films during the particle release. The entanglement and the H-bond caused cohesion of the long PEI chains, used in this approach, is strong enough to generate stable patch structures and overcome the adhesion between the polyelectrolyte and the negative PDMS stamp surface upon the particle release.
To compare the difference between linear and branched PEI, analogous experiments were conducted using BPEI with the same molecular weight and similar four film thicknesses originating from 1, 2, 3 and 4 wt% solutions. Fluorescence microscopy, SEM images and the patch diameter distribution of the 3 wt% sample are displayed in Fig. 4.
 | ||
| Fig. 4 Overview of particles with 3D patches consisting of linear and branched PEI released in acetone: fluorescence microscopy and SEM images of patchy particles printed with 1, 2, 3 and 4 wt% LPEI (a–d) and BPEI (e–h). The calculated patch diameter distribution of the 3 wt% LPEI and BPEI sample (c′ and g′). | ||
The structure of the 3D BPEI patches varied slightly from the ones obtained for LPEI. The patch diameter and thickness demonstrate the increasing dependences of the BPEI concentration in the coating solution with almost identical slopes but lower absolute values than in the case of LPEI (see Tables 1 and 2). The morphology of patches exhibited some integrity defects and rough surfaces, especially for the 3 and 4 wt% samples. Additionally, the exposed silica surface of the 4 wt% sample in Fig. 4h seems to obtain a very thin polymeric layer which was not observed for the 4 wt% LPEI sample in Fig. 4d.
As mentioned previously, the film thicknesses of PEI and the indentations on the PDMS stamp after particle release were measured and their values for the branched PEI lay below the corresponding values for its linear counterpart (see Table 2). With this, smaller patch diameter and thickness could be explained, but changes in the morphology and stability of the 3D patches can only be accounted for the branched structure of the polymer leading to lower viscosity, slightly higher solubility in the washing solvent (ethanol), due to the less pronounced interactions of the polymer chains compared to linear PEI.45
The achieved results for patch diameters and thicknesses are summarized in Tables 1 and 2 respectively. The average patch diameter increases steadily with higher PEI concentration for all systems. Switching from ethanol to acetone enhances the increase of the patch diameter even further. The shift from linear to branched PEI decreases the patch diameter slightly. Only the 1 wt% sample exhibits the same patch diameter for all systems. As mentioned earlier, this could be due to the very thin PEI film on the stamp at this concentration. The film will be completely removed despite the use of ethanol or acetone. The results displayed in Table 2 show that the thickness of 3D structures consisting of LPEI and BPEI is in good agreement with the film thicknesses of LPEI and BPEI measured by SFM on silica wafers.
The benefit of particles with novel 3D patches produced by our new approach is the possibility to selectively functionalize particle surfaces combining different physical and chemical properties:
3D patches give rise to a new dimension of utilizable space which can be used to incorporate various nanoadditives with physical functionalities into the patch volume without affecting the chemical properties of its surface. Nanoparticles with appropriate surface charge can easily be blended in the polyelectrolyte ink prior to spin coating. Upon subsequent printing, 3D patch structures with included physical and controlled structural properties can be available. Furthermore, a suitable chemical functionalization only of the patch surface can be selectively made, keeping the remaining particle surface unaffected. Independent physical and chemical modification of a single patch on an anisotropic particle can be produced.
Before further modifications were made, fluorescence microscopy experiments were conducted to exclude any factors that could lead to non-specific aggregation of the patchy particles. For this, 4 μm silica particles were printed with a 3 wt% LPEI coated stamp and labelled subsequently using FITC. These particles were brought to a solvent–air interface where they were driven into close proximity of each other due to the movement of the surface caused by surface forces. Eight images over a 4 s time period are shown in Fig. S4a (ESI†): even upon contact, the particles were still able to freely rotate showing no aggregation in any of the three possible contact scenarios, namely silica to silica, LPEI to LPEI or silica to LPEI. Especially the last possibility seems surprisingly taken into account the positive and negative charge of the contacting surfaces. Still, this interaction is apparently not strong enough compared to the kinetic energy of the microparticle motion to induce aggregation of silica particles of this size. Fig. S4b (ESI†) displays microscope pictures of patchy particles at a high concentration dispersed in water. Even under these conditions, no agglomeration of the particles is visible. Additional experiments with smaller particles suggest a critical particle size, at which assemblies emerge due to the oppositely charged patches and surfaces, which will be a part of future research.
