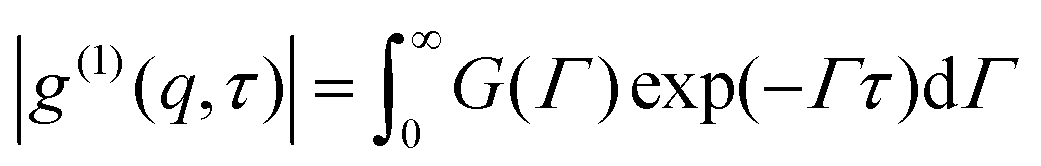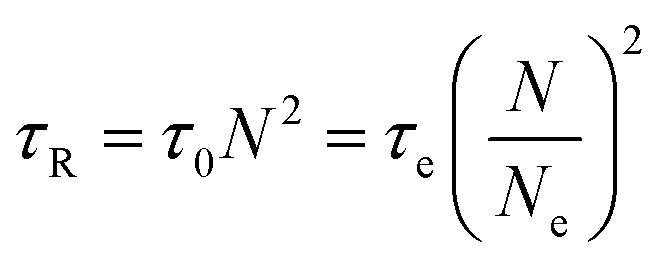DOI:
10.1039/C7SM02185B
(Paper)
Soft Matter, 2018,
14, 73-82
Viscoelastic behaviour and relaxation modes of one polyamic acid organogel studied by rheometers and dynamic light scattering†
Received
7th November 2017
, Accepted 4th December 2017
First published on 4th December 2017
Abstract
A novel polyamic acid (PAA from BAPMPO-BPDA) organogel was synthesized and characterized via dynamic light scattering (DLS), a classical rheometer, and diffusion wave spectroscopy (DWS). In situ monitoring was performed using a classical rheometer to observe the formation of the PAA organogel. The rheological curves confirm the formation of the PAA gel network and the origin of hydrogen bonding from the –NH– group (donor) and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (acceptor). The autocorrelation functions of PAA under different conditions (pure gel, gel with NaNO3, gel with formamide) are measured via DLS, and different characteristic times are obtained via the CONTIN method. Three different relaxation modes of the PAA gel, i.e., fast, intermediate and slow modes, are observed. The fast and intermediate modes show a diffusive behaviour (τ ∼ q−2), whereas the slow mode did not. When enough formamide is added into the PAA gel, the fast mode disappears; addition of enough salt (NaNO3) leads to disappearance of the slow mode. The relationship between characteristic time and diffusion vector demonstrates that the different decorrelation modes consisted of two homodyne and two heterodyne components. Two single-exponential functions and two stretched exponential functions were used, and the different decorrelation modes of the PAA gel are expressed with a non-linear function, which fits the autocorrelation function very well. And the different decorrelation modes are also discussed. DWS results in the high-frequency region not only demonstrate the formation of a PAA gel network but also indicate that the semiflexible chains of PAA are due to electrostatic interaction. The DWS results at different time scales are analyzed by applying the de Gennes’ reptation model.
O group (acceptor). The autocorrelation functions of PAA under different conditions (pure gel, gel with NaNO3, gel with formamide) are measured via DLS, and different characteristic times are obtained via the CONTIN method. Three different relaxation modes of the PAA gel, i.e., fast, intermediate and slow modes, are observed. The fast and intermediate modes show a diffusive behaviour (τ ∼ q−2), whereas the slow mode did not. When enough formamide is added into the PAA gel, the fast mode disappears; addition of enough salt (NaNO3) leads to disappearance of the slow mode. The relationship between characteristic time and diffusion vector demonstrates that the different decorrelation modes consisted of two homodyne and two heterodyne components. Two single-exponential functions and two stretched exponential functions were used, and the different decorrelation modes of the PAA gel are expressed with a non-linear function, which fits the autocorrelation function very well. And the different decorrelation modes are also discussed. DWS results in the high-frequency region not only demonstrate the formation of a PAA gel network but also indicate that the semiflexible chains of PAA are due to electrostatic interaction. The DWS results at different time scales are analyzed by applying the de Gennes’ reptation model.
1. Introduction
Gels are defined as non-fluidic colloidal networks or polymer networks that are expanded throughout their whole volume by a fluid.1 Depending on their crosslinking system, gels can be divided into chemical gels (multi-functional group crosslink) and physical gels (physical interaction crosslink). For physical gels, various types of physical crosslinks exist, such as hydrogen bonds, dipole interaction, micellar formation of hydrophobic groups, and microcrystalline junctions.2 In recent years, physical gels made from noncovalent interaction have attracted much attention because of their unique responsive behaviour and functions (hydrogen bonding interaction,3 ionic interaction,4 and hydrophobic/hydrophilic interaction5). Physical gels have been applied in the food industry, drug delivery, musculoskeletal tissue engineering, molecular filters, anion sensors, and so on.6–11 For instance, Yan et al. proposed an alternative pathway for fabrication of a collagen protein hydrogel with shear-thinning and self-healing properties based on a biomineralization triggered process, which involves electrostatic complexation between positively charged collagen chains and inorganic anionic clusters.12 The resulting hydrogels exhibit advantages as an in vivo injectable material for biomedical applications.
Gels are also classified as hydrogels (in water) or organogels (in organic solvent) depending on the medium used for fabrication. The mechanisms and applications of physical crosslinking hydrogels, especially hydrogen bond crosslinking, have been experimentally and theoretically studied. Meanwhile, organogels have attracted much attention because of their super-sensitivity to changes in their environment. Yao et al.13 synthesized a novel organogel whose colour changes from milky white to transparent with increasing temperature. Moreover, the contact angle also drastically decreases with increasing temperature. Garcia et al.14 designed a novel organogel via paraformaldehyde condensation with diamines, and the produced organogel showed high recyclability and strong thermoset abilities. However, compared with hydrogels, organogels are seldom reported in the literature.
Polyimides are one kind of advanced material that has been extensively investigated15–17 and can be applied in microelectronics, aerospace, and many areas as a type of resin, film, fiber, or foam.18–22 However, the precursor of polyimides, i.e., polyamic acid (PAA), has not been systematically studied because of its instability and its so-called “degradable” behaviour. Most studies have focused on the stability of PAA. Artem'eva et al.23 used three different structures of diamine to synthesize PAA and analyzed their intrinsic viscosity during storage at room temperature. The stability of polypyromellitamic acids in solution was determined by the length of the monomeric unit, i.e., by the content of weak o-carboxyamide units in a macromolecule. Cai et al.24 synthesized a novel PAA-ammonium salt, which has higher stability than that of PAA. The degradation of PAA has been suggested to be caused by acid-catalyzed hydrolysis in the amide group, and this process involves the intramolecular cyclization of the o-carboxyl groups onto the amide linkage with the elimination of the amine. When o-carboxyl groups in PAA are converted to amine salts, the reaction of acid-catalyzed hydrolysis is suppressed by the presence of amine salts. However, studies on the physical properties of concentrated PAA solution, especially for PAA gel, are limited.
