
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DMA study of water's glass transition in nanoscale confinement
V.
Soprunyuk
and
W.
Schranz
 *
*
University of Vienna, Faculty of Physics, Physics of Functional Materials, Boltzmanngasse 5, A-1090 Wien, Austria. E-mail: wilfried.schranz@univie.ac.at
First published on 6th August 2018
Abstract
Dynamic mechanical analysis (DMA) measurements of water confined in nanoporous silica have been performed as a function of temperature and frequency for different pore sizes (2.5–10 nm) at heating and cooling. Most of the data show three processes, P1, P2 and P3, where P1 and P2 depend on measurement frequency and P3 does not. The characteristic shift of P3 with pore size shows that this process corresponds to freezing/melting of “internal water”, i.e. in the core of the pores. Thermal expansion data indicate – in agreement with e.g. [A. Taschin, P. Bartolini and R. Torre, Meas. Sci. Technol., 2017, 28, 014009] – that in all our nanoporous systems about 2 layers of water remain liquid much below the freezing point. Dynamic elastic measurements show clear signatures of glass freezing of this supercooled water in the vicinity of P1. Extrapolating the DMA data to the timescale (103 s) of adiabatic calorimetry unveils a systematic behaviour: P1(T) shows a clear size dependence for a broad range of pore diameters, i.e. 2.5 nm ≤ d ≤ 52 nm, implying (together with the corresponding activation energy 0.5 eV) that P1 corresponds to the glass–liquid transition of a few layers of supercooled water at Tg(d). An extrapolation of Tg(d) to d → ∞ yields Tg(∞) ≈ 136 K, the traditional value for bulk water. The small (liquid like) value of Young's modulus in a temperature region above P1 is most naturally explained assuming that the supercooled water in this range is still liquid, implying that Tg values of 160 K or even 210 K – as suggested by various authors – are unlikely.
I Introduction
Water is of fundamental importance for all living organisms as well as for abiotic environments. Despite its simple molecular structure when compared with other glass forming liquids, some important thermodynamic and kinetic properties are still rather controversially discussed.1 A vexed question relates e.g. to the location of its bulk glass transition Tg. In vapour-deposited amorphous solid water (ASW)2 as well as in rapidly quenched water below 100 K (hyperquenched glassy water HGW)3,4 a glass to liquid transition was reported from calorimetric measurements to occur at Tg = 136 K, which then was assigned to the bulk glass transition temperature of water.Later, Angell et al.5,6 proposed a much higher value of bulk Tg ≈ 165 K, based on the heat-release of HGW.
Unfortunately, the glass transition of water cannot be easily studied, since water can only be supercooled at most down to 235 K, the homogeneous nucleation temperature, below which it crystallizes. A common method to avoid crystallization is to put water into nanoscale confinement. E.g. Cerveny et al.7 studied water confined in vermiculite clay and bread by dielectric and DSC measurements, arriving at the conclusion that Tg ≈ 160–165 K.
Oguni et al.8–11 studied the glass transition of water in various nanoscale confinements (silica-gel nanopores with rather irregular pores and MCM-41 with regular pore structures) by adiabatic calorimetry, suggesting11 a value of Tg = 210 K for bulk water.
Despite all efforts, even after decades of research12,13 many properties of water are still heavily discussed. An important dispute relates to the question of a possible “fragile–strong” transition, triggered by an observed crossover from super-Arrhenius to Arrhenius behaviour around TFS ≈ 225 K. For bulk supercooled water a “fragile to strong” (FS) transition was proposed at TFS ≈ 228 K14,15 based on dielectric relaxation. A similar dynamic crossover was also found16–18 by different experimental techniques for confined supercooled water and there is an ongoing discussion1 about the origin of such a crossover.
A possible scenario relates the FS-crossover19,20 to a transition21 (liquid–liquid critical point) from a high density liquid (HDL at T > TFS) to a low density liquid (LDL at T < TFS). Another interpretation17 assumes that the dynamic crossover of confined water is due to a confinement induced crossover from a cooperative α-process to a Johari–Goldstein type local β-process. A possible explanation for such a change is that at the crossover temperature – observed in dielectric data at about 180 K – the characteristic length ξ of dynamically correlated regions becomes equal to the size d of the confinement, which limits the characteristic length at a constant value ξ(TFS) = d and as a result the high-temperature super-Arrhenius dependence (Vogel–Fulcher, power-law) of τα transforms to a low-temperature Arrhenius behavior τβ. This scenario would imply that the dynamic correlation length ξ of water should be in the range of 2 nm around TFS ≈ 180 K.
In another approach some authors show22,23 that a power-law ∼(T − Tx)−γ fits the observed super-Arrhenius behaviour of viscosity η or relaxation time τ better than a Vogel–Fulcher law. Such a power-law can be interpreted in several ways. One is mode-coupling theory,24 where Tx = Tc > Tg is the mode-coupling temperature. Another explanation25,26 suspects the existence of an order–disorder transition around Tx ≈ 225 K, which however cannot be reached due to prior crystallization. The power-law would then occur due to the appearance of precursor fluctuations, which would lead to a λ-type anomaly in the specific heat at Tx. Using the Adam–Gibbs equation27τ = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(C/TSc), the strong–fragile transition would then be a direct consequence of the corresponding configurational entropy change Sc.
exp(C/TSc), the strong–fragile transition would then be a direct consequence of the corresponding configurational entropy change Sc.
An idea which then was further perpetuated26 is that the glass transition in water around 136 K is nothing else than an ergodicity breaking transition (i.e. due to freezing of some remaining orientational degrees of freedom) appearing in the tail of the order–disorder transition, quite similar to that demonstrated28,29e.g. for C60.
Another idea, which is quite consistent with the previous one, was recently proposed30 on the basis of a study of the reorientation dynamics of water molecules in different ice phases. There the authors claim that the calorimetric signatures of the amorphous ices (around 136 K for LDA) that have been interpreted as a glass-to-liquid transition, occur due to unfreezing of re-orientation of water molecules and not due to unfreezing of long range molecular diffusion. The real glass transition – corresponding to unfreezing of translational diffusion of water molecules – would then occur at higher temperature, which the authors preliminarily associate with the fragile–strong transition at TFS.
Unfortunately we cannot measure the crossover from Arrhenius to super-Arrhenius behaviour with our DMA method, since we are restricted in frequency to at most 100 Hz. But from the value of the activation energy of the order of Ea ≈ 0.5 eV (see below), which is in very good agreement with previous dielectric data,1,7,31 we can conclude that one of our damping peaks (P1) corresponds to the same process as the denoted β-process.
Below we will show that we have strong evidence that this damping peak (P1) is not a local process, but originates from a glass-to-liquid transition of water in the nanopores. We mainly infer this from the fact that we observe a very pronounced softening in the real part of the complex Young's modulus, which starts in the temperature region of the damping peak P1 and extends to temperatures near the proposed TFS.