Multifunctionality
To illustrate the versatility of our approach mentioned in the previous section, we designed an experiment utilizing the volume of 3D patches and the high density of functional groups on their surface. Fig. 5a presents schematically the pathway of this experiment.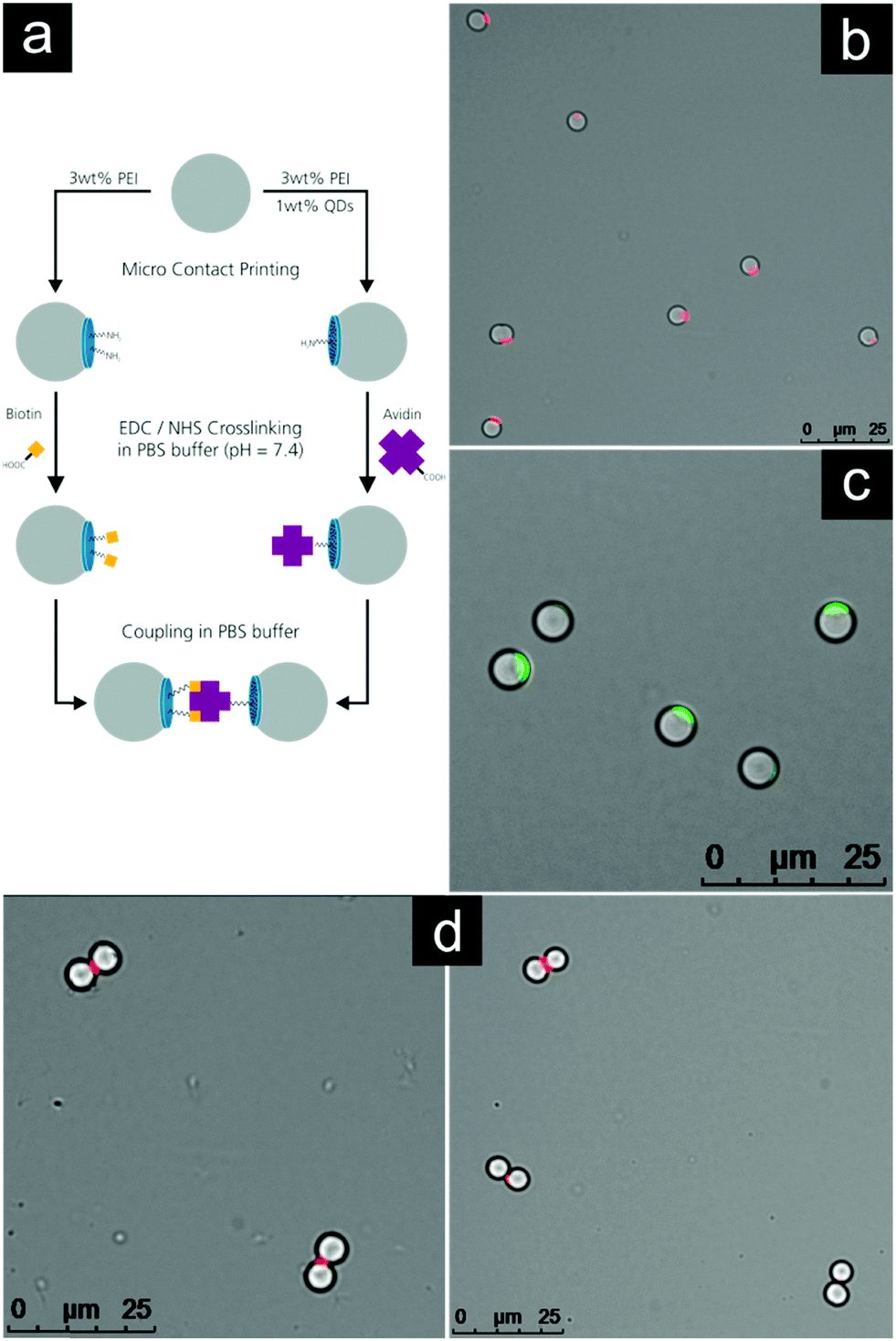 | ||
| Fig. 5 Scheme and fluorescence microscopy pictures of the self-assembly experiment of 3D patchy particles. (a) Synthesis route for particles with avidin and biotin functionalized patches and there self-assembly. (b) 3 wt% LPEI patch with 1 wt% QDs. (c) 3 wt% LPEI patch labelled with FITC. (d) Patchy particles self-assembled into heterodimers. | ||
Firstly, two different species of patchy particles were created. Both species were produced using a 3 wt% LPEI stamp and were released in acetone. One species was 3D-labelled using the volume of the patches, by blending a small amount of fluorescent quantum dots into the LPEI solution prior to spin coating. Due to a negative surface charge, the nanoparticles distributed very easily forming a stable dispersion. No differences in properties were observed for this quantum dot labelled sample comparing with the pure PEI during μCP.