In the present study, a PAA organogel was synthesized and its viscoelastic properties were investigated via classical rheometer, dynamic light scattering (DLS), and diffusion wave spectroscopy (DWS, also called microrheometer). The formation mechanism of a PAA organogel is also discussed. This study will be helpful to understand the origin of this kind of PAA gel and its viscoelastic behaviour.
2. Experimental section
2.1 Chemicals
The reactant bis(3-aminophenyl)methylphosphine oxide (BAPMPO), 4,4′-oxydianiline (4,4′-ODA) and 3,3′,4,4-biphenyl dianhydride (BPDA) were purchased from Beijing Multi Technology. The solvent N,N-dimethylacetamide (DMAc) was purchased from Beijing Multi Technology. Before preparation, all the monomers were purified by sublimation.
2.2 Synthesis of PAA
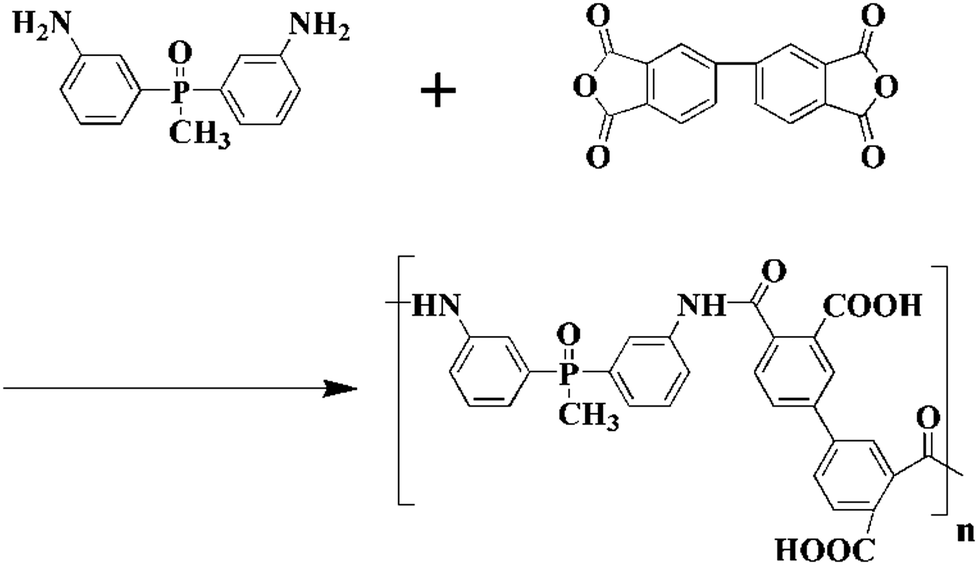
In a 500 mL round-bottomed flask, 16.92 g (0.055 mol) BAPMPO was dissolved in 200 mL of DMAc (dried in the presence of P2O5 overnight), then 16.15 g (0.055 mol) BPDA was added and the reaction was carried out at 17 °C for 48 h under stirring and dry nitrogen flow. Scheme 1 shows the synthetic route of PAA. After the reaction was finished, the sample was sealed and stored at −18 °C in a refrigerator.
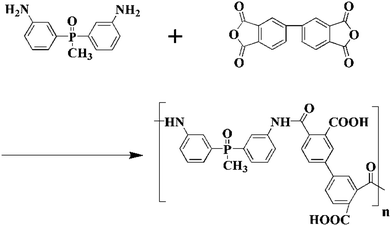 |
| | Scheme 1 Synthetic route of BAPMPO-BPDA polyamic acid. | |
2.3 Rheometer
All rheological measurements were carried out using a DHR-2 stress-controlled rheometer (TA Instruments), where a 40 mm, 2° cone and plate geometry was used. A temperature-controlled Peliter plate was used to control the temperature of the measurements. Before the experiment, all the samples were tested by oscillation amplitude to ensure the measurement was in the region of linear viscoelasticity. The oscillation frequency was varied from 10−2 to 102 rad s−1. In order to prevent the evaporation of solvent, two semicircle 60 mm iron plates were covered around the geometry, and silicone grease was filled around the gap of the iron plates.
2.4 Dynamic light scattering
Dynamic light scattering (DLS) is an effective tool in probing different relaxation processes. Using the intensity correlation function| | 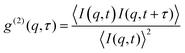 | (1) |
where t is the delay time, I(q,t) is the scattering intensity at time t, q is the scattering vector, defined as| | 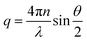 | (2) |
λ is the incident wavelength and n is the refractive index of the sample. The intensity–intensity time autocorrelation function g(2)(q,t) is related to g(1)(q,t) by the Siegert relation| | | |g(2)(q,τ)| = 1 + β[g(1)(q,τ)]2 | (3) |
β is a constant depending on the system. For a well-defined, monodispersed relaxation process, g(1)(q,t) represents an exponential decay function| | |g(1)(q,τ)| = G![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−Γτ) exp(−Γτ) | (4) |
For a polydispersed dynamic relaxation, eqn (4) may be generalized as| | 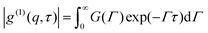 | (5) |
G(Γ)dΓ is the distribution of a dynamic relaxation process, Γ = 1/τ, the reciprocal of characteristic decay time, defined as the rate of dynamic relaxation. If the relaxation obeys the diffusion rule, it can be characterized asD is the diffusion coefficient.
The scattering field for nonergodic media, such as polymer gel, contains both time-dependent (homodyne) and time-independent (heterodyne) components. Hence, it can be expressed as:25
where
E(
t) is the total scattering field,
EF(
t) is the fluctuating scattering field component, which is time-dependent, and
Ec is the time-independent scattering field component.
The time–intensity correlation function can be given by:26
| | | g(2)(τ) − 1 = X2[g(1)F(τ)]2 + 2X(1 − X)g(1)F(τ) | (8) |
where
X is the ratio of the intensity of the fluctuating (time-dependent) component, and
g(1)F(
τ) is the autocorrelation function of the fluctuating component. In this study, the CONTIN method was used. To minimize the equation below, the different relaxation modes can be obtained:
| | 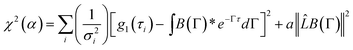 | (9) |
where
σi is the standard deviation of
g1(
τ);
a||
![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) B
B(
Γ)||
2 is the regulation term, using 0 <
a < 1 to determine the details of the solution of equation;
B(
Γ) is the distribution of decay rate. Finally, the CONTIN method was used to separate these components.