Although not so commonly used, dynamic mechanical analysis (DMA) turned out to be an excellent method32–34 for the study of confined glass forming liquids.
In the present work we show (Section 3) results of dynamic mechanical analysis (DMA) and thermomechanical (TMA) measurements of water confined in nanoporous silica, Vycor and Gelsil with pore diameters d = 10 nm (V10), 5 nm (G5) and 2.5 nm (G2), respectively, as a function of temperature (heating and cooling) and frequency (0.1–70 Hz).
Combining our data with adiabatic calorimetry data of Oguni et al.8–11 unveils a clear systematic behaviour concerning the thermal relaxation processes of water in nanopores over a pore size range of d = 1.2–52 nm, which is presented also in Section 3. In Section 2 we specify the host materials which are used to confine water and the DMA-method is briefly described. Section 4 concludes the paper.
II Experimental
Table 1 summarizes the main specifications of the SiO2-based nanoporous host materials, as obtained35–37 from N2 adsorption and SAXS measurements.| Properties | V10 | G5 | G2 |
|---|---|---|---|
| Pore size (nm) | 10 ± 0.5 | 5 ± 0.8 | 2.5 ± 1 |
| Porosity | 0.4 | 0.54 | 0.36 |
| Pore surface area/pore volume | 4.5 | 8.3 | 15 |
| Pore surface area (m2 g−1) | 90 | 510 | 590 |
| Surface fractional dimension Ds | 2.3 | 2.24 | — |
Vycor (Corning Inc., New York), is produced via phase separation within a Na2O–B2O3–SiO2 melt, and subsequent acid leaching, which leaves a 96% pure SiO2 skeleton containing interconnected cylindrical pores of random length and direction. A narrow pore size distribution and an average ratio of pore length l over pore diameter d of l/d ≈ 4 is reported.37 Gelsil samples result from a sol–gel process38 and consist of randomly formed pure fused SiO2 monodisperse spheres39 touching and penetrating each other. The voids between these spheres create a random network of inter-connected corridors and pockets and show a larger pore size distribution as compared to Vycor.
For DMA measurements macroscopic samples of a few mm3 are needed. A diamond saw was used to cut the samples of Vycor and Gelsil with typical sizes of 3 × 2 × 2 mm3. The samples were sanded to gain parallel surface plains. The geometrical accuracy of the polished samples was better than 10 μm. Then the samples were cleaned by first dropping them into a 30% H2O2 solution at 90 °C for 24 h, followed by drying at 120 °C in a high vacuum chamber also for 24 h. Filling of the samples with distilled water was done by spontaneous imbibition.40
With the effective volume of a water molecule in the liquid state (ca. 3 × 10−29 m3), a rough estimation yields a number of about 300 water molecules in 2.5 nm pores, 2400 in 5 nm pores and about 100000 water molecules in Vycor with 10 nm pores.
For thermal expansion measurements we used a TMA 4000 (Perkin Elmer) as well as a Diamond DMA (Perkin Elmer).
To study the slow dynamics of confined supercooled water, we performed Dynamic Mechanical Analysis (DMA) measurements using two devices (DMA 8000 and Diamond DMA, Perkin Elmer). With DMA, the real Y′ and imaginary Y′′ parts of the complex Young's modulus Y* = Y′ + iY′′ are determined from the measured sample strain and phase shift between the externally applied dynamic force and the sample strain. A force up to 10 N can be applied, with a resolution of 0.002 N. The resolution of the sample height is about 3 nm and the phase shift δ can be measured with an accuracy of about 0.1°. The relative accuracy of the DMA method is about 0.2–1%, but the absolute accuracy of such a measurement is usually not better than 20%. To obtain reasonable absolute values we have normalized our measured Y′-data at room temperature to the Young's modulus data previously measured35 by RUS (resonance ultrasonic spectroscopy). The Y′′ values are then obtained from the phase shift data δ using Y′′ = Y′tanδ.
The measurement frequency can be varied between 0.01 Hz and 100 Hz at temperatures between 80 K and 600 K. More details of the DMA method are given in ref. 41 and 42.
Here we present DMA and TMA results of water in Gelsil (2.5 nm, G2 and 5 nm, G5) and Vycor (10 nm, V10) (Table 1) measured at cooling and heating as a function of temperature and frequency.
Filling is done by making use of the strong capillary forces. To avoid the formation of air bubbles the sample should be attached to water only from one side. After full filling (controlled by a balance) the samples become transparent.
III Results and discussion
Thermal expansion of water confined in nanopores
Very recently, we have measured43 the mechanical relaxation behaviour of supercooled water in Vycor and Gelsil by quenching the samples down to 80 K, followed by subsequent heating at a rate of 1.5 K min−1. In the present work we use rather moderate cooling/heating rates of 2 K min−1.It is instructive to compare the data from quenching experiments with the present ones. As an example we show (Fig. 1) the thermal expansion behaviour of water in G2 for quenched samples and slowly cooled/heated (2 K min−1) samples. A very similar behaviour was also found for water confined in G5 and V10. It is evident that even in 2.5 nm pores some portion of ice is formed, most probably in the central part of the pores (see below). After quenching to about 80 K, the formation of ice takes a few minutes as can be seen in the inset of Fig. 1.