Fig. 5b shows the quantum dot labelled patchy particles. The fluorescence contrast is very good and no leakage of the quantum dots into the surrounding solvent was observed. This point is important for further steps, so that one labelled species does not lead to the contamination of a second species in spite of negatively charged quantum dots, which could bind to free PEI patches. This fluorescence functionality can easily be incorporated into the 3D patches without any modification of functional groups on its surface. Fig. 5c shows a sample of the second species labelled with FITC to demonstrate the successful printing of the particles.
Secondly, the two species were functionalized with avidin and biotin, respectively. The established EDC/NHS method was used to activate the carboxylic acid groups of avidin and biotin with a reactive NHS ester, coupling these to the amine groups of the patchy particles.70,71 Fluorescence images in PBS-buffer were taken to verify that no self-assembly or agglomeration of the two distinctly functionalized species took place at this stage (see Fig. S5 and S6, ESI†). Finally both species were joined and mixed for 10 min. After that the sample was investigated using fluorescence microscopy. The results are shown in Fig. 5d.
Due to the strong binding forces between avidin and biotin, the functionalized patchy particles were self-assembled into heterodimers. It is clearly visible that the particles bind along the line connecting their centers which is perpendicular to the quantum dot labelled avidin patch.
Conclusions
A novel version of the microcontact printing approach for simple preparation of microparticles with 2D and multifunctional 3D patches is introduced. 2D or 3D structures, their diameter and thickness can be precisely controlled by the appropriate choice of polymeric ink concentration and its solubility in a releasing solvent while keeping the mechanical parameters of the process constant. The chemical structure of polymers used in the inks provided an additional possibility to adjust the structure and morphology of resulting patches.The 3D structure of patches enables new pathways for orthogonal functionalization of microparticles including not only the chemical surface modification of patches but also their versatile physical modification by embedding the corresponding nanoadditives in their bulk.
Another advantage of the proposed method is its operational simplicity opening up ways to easy-to-use upscaling procedures and therefore to higher yields of multifunctional patchy micro- and nanoparticles.
A statistical evaluation of the coupling into heterodimers showed a binding efficiency of up to 55% (compare Fig. S7, ESI†). This is an impressive value considering that the formation of a heterodimer can only be accomplished if the patches of two particles with different functionalization encounter, which is difficult due to the Brownian motion of silica particles of this size and at this low concentration. This again underlines the high accessibility and binding ability of our functionalized 3D patches.
Materials and methods
Chemicals
Poly(dimethylsiloxane) (PDMS) was prepared from the Sylgard 184 elastomer kit obtained from Dow Corning. 50 wt% aqueous solutions of linear and branched polyethyleneimine (LPEI/BPEI) were purchased from Fluka Chemicals and Sigma Aldrich, respectively, with a molecular weight distribution from 600 to 1000 kg mol−1 for both polyelectrolytes. Dry silica particles with a size of 5 ± 0.44 μm were purchased from Bang Laboratories and with a size of 4 ± 0.11 μm from Cospheric. Ethanol was purchased from Th. Geyer (reinst), acetone from VWR (ACS reagent) and dimethylsulfoxide (DMSO) from Merck (ACS grade). Fluorescein isothiocyanate (FITC) was obtained from Sigma Aldrich. Salt free avidin was purchased from Thermo Scientific and biotin from Applichem. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) from Fluka and N-hydroxysuccinimide (NHS) from Sigma Aldrich were used. InP based core–multi-shell quantum dots were provided by the department Functional Materials and Devices of Fraunhofer IAP. The quantum dots consist of an InPZnS hybrid core covered with one monolayer of ZnSe and six monolayers of ZnS shells.72 The ligand shell consisted of mercaptopropionic acid molecules which were attached to the quantum dot surface via the thiol group. The peak emission wavelength was 618 nm with a full width half maximum of 68 nm. The quantum yield of the quantum dots was 34%.PDMS stamps
Poly(dimethylsiloxane) (PDMS) was synthesized on the basis of an elastomer kit, which consists of monomer and curing agent. To produce flat PDMS stamps a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of monomer and curing agent was poured into a clean, plane Petri dish to obtain a 3 mm thick film. The film was degassed over night under ambient conditions to remove all enclosed air bubbles and cured for 2 h at 80 °C. The crosslinked PDMS was cut into 1 × 1 cm2 pieces for further use.