A commercial LLS spectrometer (ALV CGS-3) was equipped with a multi-τ digital time correlator (ALV7000) and a cylindrical 22 mW He–Ne laser (λ0 = 632.8 nm, Uniphase) as a light source (ALV-GmbH, Germany). The temperature of the scattering vial was controlled by a thermostatic water bath (JULABO F12-ED) with a temperature precision of 0.02 °C. Each temperature was kept for at least 10 min to reach equilibrium before measurements. A gel is a nonergodic system. In order to circumvent the nonergodicity, the sample cell was rotated steadily while time-averaging. All the measurements were kept at 20 °C for 30 min to get the correlation function precisely. The range of scattering angle θ was from 70° to 140°, with a step of 10°. The decay time distribution was obtained by using the CONTIN method of the experimental correlation function.
2.5 Diffusion wave spectroscopy
Diffusion wave spectroscopy extends the applications of traditional DLS. Based on strong multiple light scattering, for the transmission method, the g(1)(τ) was given by27 (for λ ≪ l* ≪ L, λ is the wavelength of light, L is the sample thickness, strong multiple light scattering is irrelevant of scattering vector):| | 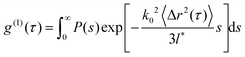 | (10) |
P(s) is the distribution length, l* is the transport mean free path of scattered light, which means that the scattered light must travel to randomize their direction, k0 is the wavevector of the light in the medium, and 〈Δr2(τ)〉 is the mean square displacement. For the backscattering method, the relationship between g(1)(τ) and 〈Δr2(τ)〉 can be expressed by27| | 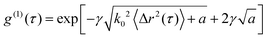 | (11) |
γ is a constant which allows low-order scattering to be quantified, and a characterizes deviations from the idealized semi-infinite slab geometry. Taking the Laplace transform, the relation between ![[G with combining tilde]](https://www.rsc.org/images/entities/i_char_0047_0303.gif) (s) and 〈Δ
(s) and 〈Δ![[small gamma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dd.gif) 2(s)〉 can be written as28
2(s)〉 can be written as28
| | 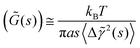 | (12) |
where
(
s) is the viscoelastic spectrum; 〈Δ
2(
s)〉 is the Laplace 〈Δ
r2(
τ)〉;
s is the Laplace frequency. And finally, using the unilateral Fourier transform, an expression for the viscoelastic modulus as a function of frequency can be obtained.
| | 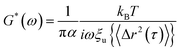 | (13) |
where
G*(
ω) is the complex modulus;
ξu{〈Δ
r2(
τ)〉} is the unilateral complex Fourier transform of 〈Δ
r2(
τ)〉.
DWS experiments were performed on a DWS RheoLab II (LS Instrument) equipped with a single photon-counting avalanche photodiode (APD) as a detector and a linear correlator with the smallest sampling time of 12 ns. The coherent source was a diode laser with a wavelength of 687 nm and power of 40 mW.
Monodispersed SiO2 nanoparticles with a radius of 270 nm purchased from Bangs Laboratories were chosen as tracer particles. 0.5 g nanoparticles were poured into 1 mL of DMAc in a 10 mL conical flask, and the mixture was ultrasonicated for at least 20 min to obtain a homogeneous dispersion. The turbid solution was added into polyamic acid gel and stirred, and stored in a refrigerator for at least one day to remove the air bubbles completely.
The width of the sample cell holder was 10 mm and the temperature of measurement was 20 °C. Before measurement, the transport mean free path l* was measured by transmission scattering. However, the method of adding the tracer particle can dilute the polyamic acid gel and influence the result. Only a small amount of tracer particles was added in and makes the ratio L/l* = 5–6 (near the lower limit of the DWS experiment).
2.6 UV-visible spectroscopy
UV-visible spectra were recorded on a Shimadzu UV-2450 double-beam recording spectrophotometer using 1 cm quartz cells (Shimadzu Corporation, Japan). The as-prepared polyamic acid solution or gel exhibits a yellow colour, Fig. S1 (ESI†) is the UV-visible spectrum and the transmittance of polyamic acid gel at 633 nm was up to 94%, indicating almost no influence on both DLS and DWS measurements.
2.7 FT-IR spectroscopy
Fourier transform infrared (FTIR) spectra were recorded on a Bruker Vertex 70 spectrometer at 64 scans with a resolution of 2 cm−1 for polyamic acid solution (Bruker Corporation).
3. Results and discussion
3.1 PAA organogel
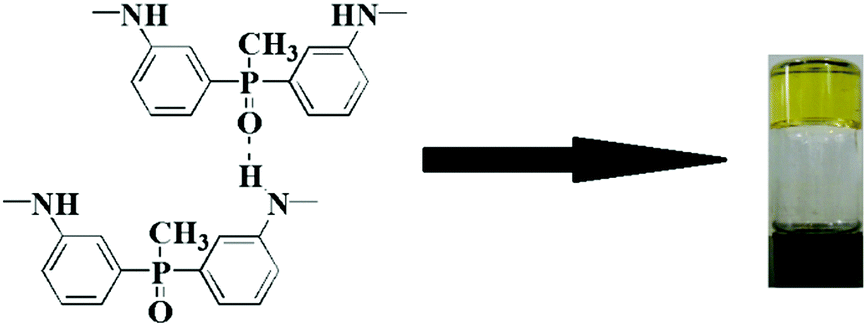
The BAPMPO-BPDA PAA in DMAc lost its mobility, which can be seen clearly in Fig. 1a. In a macroscopic sense, it indicates that the gel had been formed. Excessive solvent cannot dissolve the PAA gel (see Fig. S2, ESI†). Formamide is often applied to break the hydrogen bonds in solution. In this study, a small amount of formamide (2 wt%) and excessive DMAc as a solvent were added to the PAA gel and then stirred. As shown in Fig. 1b, the original PAA gel became transparent. This result demonstrates that the formation of hydrogen bonds results in aggregation of the PAA chains to form a gel.
 |
| | Fig. 1 Pictures taken in indoor day-time, (a) PAA gel (15 wt% in DMAc), (b) PAA gel with 2 wt% formamide and excessive solvent DMAc. | |
3.2 Formation mechanism of the gel from a classical rheometer
3.2.1
In situ monitoring on gel formation using a classical rheometer.