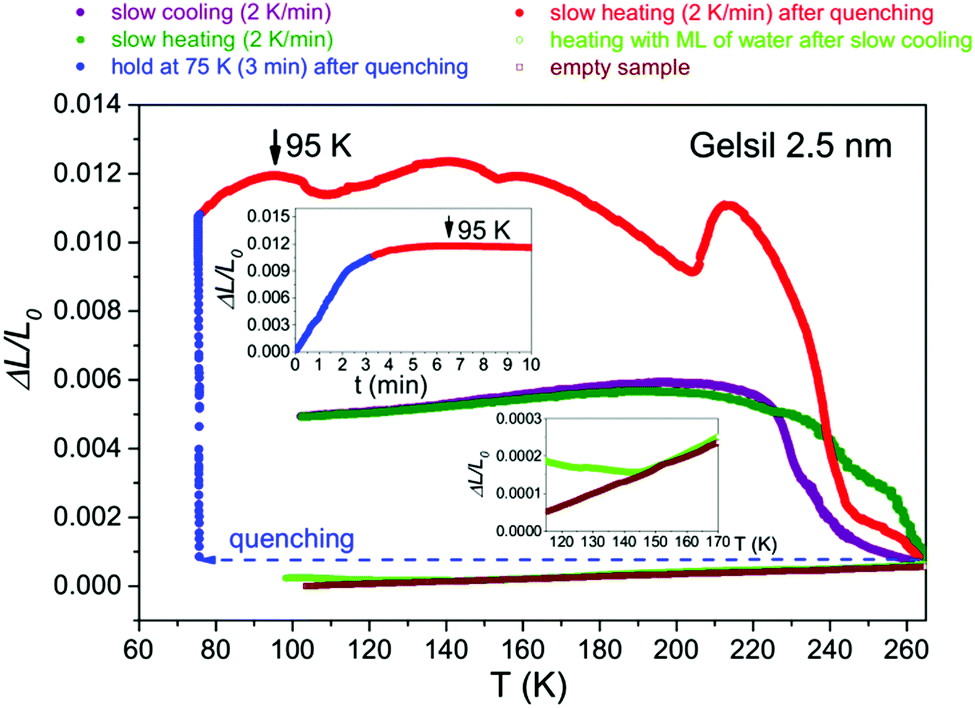 | ||
| Fig. 1 Thermal expansion of water in Gelsil G2 for quenched (red) and slowly cooled/heated (2 K min−1, green/violet) samples compared with partially filled Gelsil (light green) and empty Gelsil (brown). | ||
The big difference between quenched and slowly cooled samples in the magnitude of the anomalies suggests that in quenched samples a larger portion of water freezes to ice compared to the slowly cooled samples. A similar cooling/heating rate dependence of thermal expansion of water confined in Vycor (Corning no. 7930, pore diameter ca. 6 nm) was reported44 much earlier. The authors explained this time dependent expansion by a transport process inside the pores, which is needed to dissipate pressure generated by the ice formation. The pressure distribution of water confined in cylindrical pores of MCM41 was recently studied by molecular dynamics simulations45 in a temperature range from 298 K to 210 K. The author found that in this range water is under significant negative pressure. But the presence of ice in the core of the pores renders the situation much more complex and at present we cannot draw a final conclusion on the observed time dependence of thermal expansion and Young's moduli data.
In our recent work43 we used thermal expansion data of water confined in nanoporous silica measured at 2 K min−1 rates to estimate the relative amount of supercooled (Vsw) water for various pore sizes below the freezing point, yielding Vsw/Vpore ≈ 0.6, 0.3 and 0.1 for G2, G5 and V10, respectively.
If – in correspondence to a recent study46 of water in Vycor (4 nm pore size) – we assume that two layers of water remain liquid below the freezing temperature, and approximating the complex cavities of the present nanoporous silica matrices (Vycor and Gelsil) by cylindrical pores and the water molecules with a diameter of about 0.3 nm, we obtain Vsw/Vpore ≈ 0.7, 0.4 and 0.2 for G2, G5 and V10, respectively. Although these estimations are rather rough, they show that it is reasonable to assume that a few layers of supercooled water can take part in the glass freezing process of water in the presently used nanoporous silica. This is also in agreement with recent proton NMR spectroscopy47 of water confined in 1–10 nm sized pores.
Dynamic mechanical response of water in nanopores
In addition to thermal expansion, DMA measurements yield information about dynamical processes of supercooled water in the nanoporous silica host materials.Fig. 2 shows Y′ and Y′′ for water in Vycor and Gelsil for heating after cooling at 2 K min−1.
 | ||
| Fig. 2 Real Y′ and imaginary Y′′ parts of the complex Young's modulus of water in Gelsil 2.5 nm, 5 nm and Vycor 10 nm, measured at a heating rate of 2 K min−1 after cooling at a cooling rate of 2 K min−1. The inset shows a sketch of the different states of water according to the presumed core–shell model.46 | ||