:1 mixture of monomer and curing agent was poured into a clean, plane Petri dish to obtain a 3 mm thick film. The film was degassed over night under ambient conditions to remove all enclosed air bubbles and cured for 2 h at 80 °C. The crosslinked PDMS was cut into 1 × 1 cm2 pieces for further use.
Polymer ink films
PEI solutions were further diluted with Milli-Q water to obtain desired concentrations of 1, 2, 3 and 4 wt%. To achieve fluorescent 3D patches 1 wt% of quantum dots was added to the solution (with respect to the polymer weight). The surface of flat PDMS stamps was activated using air plasma treatment (60 s, 300 W, 0.2 mbar/PlasmaFlecto 10). The activated PDMS stamps were loaded with polymer ink by spin coating 60 μL PEI solution of the desired concentration (60 s at 4000 rpm/Laurell WS-650-23B).Silica particle monolayers
The 4 μm and 5 μm silica particles were dispersed in ethanol at concentrations of 3 wt% and 4 wt% respectively, using an ultrasonic bath for 10 min (37 kHz/Elmasonic P). Microscope slides (VWR) were cut into 1 × 1 cm2 pieces, cleaned with ethanol, dried under nitrogen and activated using air plasma treatment (60 s, 300 W, 0.2 mbar/PlasmaFlecto 10). Particle monolayers were obtained by drop casting 5 μL of particle dispersion onto the glass substrates. Possible multilayer buildups were removed during additional microcontact printing steps.Microcontact printing
The fabrication of particles with 2D or 3D patches was conducted as follows: a silica particle monolayer was formed on a glass substrate fixated horizontally using a vacuum sample holder. A loaded PDMS stamp was brought into contact with the particle monolayer and pressure was applied (∼1.3 × 104 Pa/see Fig. S8, ESI†). During separation of the stamp and glass substrate, particles were embedded into the PEI film and were removed with the PDMS. Afterwards the loaded stamp was immersed into 2 mL of the solvent, choosing ethanol for 2D and acetone for 3D patches, and the particles were released using an ultrasonic bath (37 kHz/Elmasonic P). Due to the different solubility of PEI in the chosen solvent, time needed for particle release was changed from 10 min to 25 min for ethanol and acetone, respectively. Particles with 2D or 3D patches were collected and concentrated for further applications using centrifugation.Patch functionalization
Fluorescent labelling of patchy particles for microscopy was conducted as follows: a sample of released particles was centrifuged and re-dispersed in a 1 wt% FITC ethanol solution. After an incubation time of 15 min the sample was washed three times in ethanol by centrifugation and subsequent re-dispersion using an ultrasonic bath. 3D patches were functionalized with avidin and biotin by EDC/NHS-crosslinking: 1 mg of EDC was dissolved in 10 μL of PBS-buffer (pH = 7.4), 2 mg NHS in 20 μL DMSO and 1 mg avidin or biotin was dissolved in 50 μL PBS-buffer. A released particle sample was collected and then re-dispersed in 100 μL of PBS-buffer. The EDC, NHS and avidin/biotin solution were mixed into additional 100 μL of PBS-buffer. After 15 min the particle solution was added to this mixture and the vessel was shaken for 2 h (400 rpm/Hettich Benelux). Finally, the functionalized particles were washed three times with PBS-buffer to remove remaining reagents.Characterization