The time evolution method was performed to monitor the reaction process of PAA from BAPMPO and BPDA by using a classical rheometer. In Fig. 2, two periods were observed, namely, induction and reaction stages. DMAc is a good solvent for BAPMPO and a poor solvent for BPDA. BPDA is precipitated at the bottom and needs plenty of time to react with BAPMPO completely. At the early stage (0–300 min), the PAA chains are not long enough to form a network in the system, and noisy data were obtained during the induction period. When BPDA was completely dissolved in DMAc, at about 300–350 min, the PAA chains quickly grew and the corresponding storage and loss moduli sharply increased as the PAA network began to form. At 1000 min for reaction, G′ and G′′ can reach 2.58 × 104 Pa, and 1.68 × 104 Pa, respectively.
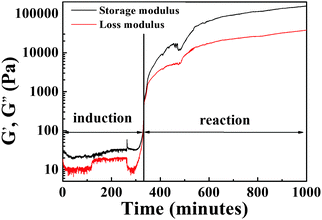 |
| | Fig. 2 Time evolution of storage and loss modulus of PAA in DMAc (15 wt%), at a strain of 1% and angular frequency of 1 rad s−1. | |
3.2.2 Acceptor of hydrogen bonds.
The results in Fig. 1b demonstrate that the formation of the PAA gel was due to hydrogen bonding. The donor certainly comes from the –NH– group. However, the acceptor can be the PO, CO, or CO–O− group. To confirm the identity of the acceptor, another diamine, 4,4′- oxydianiline (4,4′-ODA), was chosen and different contents of 4,4′-ODA and BAPMPO were copolymerized with BPDA (shown as Scheme 2).
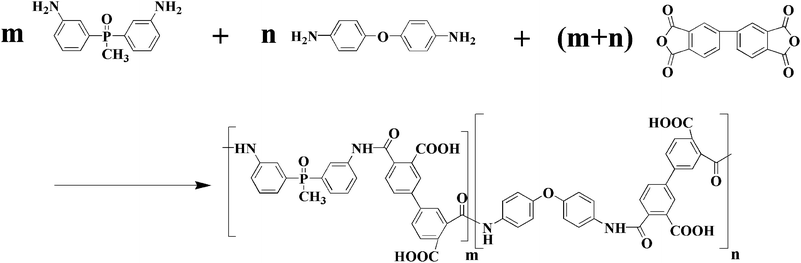 |
| | Scheme 2 Synthetic route of BAPMPO-ODA-BPDA polyamic acid. | |
As shown in Fig. 3, in the low-angular frequency region, the storage modulus of PAA without BAPMPO (i.e., ODA-BPDA) grows linearly as the frequency increases. When the ratio of BAPMPO was increased, the slope of the storage modulus decreased in the low-frequency range. The storage modulus plateaued when the ratio of BAPMPO was above 70%. This result indicates the elasticity of the PAA gel in the low-frequency range. Similar to the 100% BAPMPO PAA gel, the 70% BAPMPO PAA gel cannot be dissolved by DMAc.
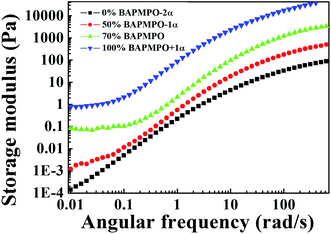 |
| | Fig. 3 Storage modulus of 15 wt% PAA with different amounts of diamine (BAPMPO:4,4′-ODA) as a function of angular frequency. In order to make the curve clearer, some are moved by α (1 decade). | |
An ideal gel has an almost pure elastic response behaviour. The elastic modulus should be much higher than the viscous modulus and is independent of frequency. However, when gel networks are formed with imperfections, the response of the polymer gel depends on frequency, i.e., both the storage and loss moduli increase with frequency. The polymer gel can be tested at various time scales by using the oscillation mode in rheological experiments. At low frequencies, both the gel and polymer sol are rearranging because of the Brownian motion. Thus, the measured properties are dominated by the elastic deformation equilibrium of the gel network. As shown in Fig. 3, when the frequency approaches zero, the elastic modulus approaches a constant value, known as the equilibrium modulus of the gel network.
Physical entanglements are created and broken quickly at low frequencies. Thus, they do not contribute drag or store elastic energy. At intermediate frequencies, both the polymer sol and the polymer chain segment between two physical crosslink points begin to relax. Thus, the storage and loss moduli obey the Maxwellian behaviour. At high frequencies, physical entanglements persist longer than oscillation frequency. Thus, the entanglements physically constrain the polymers, store elastic energy, and contribute to viscous dissipation.29 In the present study, the plateau behaviour of the storage modulus in the low frequency range indicates the appearance of the PAA gel, and the hydrogen bonding originates from the –NH– group (donor) and PO group (acceptor), as illustrated in Fig. 4.
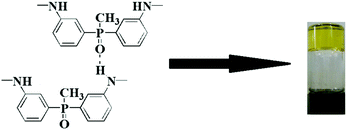 |
| | Fig. 4 Origin of hydrogen bonding in the PAA organogel. | |
Even though the weight percentage of BAPMPO-BPDA decreases to 11 wt%, the plateau of the storage modulus in the low-angular frequency range was still observed, as shown in Fig. 5. More solvent was added into the 11 wt% PAA gel, but the gel cannot absorb excessive solvent, which is consistent with the result in Fig. S2 (ESI†).
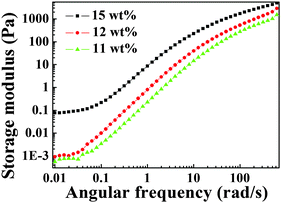 |
| | Fig. 5 Storage modulus of different weight percentages of PAA gel (BAPMPO-BPDA) as a function of angular frequency. | |
3.3 Relaxation modes from DLS results
3.3.1 Autocorrelation function of PAA under different conditions.
Fig. 8 shows the autocorrelation function of PAA under different conditions from DLS data. For pure PAA gel and the PAA gel with 8 wt% NaNO3 (eliminate the electrostatic interaction), their autocorrelation functions cannot decay to zero in finite time, as confirmed by the formation of a gel structure. Addition of formamide can break the network of the PAA gel, and the autocorrelation function of the PAA gel with 30 wt% formamide can approach to zero in finite time. Formamide should be added in excess to facilitate decay of the autocorrelation function to zero. DLS can provide information in the nano-scale as reflected in the autocorrelation function. Only the network of the gel breaks down completely, thus, the autocorrelation function decays to zero in finite time.