Three peaks are visible in Y′′, denoted as P1, P2 and P3. The general behaviour is in excellent agreement with earlier mechanical relaxation measurements44 of water in Vycor (7930, d = 6 nm), measured at about 400 Hz. Inspecting the temperature variation of Y′ in Fig. 2 one observes that at about 100 K the Young's modulus has increased by more than 70% over the room temperature value (see also Fig. 3–5), whereas at about 200 K – where a large fraction of water has frozen – Y′ has only increased by some 10%. This is another signature of the effect of the liquid film of supercooled water that surrounds the ice core in the pores and decouples (with respect to elasticity) the ice from the silicon matrix.
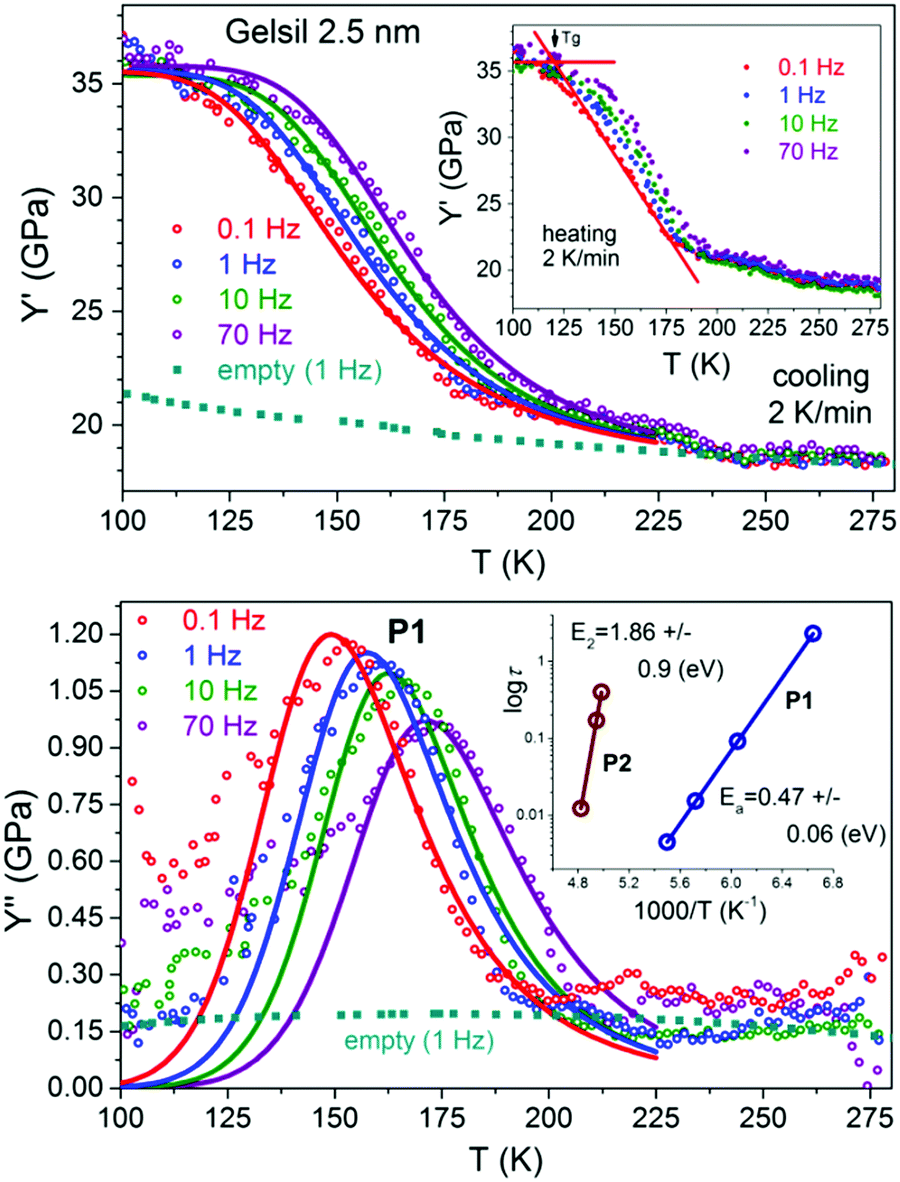 | ||
| Fig. 3 Temperature dependencies of Y′ and Y′′ of slowly cooled water in Gelsil 2.5 nm at different frequencies. The blue squares show results of empty samples. Insets show a corresponding heating run and an Arrhenius plot corresponding to P1 and P2. The activation energy E2 is determined from DMA measurements on quenched samples.43 The lines are fits using a Cole–Cole relaxation process eqn (1) and an Arrhenius dependence of the mean relaxation time, yielding Ea = 0.47 ± 0.06 eV, τ0 = 1.6 × 10−15 s, Y∞ = 36 GPa, ΔY = 18.3 ± 0.2 GPa and α = 0.28 ± 0.01. | ||
 | ||
| Fig. 4 Temperature dependencies of Y′ and Y′′ of slowly cooled water in Gelsil 5 nm measured at various frequencies. The blue squares show results of empty samples. The insets show heating runs at various frequencies and an Arrhenius plot corresponding to P1 and P2. The lines are fits using a Cole–Cole relaxation process eqn (1) and an Arrhenius dependence of the mean relaxation time, yielding Ea = 0.49 ± 0.07 eV, τ0 = 3.8 × 10−18 s, Y∞ = 37.5 GPa, ΔY = 27 ± 0.3 GPa and α = 0.22 ± 0.01. | ||
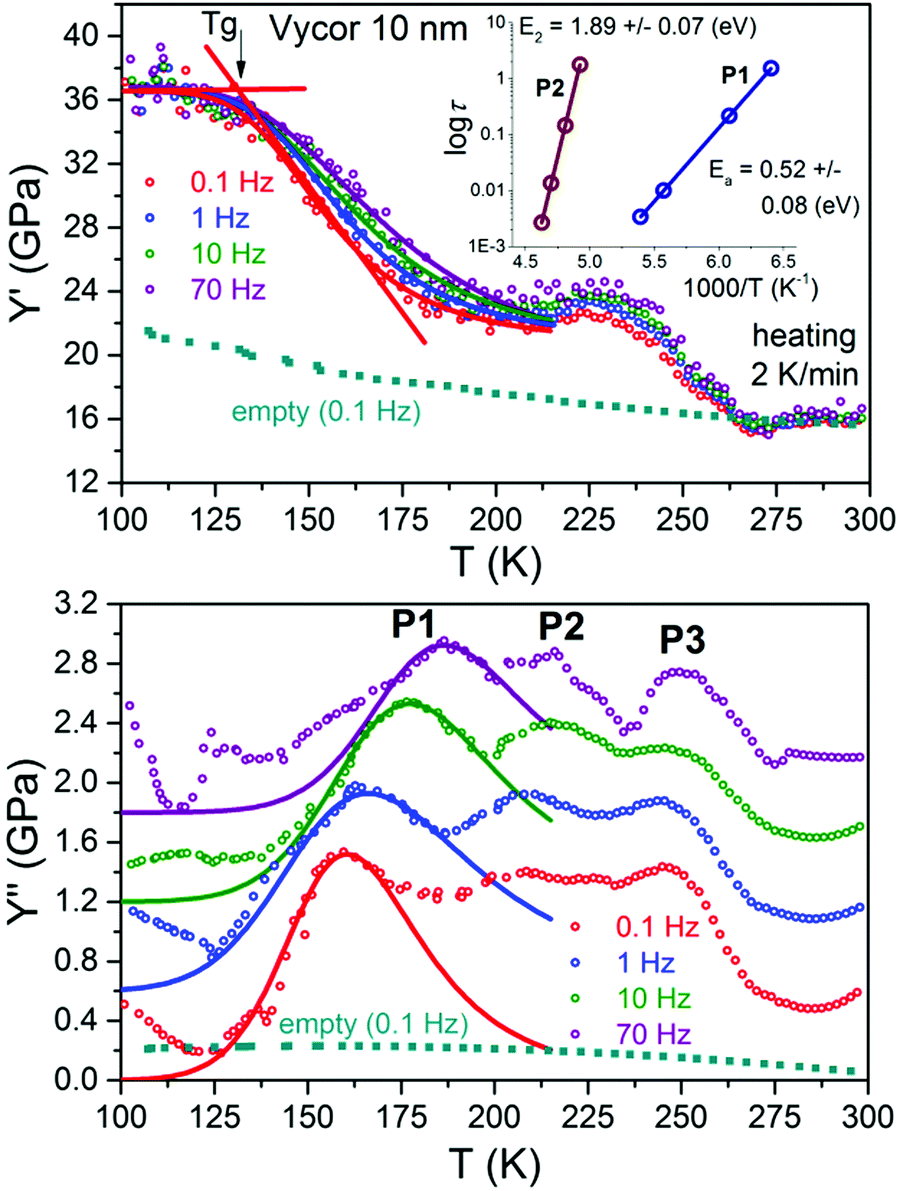 | ||
| Fig. 5 Temperature dependencies of Y′ and Y′′ of slowly cooled water at heating with a rate of 2 K min−1 in Vycor 10 nm measured at various frequencies. Note that in Y′′ the curves for 1, 10 and 70 Hz are shifted from the original 0.1 Hz data for clarity. The blue squares show results of empty samples. The inset shows Arrhenius plots corresponding to P1 and P2. The lines are fits using a Cole–Cole relaxation process eqn (1) and an Arrhenius dependence of the mean relaxation time, yielding Ea = 0.52 ± 0.08 eV, τ0 = 3 × 10−19 s, Y∞ = 36.3 GPa, ΔY = 14.2 ± 0.2 GPa and α = 0.2 ± 0.02. | ||
To get additional information on the dynamic processes of supercooled water in the pores we performed measurements at various frequencies. Fig. 3 shows the data for water in Gelsil 2.5 nm at frequencies between 0.1 and 70 Hz. Interestingly enough, the re-hardening process which was observed43 in the quenched samples prior to melting is strongly suppressed (for V10) or even absent (for G5 and G2) in slowly cooled samples. This has the advantage that the elastic softening in the vicinity of P1 becomes now more clearly visible, since it is no longer obscured by the re-hardening process of water in quenched samples. Similar to the quenched samples,43 the low temperature peak P1 shifts to higher temperatures with increasing frequency.