Scanning electron microscopy (SEM) images were taken using a JSM6330F from JOEL at an acceleration voltage of 5 kV. Before measuring, the samples were sputtered with platinum (4 nm thickness). Fluorescence microscopy (FL) pictures were taken using DMi8 from Leica at different magnifications (20×/40×/63× dry and 100× oil objective). For image processing LAS X software (Version 2.0.0) from Leica was used. Scanning force microscopy (SFM) images were taken with a Bruker Dimension Icon using Tapping Mode with OTESPA tips (k = 42 N m−1, f0 = 300 kHz). Nanoscope (Version 9.1) and Nanoscope Analysis (Version 1.5) were used for measurements and for the image processing, respectively. X-ray photoelectron spectroscopy (XPS) measurements were carried out using an AXIS165 instrument (Kratos Analytical, UK). Monochromatic AlKα radiation (300 W) was used for excitation. The instrument was run in electrostatic mode and thermal electrons from a filament were used to neutralize the sample charges. CASA-XPS software (2.3.16) was used for data processing. All quantification was carried out after subtracting a Shirley background and Gaussian–Lorentzian functions (30% Lorentz) were used for peak fitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank S. Grunst and Dr M. Pinnow for help with the SEM measurements, Dr A. Holländer for the XPS measurements and the department of Functional Materials and Devices of Fraunhofer IAP for providing the used quantum dots. The research leading to these results has received funding by the European Research Council (Replicoll; Project 648365) and the Volkswagen foundation (FR: 91 791).References
- A. van Blaaderen, Nature, 2006, 439, 545–546 CrossRef CAS PubMed.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- S. Jiang, Q. Chen, M. Tripathy, E. Luijten, K. S. Schweizer and S. Granick, Adv. Mater., 2010, 22, 1060–1071 CrossRef CAS PubMed.
- Q. Chen, S. C. Bae and S. Granick, Nature, 2011, 469, 381–384 CrossRef CAS PubMed.
- Y. Iwashita and Y. Kimura, Sci. Rep., 2016, 6, 27599 CrossRef CAS PubMed.
- Z. Zhao, Z. Shi, Y. Yu and G. Zhang, Langmuir, 2012, 28, 2382–2386 CrossRef CAS PubMed.
- K. B. Shepard, D. A. Chrstie, C. L. Sosa, C. B. Arnold and R. D. Priestley, Appl. Phys. Lett., 2015, 106, 1–5 CrossRef.
- Y. Wang, A. D. Holingsworth, S. K. Yang, S. Patel and D. J. Pine, J. Am. Chem. Soc., 2013, 135, 14054–14067 Search PubMed.
- E. Elacqua, X. Zheng and M. Weck, ACS Macro Lett., 2017, 6, 1060–1065 CrossRef CAS.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247–251 CrossRef PubMed.
- I. Schick, S. Lorenz, D. Gehrig, A.-M. Schilmann, H. Bauer, M. Panthöfer, K. Fischer, D. Stand, F. Laquai and W. Tremel, J. Am. Chem. Soc., 2014, 136, 2473–2483 CrossRef CAS PubMed.
- D. J. Kraft, R. Ni, F. Schmallenburg, M. Hermes, K. Yoon, D. A. Weitz, A. van Blaaderen, J. Groenewald, M. Dijkstra and W. K. Kegel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10787–10792 CrossRef CAS PubMed.
- R. Brüx, S. Hiltl, V. Schröder, C. von Essen and A. Böker, Part. Part. Syst. Charact., 2014, 31, 871–878 CrossRef.
- Y. K. Takahara, S. Ikeda, S. Ishino, K. Tachi, K. Ikeue, T. Sakata, T. Hasegawa, H. Mori, M. Matsumura and B. Ohtani, J. Am. Chem. Soc., 2005, 127, 6271–6275 CrossRef CAS PubMed.
- S. Jiang and S. Granick, Langmuir, 2008, 24, 2438–2445 CrossRef CAS PubMed.
- S.-H. Hu and X. Gao, J. Am. Chem. Soc., 2010, 132, 7234–7237 CrossRef CAS PubMed.
- M. Sabapathy, S. D. C. Pushpam, M. G. Basavaraj and E. Mani, Langmuir, 2015, 31, 1255–1261 CrossRef CAS PubMed.