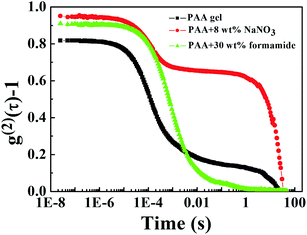 |
| | Fig. 8 Autocorrelation functions of PAA under different conditions. | |
3.3.2 Relaxation modes of PAA under different conditions.
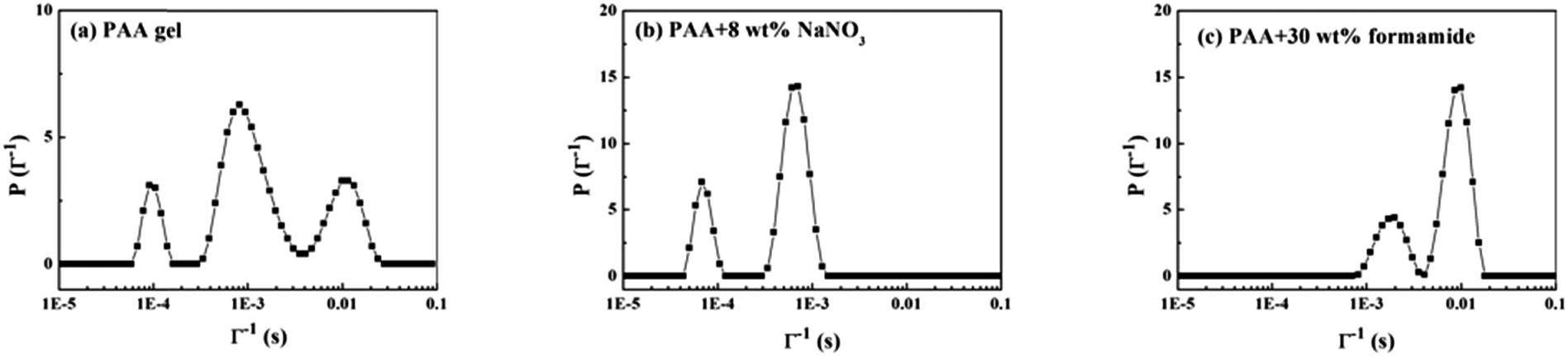
Fig. 9a shows the CONTIN result of the pure PAA gel. Three modes of decorrelation exist: τf (fast mode), ∼10−4 s; τi (intermediate mode), ∼10−3 s; and τs (slow mode), ∼10−2 s. Actually, four modes of decorrelation exist, the fourth mode reflects the bulk of the PAA gel, but its characteristic time is beyond the scale of measurement. We will not discuss it here but will analyze it later. Fig. 9b shows the PAA gel with 8 wt% NaNO3 by isolating the electrostatic interaction between the –COOH and –NH– groups. The slow mode disappeared. Fig. 9c shows the PAA with 30 wt% formamide by completely breaking the physical gel network. Only two modes are retained. Meanwhile, breaking the gel network can also destroy the fourth mode of decorrelation. It should be noted that adding enough formamide makes the system become ergodic, and we cannot discuss the CONTIN results in a similar way as a gel system. Thus, the DLS results of the pure gel system are discussed and analyzed emphatically.
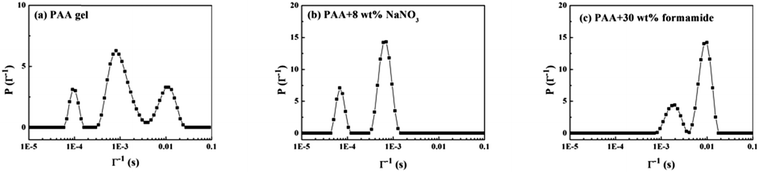 |
| | Fig. 9 CONTIN results of PAA under different conditions. (a) PAA gel, (b) PAA + 8 wt% NaNO3, (c) PAA + 30 wt% formamide. The scattering angle is 90°. | |
To discuss the autocorrelation function with several decorrelation modes, a combination of several stretched exponential functions should be used to fit the correlation function:31,32
| | g(2)(τ) − 1 = σI2(A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Γ1τ)δ1 + A2exp(−Γ2τ)δ2+…) exp(−Γ1τ)δ1 + A2exp(−Γ2τ)δ2+…) | (14) |
where
σI2 is a parameter reflecting the depression of the initial amplitude, and
A1 and
A2 are the intensity contributions of different decorrelation modes. If the decorrelation mode is time-dependent, then the stretched exponent
δi is equal to 1 and the decorrelation mode obeys the diffusion rule. If the decorrelation mode is time-independent, then the stretched exponent
δi deviates from 1. The dependence of the characteristic time
τ of each mode from the autocorrelation function on the diffusion vector
q was measured to analyze the nature (time-dependent and time-independent) of the three decorrelation modes as shown in
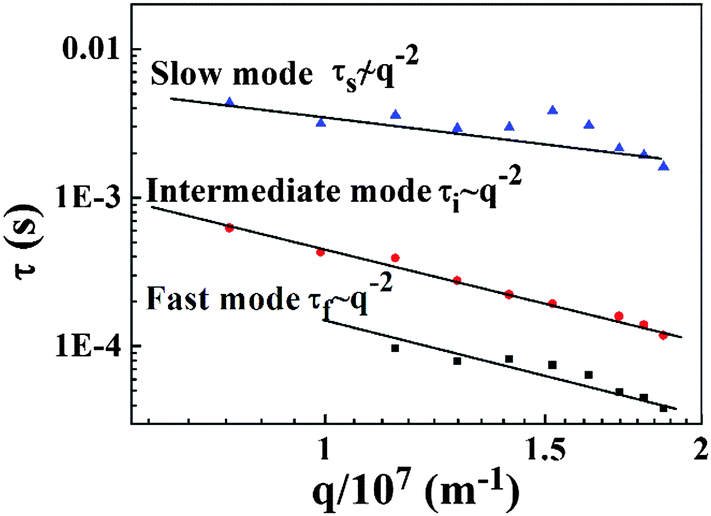
Fig. 10. The characteristic times of the fast and intermediate modes vary as
τ ∼
q−2 and obey the diffusion rule. When the scattering angle is below 80°, the intensity of the signal of the fast mode is very low. Thus, the result is unreliable, and the data of the fast mode at low angle are ignored. However, the slope of characteristic time for the slow mode
versus the diffusion vector deviated far from −2, which denoted a time-independent decorrelation mode.