Before we discuss different possible origins of the relaxation peak P1 in more detail, let us see which relaxation function fits the data well. Following previous dielectric measurements of confined water7,48 we use a symmetric Cole–Cole (C–C) function
 | (1) |
exp(Ea/kBT).
It turns out that the Cole–Cole relaxation (1) fits the data as well as it can be expected. The real part of the complex Young's modulus is nearly perfectly described in a broad temperature range around P1. For the imaginary part deviations below and above P1 are visible, but also Y′′ can be well fitted for a considerable temperature range around the peak maximum.
The same quality of the fits has been found for G5 (Fig. 4) and V10 (Fig. 5). The agreement with the fits from dielectric data48 of confined water is remarkable. E.g.Table 1 of ref. 48 shows fit parameters for dielectric measurements of water confined in MCM-41 with pore diameters of 2.1 nm (C10) and 3.6 nm (C18). Our P1 should be identified with their process 2, which for C10 (2.1 nm) yielded an activation energy (for T ≤ 170 K) of Ea = 0.47–0.49 eV with τ0 = 2 × 10−18 s and a broadening parameter α = 0.45–0.48. Our values are nearly identical. Only our broadening parameter is a little smaller, which is not surprising, since Vycor and Gelsil have a broader pore size distribution compared to MCM-41.
To obtain the temperature dependence of the corresponding relaxation time, we used two different procedures, i.e. based on a simple analysis of the peak shifts with frequency and by fitting the data with a Cole–Cole relaxation function eqn (1). Since the Cole–Cole relaxation is symmetric, the mean relaxation time τ of eqn (1) can be determined either from fitting the data with eqn (1), or from the inverse of the peak angular frequency ωP−1 of Y′′.
With τP1 = τ01exp(Ea/kBT) and fP1 = 1/2πτP1 we obtain from the corresponding Arrhenius plot (inset of Fig. 3) an activation energy of Ea = 0.47 ± 0.06 eV. Thus P1 can be identified with the low temperature main dielectric relaxation of confined water which also shows Arrhenius behaviour with the same activation energy, regardless of the type of confinement (see e.g. Fig. 1 of ref. 7).
Fig. 4 and 5 show cooling/heating runs at 2 K min−1 at various frequencies for water confined in Gelsil 5 nm and Vycor 10 nm, respectively, and corresponding fits with eqn (1), yielding increasing activation energies with increasing pore size.
Inspecting Fig. 2–5 we find that the main difference between the quenching experiments43 and the slow cooling/heating runs occurs in Y′ in the temperature region between ≈180 K and the freezing/melting transition. However, the region of the glass transition is not influenced by the different cooling rates. From the frequency dependence of P1 we obtain the same activation energies around 0.5 eV and the same pore size dependence of P1 as for the quenched case.
Although the present data are very well reproducible and meaningful, due to the complexity of the problem interpretation of the data is not unambiguous and it is instructive to discuss them in the light of previous experiments and interpretations.
Comparison with adiabatic calorimetry
The glass transition temperature Tg is usually defined from differential scanning calorimetry (DSC) measurements by the onset of the heat capacity rise, which corresponds to the onset of the decay in Young's modulus Y′′ (see insets in Fig. 3–5). However, if the jump in heat capacity around Tg is small – as it is the case for water50 – other methods are more appropriate. Adiabatic calorimetry turned out to be a very powerful tool for the identification of glass transition temperatures of confined water.A comparison of our data with adiabatic calorimentry8–11 of water in various nanoporous systems in addition to the comparison with dielectric studies7,12,18,48 turns out to be very helpful for a discussion of the behaviour of water in nanopores. Oguni et al.8–11 measured the enthalpy relaxation dH/dt of water in two different confining systems. They used silica-gels8,9 with rather irregular pore structures and broad pore size distributions with average diameters† of 3, 6, 12 and 52 nm and MCM-4110,11 having regular cylindrical pores with diameters between 1.5 and 5 nm. The technique can be considered as a very low frequency spectroscopic method,51 where timescales of relaxation can be detected between 102 s and 106 s. The glass transition temperature is then determined at the point at which −dH/dt changes sign (for rapidly cooled samples) or shows a minimum (for slowly cooled samples). The so determined Tg value corresponds8 to a relaxation time τ = 103 s.
Very similar to our results, the authors observed up to three peaks, which we identify with our three peaks P1, P2 and P3, observed in Y′′ as follows (Fig. 6):
 | ||
| Fig. 6 Pore size dependencies of the processes P1, P2 and P3 from DMA measurements and adiabatic calorimetry data.8–11 | ||
P3 of our data results from the freezing/melting process of water/ice in the core of the pores. This can be verified e.g. by the observed downshift of P3 with decreasing pore size (in agreement with ref. 52 and 56), the observed anomalies in thermal expansion (ref. 43 and Fig. 1 of the present work) and the fact that P3 does not shift with frequency, as expected for a (first order) freezing/melting phase transition.
The processes P1 and P2 are also observed in adiabatic calorimetry.8–11 Our frequency dependent measurements (Fig. 3–5) show that both processes are thermally activated. As already mentioned above, the process P1, which for d < 2.5 nm occurs around 118 K, corresponds to the main (“universal”) dielectric relaxation of confined water, which by many authors12 is identified with the glass-to-liquid transition of supercooled water.
The frequency shift of P2 can also be fitted with an Arrhenius dependence of the relaxation time, i.e. τP2 = τ02exp(E2/kBT), but yields a much higher activation energy E2 ≈ 2–2.5 eV (Fig. 4 and 5) compared to P1. Moreover, its position is independent of pore size.
Extrapolating the relaxation times τPi to 103 s, we find that our P1 corresponds to the lower temperature peak (faster process) of −dH/dt, whereas P2 can be identified with the higher temperature peak (slower process) at ≈160 K (Fig. 6) of the adiabatic calorimetry data.8–11 This identification is not obvious from a first glance, because all peak positions depend strongly on the measurement time scale and the extrapolation to 103 s shifts our peaks P1 and P2 to lower temperatures.
In this way we find – by including the data of adiabatic calorimetry – a very interesting systematic behaviour (Fig. 6) of the processes P1, P2 and P3 with varying pore size.
As Fig. 6 shows, P3 definitely corresponds to the freezing/melting process of internal water/ice in nanopores, i.e. the slope dTf/d(1/d) ≈ 110 K nm fits well with previous findings.49,52 For pore sizes d > 2.5 nm also Oguni et al. identified this process (P3) with freezing/fusion of internal water/ice in the pores. But for pore sizes below ca. 2.5 nm, Oguni et al.10,11 found three broad peaks in −dH/dt, where the high temperature peak around 210 K led them to the conclusion that Tg = 210 K for bulk water. Inspecting Fig. 6, we strongly doubt this conclusion. Since these peaks around 210 K (see encircled data points of Fig. 6) are located at the extrapolated freezing line Tf(d), we think that the observed peak around 210 K results from freezing of water in small pores rather than a glass transition. Moreover, combining the available data of the two other processes, we find that P2 (extrapolated to 103 s) of our DMA measurements corresponds to the calorimetric anomaly observed at ≈160 K (Fig. 6).