- O. Cayre, V. N. Paunov and O. D. Velev, J. Mater. Chem., 2003, 13, 2445–2450 RSC.
- T. T. Chastek, S. D. Hudson and V. A. Hackley, Langmuir, 2008, 24, 13897–13903 CrossRef CAS PubMed.
- S. Sacanna, L. Rossi and D. J. Pine, J. Am. Chem. Soc., 2012, 134, 6112–6115 CrossRef CAS PubMed.
- B. Bharti and O. D. Velev, Langmuir, 2015, 31, 7897–7908 CrossRef CAS PubMed.
- K. Chaudhary, J. J. Juárez, Q. Chen and S. Granick, Soft Matter, 2014, 10, 1320–1324 RSC.
- S. Gangwal, A. Pawar, H. Kretzschmar and O. D. Velev, Soft Matter, 2010, 6, 1413–1418 RSC.
- C. W. Shields, S. Zhu, Y. Yang, B. Bharti, J. Liu, B. B. Yellen, O. D. Velev and G. P. López, Soft Matter, 2013, 9, 9219–9229 RSC.
- T. Kaufmann, M. T. Gokmen, C. Wendeln, M. Schneiders, S. Rinnen, H. F. Arlinghaus, S. A. F. Bon, F. D. Prez and B. J. Ravoo, Adv. Mater., 2011, 23, 79–83 CrossRef CAS PubMed.
- T. Kaufmann and B. J. Ravoo, Polym. Chem., 2010, 1, 371–387 RSC.
- S. Sagebiel, L. Stricker, S. Engel and B. J. Ravoo, Chem. Commun., 2017, 53, 9296–9299 RSC.
- T. Tigges, D. Hoenders and A. Walther, Small, 2015, 11, 4540–4548 CrossRef CAS PubMed.
- S. Jiang and S. Granick, Langmuir, 2009, 25, 8915–8918 CrossRef CAS PubMed.
- B. Chen, Y. Jia, Y. Gao, L. Sanchez, S. M. Anthony and Y. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 18435–18439 CAS.
- Y. Gao and Y. Yu, Langmuir, 2015, 31, 2833–2838 CrossRef CAS PubMed.
- L. Hong, S. Jiang and S. Granick, Langmuir, 2006, 22, 9495–9499 CrossRef CAS PubMed.
- H. Bao, W. Peukert and R. N. K. Taylor, Adv. Mater., 2011, 23, 2644–2649 CrossRef CAS PubMed.
- P. D. J. Oostrum, M. Hejazifer, C. Niedermayer and E. Reimhult, J. Phys.: Condens. Matter, 2015, 27, 234105 CrossRef PubMed.
- D. B. Hall, P. Underhil and J. M. Torkelson, Polym. Eng. Sci., 1998, 38, 2039–2045 CAS.
- M. J. Owen and P. J. Smith, J. Adhes. Sci. Technol., 1994, 8(10), 1063–1075 CrossRef CAS.
- A. Kumar, N. L. Abbott, E. Kim, H. A. Biebuyck and G. M. Whitesides, Acc. Chem. Res., 1995, 28, 219–226 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- D. M. Yanker and J. A. Maurer, Mol. BioSyst., 2008, 4, 502–504 RSC.
- L. Tang and N. Y. Lee, Lab Chip, 2010, 10, 1274–1280 RSC.
- K. Efimenko, W. E. Wallace and J. Genzer, J. Colloid Interface Sci., 2002, 254, 306–315 CrossRef CAS PubMed.
- Y. Zhang, L. Ren, Q. Tu, X. Wang, R. Liu, L. Li, J.-C. Wang, W. Liu, J. Xu and J. Wang, Anal. Chem., 2011, 83, 9651–9659 CrossRef CAS PubMed.
- H. Hillborg and U. W. Gedde, Polymer, 1997, 39, 1991–1999 CrossRef.
- J. Genzer and J. Groenewald, Soft Matter, 2006, 2, 310–323 RSC.
- W. T. S. Huck, N. Bowden, P. Onck, T. Pardoen, J. W. Hutchinson and G. M. Whitesides, Langmuir, 2000, 16, 3497–3501 CrossRef CAS.
- B. A. Langowski and K. E. Uhrich, Langmuir, 2005, 21, 6366–6372 CrossRef CAS PubMed.