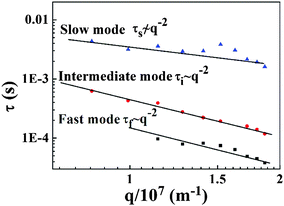 |
| | Fig. 10 Three characteristic times of the PAA gel versus diffusion vector. | |
The characteristic line width (Γ = 1/τ) versus the square scattering vector (q2) is plotted in Fig. 11. The relationship between fast mode, intermediate mode and scattering vector are a linear function of q2, and the line passes through the origin, which indicates that these two modes are diffusive control.33
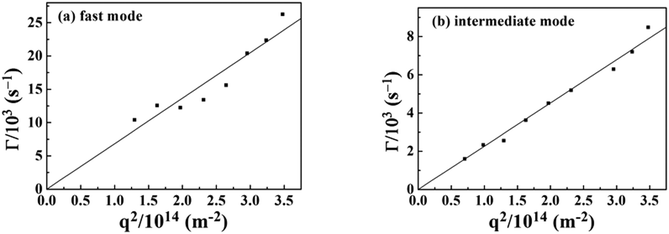 |
| | Fig. 11 Characteristic line widths (Γ) versus square scattering vector (q2) for the PAA gel, (a) fast mode, (b) intermediate mode. | |
3.3.3 Nonlinear fitting of the autocorrelation function of the PAA gel.
Nonlinear fitting was applied for two single-exponential functions and two stretched exponential functions to express different decorrelation modes and to analyze the autocorrelation function, as shown in Fig. 12. The nonlinear function is expressed as:| | 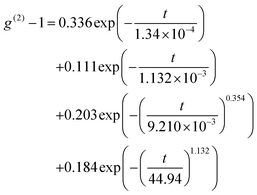 | (15) |
The characteristic times of different modes from the function are 9.89 × 10−5 s (fast mode), 1.08 × 10−3 s (intermediate mode), and 1.11 × 10−2 s (slow mode). These values are consistent with the results obtained from the CONTIN method, as shown in Fig. 9a.
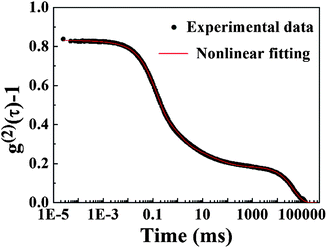 |
| | Fig. 12 Nonlinear fitting of the autocorrelation function of the PAA gel. | |
3.3.4 Origin of different decorrelation modes of the PAA gel.
From the autocorrelation function of a pure PAA gel and PAA with 8 wt% NaNO3, it can be seen that both of them cannot decay to zero in finite time. Thus, both of them can be regarded as a gel (nonergodic) and they can be discussed in a similar way. However, the autocorrelation function of PAA with 30 wt% formamide can decay to zero in finite time, which reflects that the system becomes ergodic. This should be discussed separately.
Compared with the CONTIN results of the pure PAA gel, the slow mode for PAA with 8 wt% NaNO3 disappears. Moreover, in Fig. 10, the slow mode τsversus scattering vector q does not show diffusive behaviour, which reflects the nonergodic behaviour. Thus, the electrostatic interaction causes the agglomeration of PAA chain segments (crosslink) and is the origin of the nonergodic slow mode. (The variation of the plateau MSD from the DWS results also demonstrates this. Isolating the electrostatic interaction enlarges the size of the gel network, which makes the escape of the tracer particles from the network much easier. And the time of cut-off of the plateau becomes much shorter, as shown in Fig. 13.) Both the fast mode τf and the intermediate mode τi obey the diffusion rule. Owing to the appearance of the hydrogen bonding crosslinking point in the PAA gel network, the diffusion of PAA chain segments between crosslinking points is different in different directions. Thus, the origin of the fast mode τf and the intermediate mode τi is the movement of PAA chain segments in different directions.
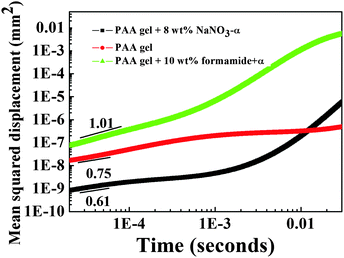 |
| | Fig. 13 Mean squared displacement as a function of time for PAA (15 wt%) under different conditions. In order to observe the curves clearly, some curves are moved by α (1 decade). | |
Wu et al.33 synthesized a novel polymer that can change from being charged to neutral by formation of different gases (bubbles). They found that the neutral polymer only had one decorrelation mode, but when it became charged, the sole decorrelation mode was split into two modes, namely, fast and slow. Both of them obey the diffusion rule, and the coupling of the diffusion of the chains and the fluctuation of chain segments nearby the counterions account for the results mentioned earlier. For PAA with 30 wt% formamide, due to the breakage of the PAA network and the appearance of –COOH and –NH– groups, the solution behaviour can be regarded as a concentrated charged polymer solution. Thus, the origin of the two modes is similar to Wu's results.33
3.4 Viscoelastic behaviour from DWS results
Above 100 rad s−1, the classical rheometer cannot be used to determine the storage and loss moduli precisely because of inertia and instrument limitation. Although time–temperature superposition can be applied to obtain precise information in extremely low- and high-angular frequency ranges, the PAA gel cannot be detected in the high-frequency range because of two reasons: the PAA gel degrades very fast when the temperature is above 40 °C,34 and water vapour condenses on the Peliter plate at temperatures below 15 °C. These problems can be avoided in the DWS experiments, and information about the PAA gel at high-frequency regions can be obtained.
The non-affinity of the gel network is considered, and interaction (depletion, absorption, and so on) between tracer particles and the gel network possibly exists.35 The result of the DWS experiment cannot fit those from a classical rheometer. Thus, only the behaviour of the PAA gel in the high-frequency region was qualitatively discussed.
Fig. 13 shows the mean squared displacement (MSD) as a function of time in the backscattering geometry. The relationship between MSD and time (〈Δr2(τ)〉 ∼ τα) can reflect the property of the medium.36 When the time is short, the tracer particle is trapped in the network of the polymer and cannot “escape” from the network. Given that the tracer particles are confined in the PAA network and cannot move further than the size of the network, a constant value of MSD was observed. We divided the time into two scales and discussed them separately.