Oguni et al. related their higher T process P2 around 160 K to the effect of glass freezing of “internal water” (water molecules surrounded only by water molecules inside the pores) in the pores, and the process P1 at lower temperature to “interfacial water”, whose molecules are bonded to the silanol groups of pore walls. Based on this interpretation, they concluded8 that the glass transition temperature Tg of bulk water is around 160 K or above and later they suggested10,11Tg of bulk water is even around 210 K, as already mentioned above.
However, inspecting Fig. 6, it is obvious that Oguni's data have to be reinterpreted: as already mentioned above, it is most likely that the data of Oguni et al. around 210 K (encircled data in Fig. 6) correspond to the freezing/melting process of internal water in small pores and are not related to the glass transition of water.
Moreover, based on the present data and recent computer simulations,53–55 showing that the dynamics of water molecules is dramatically slowed down with decreasing distance to the pore walls, we interpret the processes corresponding to P1 and P2 quite opposite to Oguni et al. We claim that P2 (at about 160 K) – whose corresponding τ has a much higher activation energy (ca. 2 eV) than P1 (ca. 0.5 eV) – corresponds to a relaxation dynamics of water molecules which are bound to silanol groups of pore walls and therefore considerably slowed down compared to water molecules away from the pore walls which are surrounded by other water molecules and thus experiencing much faster dynamics.53–55 It should be noted that Ngai [ref. 12, p. 470] came to the same conclusion. However, he argued – in agreement with other authors – that the process P1 around 115 K, which is caused by the relaxation of internal water, is not sensitive to the form of confinement or mixtures. Inspecting Fig. 6, one clearly observes that this is in fact the case only for pore sizes up to about 2.5 nm. For pore sizes above 2.5 nm a systematic pore size dependence of Tg ∼ T∞g − 1/d is found, which extrapolates to T∞g ≈ 136 K for d → ∞.
Comparison with dielectric spectroscopy and neutron scattering
Broadband dielectric spectroscopy was intensively used to study the dynamics of supercooled water in nanopores as well as in aqueous solutions. Ample literature can be found e.g. in ref. 1 and 13. An important finding of these experiments is that around Tcross ≈ 190 ± 20 K a dynamic crossover1,13,20 of the water dynamics of supercooled confined water from Arrhenius (β-process for T < Tcross) to Vogel–Fulcher (α-process for T > Tcross) behaviour is observed in dielectric spectroscopy and in neutron scattering (QENS). Most QENS studies19,20 of supercooled water locate this crossover at a temperature Tcross of about 225 K. Despite an enormous amount of work, the physical origin of this dynamic crossover is not yet clarified.13 Some possible interpretations including the assumption of a “fragile-to-strong transition” have already been discussed in Section 1. Many of the proposed scenarios (freezing in the tail of a not-completed order–disorder transition, freezing of dipole reorientation dynamics, etc.) imply that water is macroscopically solid below Tcross. Due to the restricted frequency range of the DMA-method (0.1–100 Hz) we can follow the main relaxation of water in nanopores only at temperatures below ca. 200 K, where the dynamics shows Arrhenius behaviour and we cannot access the change in dynamics up to the super-Arrhenius regime. However, we think that any interpretation which involves water behaving as a macroscopic solid below Tcross is in contradiction with our data for the following reason: inspecting Fig. 3–5 one observes that the real part Y′ of the complex Young's modulus decreases nearly to the background value of the silica matrix/water composite at about 225 K, implying that water in the pores cannot be solid just below Tcross. Moreover, it is hard to imagine that freezing of a local β-process can lead to such a big change in Young's modulus (see Section 4 for further discussion).Another proposed interpretation relates the observed α → β crossover of water in confinement to a finite size effect. Meanwhile, it is well established34,57,58 that glass transitions are accompanied by a growing length scale ξ∼ξ0/(T − TVF)γ of dynamically correlated regions. The idea is that the vanishing of the α-relaxation and the related crossover appears to be due to the fact that the size ξ of dynamically correlated regions reaches the confinement size d at Tcross, i.e. ξ(Tcross) = d. This implies that below Tcross no correlated motion over a distance larger than d is possible and as a result the high-temperature VFT dependence of τ = τ0exp[ξ3Δ/kBT] = τ0exp[ξ03Δ/(T − TVF)3γkBT] transforms naturally to a low-temperature Arrhenius behavior τ = τ0exp[d3Δ/kBT]. This scenario would imply that the dynamic correlation length ξ of water should be in the range of a few nanometers around Tcross ≈ 200 K.
Such an interpretation of the observed α–β crossover would be consistent with our present data, since it explains the liquid like behaviour of the Young's modulus extending to temperatures much below 200 K, despite the fact that the α-relaxation disappears. In this picture, the Arrhenius behaviour occurs due to a finite size effect (the cooperative motion cannot grow larger than the pore size) and not due to the fact that the motion is local. For water there exist no data about the size of dynamical correlations, but values of ξ ≈ 1–3 nm in the vicinity of Tg have been reported34,59,60 for a number of other glass forming liquids.
IV Summary and conclusions
In the present work we have performed extensive dynamical mechanical measurements of water confined in nanoporous silica of various pore sizes (2.5, 5 and 10 nm), with heating and cooling at a rate of 2 K min−1 and at frequencies between 0.1 Hz and 70 Hz. The results are in excellent agreement with earlier DMR (dynamic mechanical resonance) measurements44 which have been performed on water confined in large bars (200 × 8 × 4.4 mm3) of Vycor samples, vibrating in free mode at a frequency of about 400 Hz. Due to the higher measurement frequency the authors find a tanδ peak (corresponding to our P1-peak in Y′′) at a higher temperature (i.e. at 188 K, Fig. 5 of ref. 44), in perfect agreement with the Arrhenius dependence of the relaxation time with Ea ≈ 0.5 eV. Quite similar to our data, these authors measured also a weakly increasing Young's modulus with decreasing temperature due to freezing of water in the core of the pores (which they call the “capillary transition”) and a much stronger increase with decreasing temperature in the vicinity of the P1 peak. In a quite consistent explanation, the authors assume that ice forms in the centre of the pores and a film (a few layers) of adsorbed water remains liquid and separates the ice from the silicon matrix. Such a core–shell ice-water structure in nanoporous systems is also confirmed by NMR47 and THz optical spectroscopy,46etc.