- S. Kobayashi, H. Shirasaka, K. D. Suh and H. Uyama, Polym. J., 1990, 22, 442–446 CrossRef CAS.
- C. K. Choudhury and S. Roy, Soft Matter, 2013, 9, 2269–2281 RSC.
- S. M. Notley and Y. K. Leong, Phys. Chem. Chem. Phys., 2010, 12, 10594–10601 RSC.
- R. Meszaros, L. Thompson, M. Bos and P. de Groot, Langmuir, 2002, 18, 6164–6189 CrossRef CAS.
- H.-J. Yun, H. Hong, J. Lee and C.-J. Choi, Mater. Trans., 2014, 55, 801–805 CrossRef CAS.
- C.-S. Cho, ISRN Mater. Sci., 2012, 2012, 1–24 CrossRef.
- S. D. Gioia and M. Conese, Drug Des., Dev. Ther., 2008, 2, 163–188 Search PubMed.
- J. Shi, B. Chou, J. L. Choi, A. L. Ta and S. H. Pun, Mol. Pharmaceutics, 2013, 10, 2145–2156 CrossRef CAS PubMed.
- Y. Ling, F. Qu, Q. Zhou, T. Li, Z. F. Gao, J. L. Lei, N. B. Li and H. Q. Luo, Anal. Chem., 2015, 87, 8679–8686 CrossRef CAS PubMed.
- I. N. Kurniasih, J. Keilitz and R. Haag, Chem. Soc. Rev., 2015, 44, 4145–4164 RSC.
- A. von Harpe, H. Petersen, Y. Li and T. Kissel, J. Controlled Release, 2000, 69, 309–322 CrossRef CAS PubMed.
- K. A. Curtis, D. Miller, P. Millard, S. Basu, F. Horkay and P. L. Chandran, PLoS One, 2016, 11(9), 1–20 Search PubMed.
- C. Y. Jung, J. S. Kim, H. Y. Kim, J. M. Ha, Y. H. Kim and S. M. Koo, J. Colloid Interface Sci., 2012, 367, 67–73 CrossRef CAS PubMed.
- H.-S. Jung, D.-S. Moon and J.-K. Lee, J. Nanomater., 2012, 2012, 1–8 Search PubMed.
- R. D. Badley, W. T. Ford, F. J. McEnroe and R. A. Assink, Langmuir, 1990, 6, 792–801 CrossRef CAS.
- K.-M. Kim, H. M. Kim, W.-J. Lee, C.-W. Lee, T. il Kim, J.-K. Lee, J. Jeong, S.-M. Paek and J.-M. Oh, Int. J. Nanomed., 2014, 9, 29–40 Search PubMed.
- J. Lee, J. S. Han and B. R. Yuo, J. Korean Chem. Soc., 2013, 34, 3805–3810 CrossRef CAS.
- S. A. McCarthy, G.-L. Davies and Y. K. Gun'ko, Nat. Protoc., 2012, 7, 1677–1693 CrossRef CAS PubMed.
- Q. Zhang, T. Zhang, J. Ge and Y. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS PubMed.
- M. Kobayashi, F. Juillerat, P. Galletto, P. Bowen and M. Borkovec, Langmuir, 2005, 21, 5761–5769 CrossRef CAS PubMed.
- X. Li, O. Niitsoo and A. Couzis, J. Colloid Interface Sci., 2013, 394, 26–35 CrossRef CAS PubMed.
- T. Kaufmann, M. T. Gokmen, S. Rinnen, H. F. Arlinghaus, F. D. Prez and B. J. Ravoo, J. Mater. Chem., 2012, 22, 6190–6199 RSC.
- N. Bowden, W. T. S. Huck, K. E. Paul and G. M. Whitesides, Appl. Phys. Lett., 1999, 75, 2557–2559 CrossRef CAS.
- S. K. Vashist, Diagnostics, 2012, 2, 23–33 CrossRef CAS PubMed.
- S. Hu and X. Gao, Adv. Funct. Mater., 2010, 20, 3721–3726 CrossRef CAS PubMed.
- C. Ippen, T. Greco and A. Wedel, J. Inf. Disp., 2012, 13, 91–95 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sm00163d |
| This journal is © The Royal Society of Chemistry 2018 |