3.4.1 Short-time scale.
For the PAA gel, as shown in Fig. 13, in the range of 2 × 10−5–7 × 10−5 s the power law between MSD and time is 0.75, which is consistent with the theory for a semi-flexible network.37 When enough salt (such as NaNO3) is added to weaken the electrostatic interaction, the exponent decreases to 0.61, which is between 1/2 (the Rouse model of flexible chain theories38) and 2/3 (the Zimm model of flexible chain theories39). This result demonstrates that electrostatic interaction stretches the PAA chain to become semi-flexible. The exponent increases to 1 when the network of the PAA gel is completely destroyed through the addition of formamide; this phenomenon is similar to a homogeneous polymer solution.40
3.4.2 Long-time scale.
The plateau value of MSD for the PAA gel in Fig. 13 was observed between 10−3 s and 10−2 s, which is consistent with the result in Fig. 9a (between the intermediate τi and slow τs characteristic times). However, the plateau value of MSD for the PAA gel with enough salt was observed between 10−4 s and 10−3 s, which is consistent with the result in Fig. 9b (between the fast characteristic time τf and intermediate characteristic time τi). Based on the DLS results, weakening the electrostatic interaction could result in the disappearance of the slow decorrelation mode, and the plateau appears between the fast characteristic time τf and intermediate characteristic time τi. Adding enough formamide could result in the disappearance of the fast mode τf and the extremely slow mode because of the breakage of the network of the PAA gel. This result implies that the electrostatic interaction causes the agglomeration of the PAA chain segments, which shrinks the size of the gel network, resulting in the appearance of the plateau of MSD between τi and τs.
For the PAA gel with salt, when the time scale is longer than the intermediate characteristic time τi, the MSD increases. Thus, the trapped tracer particle “escaped” from the network of the PAA gel. For pure PAA gel, the same phenomenon occurs on a time scale longer than the slow characteristic time τs, but the relationship between MSD and time in a prolonged time scale was not determined because of measurement limits.
3.4.3 Viscoelastic properties of the PAA gel.
Based on eqn (13), MSD can be transformed into a complex modulus, as shown in Fig. 14. The slope between the complex modulus and angular frequency is 0.75 in the high-frequency range (8000–30000 rad s−1), which is consistent with the characteristics of semi-flexible chains.41
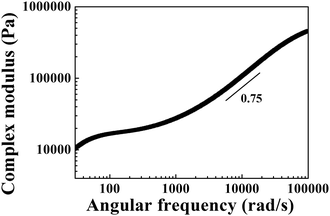 |
| | Fig. 14 The complex modulus as a function of angular frequency measured with DWS for the PAA gel (15 wt%). | |
The results in Fig. 14 can also be transformed into the storage and loss moduli as a function of angular frequency, as shown in Fig. 15. By applying the de Gennes’ reptation model,42 the results of DWS can be divided into different time scales: reptation time τrep, Rouse time of the chain τR, Rouse time of an entanglement strand τe, and the monomer motion time τ0.42τrep, τe, and τ0 can be obtained from the three intersections between G′ and G′′, and τR can be calculated from the relationship between these variables.43
| | 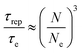 | (16) |
| | 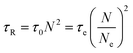 | (17) |
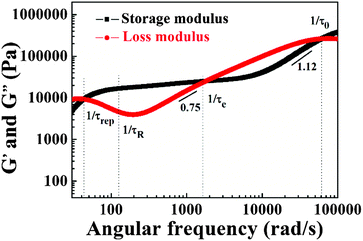 |
| | Fig. 15 The storage modulus and the loss modulus as a function of angular frequency measured with DWS for the PAA gel (15 wt%). | |
The power law between G′, G′′, and angular frequency at the high-frequency region can be used to confirm the results. For the Rouse model, the exponent is 1/2 and that of the Zimm model is 2/3.44 However, between 600 rad s−1 and 1/τe, for the PAA gel, the power law between the loss modulus and angular frequency is G′′ ∼ ω0.75 because the electrostatic interaction stretches the polymer chains, thereby deviating from the Rouse and Zimm model. The results are in accordance with the theory of internal bending modes for single Kuhn segments.40 Nevertheless, between 20000 rad s−1 and 1/τ0, the power law between storage modulus and angular frequency does not match the theoretical value. Willenbacher et al. reported the scaling relationship G′′ ∼ ω0.75 for ω > 104 rad s−1 and indicated the onset of G′ ∼ ω0.75 for ω > 5 × 105 rad s−1 for worm-like micelle solutions.45 However, the inertia of tracer particles influences the result in the extremely high-angular frequency range. Moreover, 5 × 105 rad s−1 is beyond the measurement range of the current DWS instrument. The ω0.75 scaling was also not solved by Buchanan et al.46 because their experiments were limited to frequencies below 105 rad s−1, which is similar to the present case. Thus, G′ at extremely high angular frequencies cannot be obtained.
4. Conclusions
A novel PAA organogel was synthesized and characterized via classical rheometer, dynamic light scattering, diffusion wave spectroscopy, and FTIR spectroscopy. The main results are summarized as follows.
(i) In situ monitoring results via classical rheometer and FTIR spectroscopy confirm that when the length of the PAA chains grows to a certain length, the hydrogen bonds between PO and N–H groups induce the formation of a PAA gel network. Such a PAA gel network can be destroyed by adding formamide.
(ii) In the DLS experiments, three different relaxation modes of the PAA gel, i.e., fast, intermediate and slow modes, are observed. The fast and intermediate mode shows a diffusive behaviour (τ ∼ q−2), whereas the slow mode did not. When enough formamide is added into the PAA gel, the fast mode disappears. However, when enough salt (NaNO3) is added into the PAA gel, the slow mode disappears. The origin of different modes is determined: the fast mode and the intermediate mode come from the movement of PAA chain segments between a crosslinking point in different directions, and the slow mode is the agglomeration of PAA chain segments due to electrostatic interaction. Two single-exponential functions and two stretched exponential functions were used to express the different decorrelation modes with a nonlinear function. The results fit the autocorrelation function very well.
(iii) Based on the DWS experiments, the relationship between MSD and time is determined under different conditions, and electrostatic interaction causes the PAA chains to be stretched and become semi-flexible. By applying the de Gennes’ reptation model, the DWS result was divided into different time scales. The power law of the loss modulus and angular frequency fits the theory of the internal bending modes for single Kuhn segments (G′′ ∼ ω0.75).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the financial support from the National Basic Research Program of China (2014CB643604) and the National Natural Science Foundation of China (51173178). We appreciate the help of Prof. Quan Chen for his valuable suggestion on rheological data.
References
-
G. J. Richard, K. Jaroslav, S. Robert, S. W. Edward, H. Michael, K. Tatsuki and W. V. Metanomski, Compendium of Polymer Terminology and Nomenclature, 2nd edn, Royal Society of Chemistry, 2009 Search PubMed.
-
F. Tanaka, Polymer Physics: Applications to Molecular Association and Thermoreversible Gelation, Cambridge University, 2011 Search PubMed.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193–6201 CrossRef CAS.
- G. Miquelard-Garnier, S. Demoures, C. Creton and D. Hourdet, Macromolecules, 2006, 39, 8128–8139 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–476 CrossRef CAS.