In this scenario, decreasing temperature leads to an increase in the viscosity of the adsorbed film, until at very low temperatures (around P1) the adsorbed water layer freezes and effectively cements the silicon matrix and the ice together. We think that this model is very convincing and explains also the main body of our measurements. As already mentioned above, most authors agree that not all water crystallizes to ice47,61 in nanoporous systems, but a film of a few layers remains liquid down to very low temperature.
However, some discrepancy appears in the interpretation of the increase in the viscosity of the adsorbed film with decreasing temperature. Many researchers (including ourselves) attribute the freezing transition around P1 of the adsorbed water to the glass transition of confined supercooled water. But already in ref. 62 the authors follow an opposite string of arguments. Based on mechanical relaxation measurements63 of pure and doped ice, the authors interpret the observed relaxation (with Ea ≈ 0.5 eV) due to proton movement at lattice defects of the Bjerrum64 type. Very recently, Shephard and Salzmann30 came up with a similar interpretation. They state that the anomalies of amorphous ices – which are usually interpreted as glass to liquid transitions – are in fact due to unfreezing of dipole reorientations. The real glass-to-liquid transition would then occur at higher temperature, where long range diffusion of water molecules sets in.
We think that such a scenario30 of unfreezing of the dipole reorientation dynamics upon heating can be hardly ruled out. Especially if one takes into account the similarities of activation energies65 between ice (Ea = 0.57 eV, determined by mechanical and dielectric relaxation measurements) and confined supercooled water (see e.g.Fig. 3–5 or ref. 7 and 12). However, the mechanical measurements63 on ice have shown that the strain produced by proton reorientation is small and does not produce large changes in the elastic constants. This is a rather general result for orientationally active materials, e.g. we have previously measured similar effects29 in C60, where we have detected a pronounced tanδ peak in the vicinity of the orientational glass transition, accompanied by small changes in Young's modulus. Thus we believe that the observed large changes in Young's modulus Y′ (Fig. 2–5, and ref. 44 and 62) accompanying the relaxation peak P1 in Y′′ are caused by a glass-to-liquid transition in the supercooled film of adsorbed water due to unfreezing of translational diffusion, rather than by reorientation dynamics of water molecules.
Let us state it in another way: if the large softening detected previously44,62 and in the present work in Young's modulus (Fig. 3–5) was due to unfreezing of proton reorientations and not to long range diffusion, the adsorbed water film should have some pseudo-crystalline structure with an extremely small Young's modulus. Frankly speaking this would be a very unusual material behaviour. We think that the elastic measurements here are more exploratory in comparison to dielectric measurements, where the polarization produced by bond orientation in an electric field is large and thus cannot be easily used to discriminate between a liquid and a solid material.
If we accept that the relaxation around P1 originates from a glass-to-liquid transition of supercooled water, we can draw the following conclusions:
Extrapolating our data to the timescale (103 s) of adiabatic calorimetry, we obtain the following systematic behaviour (Fig. 6) of water anomalies with varying pore size.
– The anomaly around 160 K is independent of pore size. It results most probably from freezing of “interfacial water”, i.e. water molecules that are bound to the surface of the pore walls.
– The anomaly around 115 K (for pore sizes below 2.5 nm) results from glass freezing of water molecules that are surrounded by other water molecules. This process – which corresponds to the “universal” relaxation of confined supercooled water – is independent of pore size only for pore sizes below ca. 2.5 nm. Above 2.5 nm it becomes strongly dependent on pore size, i.e. ΔTg(d)∼1/d, leading to Tg(d = ∞) ≈ 136 K. As already mentioned above the pronounced softening of the real part of the complex Young's modulus (Fig. 3–5) starting in the vicinity of the P1 peak strongly supports the conjecture that the anomaly around 115–136 K corresponds to a transition from glass to liquid and not to a local β-relaxation.
We finally note that – since our measurements have been performed on water confined in nanopores – we cannot definitely conclude that the extrapolated (to d → ∞) glass transition temperature is identical to Tg of bulk water. And in this sense, the question of Tg of bulk water still remains an open issue.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Austrian Science Fund (FWF) Grant No. P 28672-N36.Notes and references
- S. Cerveny, F. Mallamace, J. Swenson, M. Vogel and L. Xu, Chem. Rev., 2016, 116, 7608 CrossRef PubMed.
- A. Hallbrucker, E. Mayer and G. P. Johari, J. Chem. Phys., 1989, 93, 4986 CrossRef.
- G. P. Johari, A. Hallbrucker and E. Mayer, Nature, 1987, 330, 552 CrossRef.
- I. Kohl, L. Backmann, A. Hallbrucker, E. Mayer and T. Loerting, Phys. Chem. Chem. Phys., 2005, 7, 3210 RSC.
- V. Velikov, S. Borick and C. A. Angell, Science, 2001, 294, 2335 CrossRef PubMed.
- Y. Yue and C. A. Angell, Nature, 2004, 427, 717 CrossRef PubMed.
- S. Cerveny, G. A. Schwartz, R. Bergman and J. Swenson, Phys. Rev. Lett., 2004, 93, 245702 CrossRef PubMed.
- M. Oguni, S. Maruyama, K. Wakabayashi and A. Nagoe, Chem. – Asian J., 2007, 2, 514 CrossRef PubMed.
- S. Maruyama, K. Wakabayashi and M. Oguni, in CP708 “Slow Dynamics in Complex Systems: 3rd International Symposium”, ed. M. Tokuyama and I. Oppenheim, 2004, p. 675 Search PubMed.
- M. Oguni, Y. Kanke and S. Namba, in CP982 “Complex Systems: 5th International Workshop on Complex Systems”, ed. M. Tokuyama, I. Oppenheim and H. Nishiyama, 2008, p. 34 Search PubMed.
- M. Oguni, Y. Kanke, A. Nagoe and S. Namba, J. Phys. Chem. B, 2011, 115, 14023 CrossRef PubMed.
- K. L. Ngai, Relaxation and Diffusion in Complex Systems, Springer Science + Business Media, LLC, 2011, DOI:10.1007/978-1-4419-7649-9.
- J. Swenson and S. Cerveny, J. Phys.: Condens. Matter, 2015, 27, 033102 CrossRef PubMed.
- G. P. Johari, J. Chem. Phys., 1996, 105, 7079 CrossRef.
- K. Ito, C. G. Moynihan and C. A. Angell, Nature, 1999, 398, 492 CrossRef.
- R. Bergman and J. Swenson, Nature, 2000, 403, 283 CrossRef PubMed.
- J. Sjostrom, J. Swenson, R. Bergman and S. Kittaka, J. Chem. Phys., 2008, 128, 154503 CrossRef PubMed.
- H. Jansson and J. Swenson, Eur. Phys. J., 2003, E12, S51 Search PubMed.
- F. Mallamace, M. Broccio, C. Corsaro, A. Faraone, U. Wanderlingh, L. Liu, C. Y. Mou and S. H. Chen, J. Chem. Phys., 2006, 124, 161102 CrossRef PubMed.
- A. Faraone, L. Liu, C. Y. Mou, C. W. Yen and S. H. Chen, J. Chem. Phys., 2004, 121, 10843 CrossRef PubMed.
- L. Xu, P. Kumar, S. V. Buldyrev, S. H. Chen, P. H. Poole, F. Sciortino and H. E. Stanley, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16558 CrossRef PubMed.
- F. Mallamace, C. Branca, C. Corsaro, N. Leone, J. Spooren, S.-H. Chen and H. E. Stanley, PNAS, 2010, 107, 22457 CrossRef PubMed.
- A. Dehaoui, B. Issenmann and F. Caupin, PNAS, 2015, 112, 12020 CrossRef PubMed.
- W. Götze and L. Sjogren, Rep. Prog. Phys., 1992, 55(3), 241 CrossRef.
- R. J. Speedy and C. A. Angell, J. Chem. Phys., 1976, 65, 851 CrossRef.
- C. A. Angell, Science, 2008, 319, 582 CrossRef PubMed.
- G. Adam and J. H. Gibbs, J. Chem. Phys., 1965, 43, 139 CrossRef.
- P. Mondal, P. Lunkenheimer and A. Loidl, Z. Phys. B: Condens. Matter, 1996, 99, 527 CrossRef.
- W. Schranz, A. Fuith, P. Dolinar, H. Warhanek, M. Haluska and H. Kuzmany, Phys. Rev. Lett., 1993, 71, 1561 CrossRef PubMed.
- J. J. Shephard and C. G. Salzmann, J. Phys. Chem. Lett., 2016, 7, 2281 CrossRef PubMed.
- P. Pissis, J. Laudat, D. Daoukaki and A. Kyritsis, J. Non-Cryst. Solids, 1994, 171, 201 CrossRef.
- W. Schranz, M. R. Puica, J. Koppensteiner, H. Kabelka and A. V. Kityk, Europhys. Lett., 2007, 79, 36003 CrossRef.
- J. Koppensteiner, W. Schranz and M. R. Puica, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 054203 CrossRef.
- J. Koppensteiner, W. Schranz and M. A. Carpenter, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 024202 CrossRef.
- J. Koppensteiner, The glass transition in nanoscale confinement probed by dynamic mechanical spectroscopy, PhD thesis, University of Vienna, 2009 Search PubMed.
- A. Ghaffar, Confinement induced structural changes of alkali metals in nanoporous systems, PhD thesis, University of Vienna, 2014 Search PubMed.
- P. Levitz, G. Ehret, S. K. Sinha and J. M. Drake, J. Chem. Phys., 1991, 95, 6151 CrossRef.
- J.-L. R. Nogues and W. Moreshead, J. Non-Cryst. Solids, 1990, 121, 136 CrossRef.
- N. Eschricht, E. Hoinkis, F. Mädler and P. Schubert-Bischoff, Stud. Surf. Sci. Catal., 2002, 144, 355 CrossRef.
- S. Gruener, T. Hofmann, D. Wallacher, A. V. Kityk and P. Huber, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 067301 CrossRef PubMed.
- W. Schranz, Phase Transitions, 1997, 64, 103 CrossRef.
- E. K. H. Salje and W. Schranz, Z. Kristallogr., 2011, 226, 1 CrossRef.
- V. Soprunyuk, W. Schranz and P. Huber, Europhys. Lett., 2016, 115, 46001 CrossRef.
- E. J. Sellevold and F. Radjy, J. Mater. Sci., 1976, 11, 1927 CrossRef.
- A. K. Soper, Chem. Phys. Lett., 2013, 590, 1 CrossRef PubMed.
- A. Taschin, P. Bartolini and R. Torre, Meas. Sci. Technol., 2017, 28, 014009 CrossRef.
- Y. Fukatsu, K. Morikawa, Y. Ikeda and T. Tsukahara, Anal. Sci., 2017, 33, 903 CrossRef PubMed.
- J. Sjöström, S. Swenson, R. Bergman and S. Kittaka, J. Chem. Phys., 2008, 128, 154503 CrossRef PubMed.
- A. Schreiber, I. Ketelsen and G. H. Findenegg, Phys. Chem. Chem. Phys., 2001, 3, 1185 RSC.
- C. A. Angell, Science, 2008, 319, 582 CrossRef PubMed.
- H. Suga and S. Seki, Faraday Discuss. Chem. Soc., 1980, 69, 221 RSC.
- G. H. Findenegg, S. Jähnert, D. Akcakayiran and A. Schreiber, ChemPhysChem, 2008, 9, 2651 CrossRef PubMed.
- F. Klameth and M. Vogel, J. Phys. Chem. Lett., 2015, 6, 4385 CrossRef PubMed.
- C. Allolio, F. Klameth, M. Vogel and D. Sebastiani, ChemPhysChem, 2014, 15, 3955 CrossRef PubMed.
- F. Klameth and M. Vogel, J. Chem. Phys., 2013, 138, 134503 CrossRef PubMed.
- K. Morishige and K. Kawano, J. Chem. Phys., 1999, 110, 4876 CrossRef.
- S. Albert, Th. Bauer, M. Michl, G. Biroli, J.-P. Bouchoud, A. Loidl, P. Lunkenheimer, R. Tourbot, C. Wiertel-Gasquet and F. Ladieu, Science, 2016, 352, 1308 CrossRef PubMed.
- Th. Bauer, P. Lunkenheimer and A. Loidl, Phys. Rev. Lett., 2013, 111, 225702 CrossRef PubMed.
- E. Hempel, G. Hempel, A. Hensel, C. Schick and E. Donth, J. Phys. Chem. B, 2000, 104, 2460 CrossRef.
- E. Donth, H. Huth and M. Beiner, J. Phys.: Condens. Matter, 2001, 13, L451 CrossRef.
- R. T. Pearson and W. Derbyshire, J. Colloid Interface Sci., 1973, 46, 232 CrossRef.
- F. Radjy and E. J. Sellevold, Nature, 1973, 241, 133 CrossRef.
- D. Kuriowa, US Army Cold Regions Res. Eng. Lab., Res. Rep. 131, US Army Material Command, Hannover, 1965 Search PubMed.
- D. Eisenberg and W. Kauzmann, The Structure and Properties of Water, Oxford University Press, London, 1969, p. 116 Search PubMed.
- P. V. Hobbs, Ice Physics, Oxford University Press, London, 1974, p. 272 Search PubMed.
Footnote |
| † There is some confusion in the pore sizes presented in various papers of Oguni et al. In ref. 8, Fig. 6a the pore diameter is given as 1.1 nm. Exactly the same curve can be found in ref. 9 with a pore diameter of 3 nm. According to the present DMA data the pore diameter of Fig. 6a in ref. 8 is most likely 3 nm. |
| This journal is © The Royal Society of Chemistry 2018 |