- Y. Tanaka, J. P. Gong and Y. Osada, Prog. Polym. Sci., 2005, 30, 1–9 CrossRef CAS.
- S. Varghese and J. H. Elisseeff, Adv. Polym. Sci., 2006, 203, 95–144 CrossRef CAS.
- G. Tummala, T. Joffre, R. Rojas, C. Persson and A. Mihranyan, Soft Matter, 2017, 13, 3936–3945 RSC.
- N. Malviya, M. Das, P. Mandal and S. Mukhopadhyay, Soft Matter, 2017, 13, 6243–6249 RSC.
- H. Guo, T. Jiao, Q. Zhang, W. Guo, Q. Peng and X. Yan, Nanoscale Res. Lett., 2015, 10, 272 CrossRef PubMed.
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS PubMed.
- X. Yao, J. Ju, S. Yang, J. Wang and L. Jiang, Adv. Mater., 2014, 26, 1895–1900 CrossRef CAS PubMed.
- J. M. Garcia, G. O. Jones, K. Virwani, B. D. McCloskey, D. J. Boday, G. M. Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb, A. M. S. Alabdulrahman, F. Alsewailem, H. A. A. Almegren and J. L. Hedrick, Science, 2014, 344, 732–735 CrossRef CAS PubMed.
- M. Ding, Prog. Polym. Sci., 2007, 32, 623–668 CrossRef CAS.
- E. Zhang, X. Dai, Z. Dong, X. Qiu and X. Ji, Polymer, 2016, 84, 275–285 CrossRef CAS.
- E. Zhang, H. Chen, X. Dai, X. Liu, W. Yang, W. Liu, Z. Dong, X. Qiu and X. Ji, J. Polym. Res., 2017, 24, 47 CrossRef.
- A. Takuma and K. Hiroyoshi, Polymer, 2012, 53, 2217–2222 CrossRef.
- C. Zhang, Q. Zhang, Y. Xue, G. Li, F. Liu, X. Qiu, X. Ji and L. Gao, Chem. Res. Chin. Univ., 2014, 30, 163–167 CrossRef.
- R. Lei, L. Gao, R. Jin and X. Qiu, Chin. J. Polym. Sci., 2014, 32, 941–952 CrossRef CAS.
- B. Li, Y. Pang, C. Fang, J. Gao, X. Wang, C. Zhang and X. Liu, J. Appl. Sci., 2014, 131, 40498 Search PubMed.
- Y. Zhao, G. Li, X. Dai, F. Liu, Z. Dong and X. Qiu, Chin. J. Polym. Sci., 2016, 34, 1469–1478 CrossRef CAS.
- V. N. Artem'eva, V. V. Kudryavtsev, E. M. Nekrasova, V. P. Sklizova, N. G. Belnikevich, L. M. Kaluzhnaya, I. G. Silinskaya, O. P. Shkurko and V. P. Borovik, Russ. Chem. Bull., 1993, 42, 1673–1676 CrossRef.
- D. Cai, J. Su, M. Huang, Y. Liu, J. Wang and L. Dai, Polym. Degr. Stab., 2011, 96, 2174–2180 CAS.
- T. Norisuye, Q. Tran-Cong-Miyata and M. Shibayama, Macromolecules, 2004, 37, 2944–2953 CrossRef CAS.
- J. C. Mccoy and M. Muthukumar, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 2193–2206 CrossRef CAS.
-
D. A. Weitz and D. J. Pine, Dynamic Light Scattering, Oxford University Press, 1993 Search PubMed.
- T. G. Mason, G. Hu and D. A. Weitz, J. Opt. Soc. Am. A, 1997, 14, 139–149 CrossRef CAS.
-
J. D. Vicente, Rheology, 1st edn, InTech Publish, 2012 Search PubMed.
- R. Xing, W. Wang, T. Jiao, K. Ma, Q. Zhang, W. Hong, H. Qiu, J. Zhou, L. Zhang and Q. Peng, ACS Sustainable Chem. Eng., 2017, 5, 4948–4956 CrossRef CAS.
- T. Hiroi, M. Ohl, T. Sakai and M. Shibayama, Macromolecules, 2014, 47, 763–770 CrossRef CAS.
- J. Li, T. Ngai and C. Wu, Polym. J., 2010, 42, 609–625 CrossRef CAS.
- K. Zhou, J. Li, Y. Lu, G. Zhang, Z. Xie and C. Wu, Macromolecules, 2009, 42, 7146–7154 CrossRef CAS.
- E. Zhang, X. Dai, X. Liu, W. Yang, W. Liu, Z. Dong, X. Qiu and X. Ji, Acta Polym. Sin., 2017, 9, 1497–1505 Search PubMed.
- B. R. Dasgupta and D. A. Weitz, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 021504 CrossRef PubMed.
- R. R. Vincent and M. A. K. Williams, Carbohydr. Res., 2009, 344, 1863–1871 CrossRef CAS PubMed.
- D. C. Morse, Macromolecules, 1998, 31, 7044–7067 CrossRef CAS.
- P. E. Rouse and K. Sittel, J. Appl. Phys., 1953, 24, 690–696 CrossRef CAS.
- J. Galvan-Miyoshi, J. Delgado and R. Castillo, Eur. Phys. J. E: Soft Matter Biol. Phys., 2008, 26, 369–377 CrossRef CAS PubMed.
- C. J. Kloxin and J. H. V. Zanten, J. Chem. Phys., 2009, 131, 134904 CrossRef PubMed.
- F. Gittes and F. C. MacKintosh, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 1241–1244 CrossRef.
-
P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, 1979 Search PubMed.
-
M. Rubinstein and R. H. Colby, Polymer Physics, 3rd edn, Clarendon Press, Oxford, 2003 Search PubMed.
-
M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Clarendon Press, Oxford, 1986 Search PubMed.
- N. Willenbacher, C. Oelschlaeger, M. Schopferer, P. Fischer, F. Cardinaux and F. Scheffold, Phys. Rev. Lett., 2007, 99, 068302 CrossRef CAS PubMed.
- M. Buchanan, M. Atakhorrami, J. F. Palierne, F. C. MacKintosh and C. F. Schmidt, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 011504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sm02185b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yong
Zhao
b,
Wenke
Yang
a,
Hongxiang
Chen
a,
Wei
Liu
a,
Xuemin
Dai
b,
Xuepeng
Qiu
a,
Yong
Zhao
b,
Wenke
Yang
a,
Hongxiang
Chen
a,
Wei
Liu
a,
Xuemin
Dai
b,
Xuepeng
Qiu