
Coordination-driven micellelization of block copolymers with gold(I) complexes induces remarkable phosphorescence enhancements with reversible mechanochromism†
Jun
Wang
,
Qun
He
,
Chen
Wang
and
Weifeng
Bu
 *
*
Key Laboratory of Nonferrous Metals Chemistry and Resources Utilization of Gansu Province, State Key Laboratory of Applied Organic Chemistry and College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: buwf@lzu.edu.cn
First published on 5th December 2017
Abstract
The coordination-driven self-assembly of block copolymers with gold(I) complexes leads to the formation of spherical micelles with remarkable phosphorescence enhancements. The rising luminescence level increases unexpectedly with increasing the degree of polymerization of the noncoordinated block. Furthermore, one of the gold(I)-containing metallopolymers shows reversible mechanochromic luminescence.
Metallopolymers represent an intriguing class of functional soft materials bearing metal centers.1 They are easily processable and exhibit various novel properties and functions associated with the metal centers, and have demonstrated great potential in the fields of catalysis, sensors, and optoelectronic devices. On the other hand, gold(I) complexes have been proposed to be utilized as catalysts,2a anticancer drugs,2b and light-emitting materials.2c The last function is frequently related to aurophilic Au(I)⋯Au(I) interactions.2d However, gold(I)-containing metallopolymers have been explored in only a few limited cases. For example, [Au(CN)2]− anions can be spatially arranged along cationic polyelectrolytes through electrostatic and Au(I)⋯Au(I) interactions, demonstrating notable luminescence enhancements.3a,b In gold(I) complexes, weakly bound ligands such as tetrahydrothiophene (THT) and trifluoroacetate can be substituted by polymers containing strong-field ligands such as poly(4-vinylpyridine) and polyisoprene-block-poly(methylenephosphine). The former polymer with [Au(C6F5)(THT)],3c [Au(C6Cl5)(THT)],3c and [Au(I)2(O2CCF3)2{μ-Ph2P(CH2)x}PPh2]3d leads to intensely phosphorescent metallopolymers associated with Au(I)⋯Au(I) interactions, while the latter with [AuCl(THT)] self-assembles into micelle-like nanostructures.3e
Herein, we report on the preparation of gold(I)-containing metallopolymers by the coordination reaction of polystyrene-block-poly(4-vinylpyridine) (Sn-b-Vm, n = 106, m = 38, PDI = 1.09; n = 576, m = 38, PDI = 1.10; n = 1114, m = 38, PDI = 1.07, Table S1, ESI†) with [Au(C6F5)(THT)]4a and [AuCl(THT)]4b and their self-assembly into spherical micelles with a remarkable enhancement of green luminescence (Fig. 1). Meanwhile, the degree of enhancement actually increases with increasing molecular weight of the noncoordinated polystyrene block. In addition, the gold(I)-containing metallopolymer formed by [Au(C6F5)(THT)] and S106-b-V38 shows reversible mechanochromic luminescence.
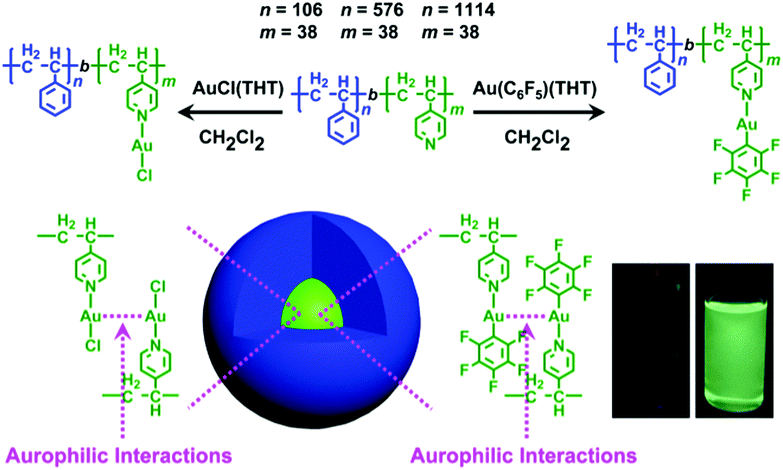 | ||
Fig. 1 Coordination-driven self-assembly of Sn-b-Vm and gold(I) complexes formed spherical micelles with remarkable enhancements of green phosphorescence. THT was weakly bound in the gold(I) complexes and can be replaced by stronger polymeric ligands of the Vm block. The dichloromethane solution of [Au(C6F5)(THT)] was totally nonemissive. In sharp contrast, intense green emission was clearly observed at an [Au(C6F5)(THT)]/V molecular ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the case of [Au(C6F5)(THT)]/S1114-b-V38 (SVAu-3, 0.4 mmol L−1 for the 4-vinylpyridine unit). :1 in the case of [Au(C6F5)(THT)]/S1114-b-V38 (SVAu-3, 0.4 mmol L−1 for the 4-vinylpyridine unit). | ||
The coordination-driven self-assembly process was monitored by using UV-vis absorption (Fig. 2a–c) and emission spectra (Fig. 2d–f), where a concentrated solution of [Au(C6F5)THT] (80 mmol L−1) was added dropwise to a dichloromethane solution of Sn-b-Vm (0.4 mmol L−1). The UV-vis absorption spectra of neat Sn-b-Vm and [Au(C6F5)(THT)] showed strong absorption bands ranging from 240 to 280 nm, while no absorption bands were observed at ≥ 300 nm. Upon treating Sn-b-Vm with [Au(C6F5)(THT)], a new low-energy shoulder band at ca. 350 nm emerged at a molar ratio of ≥0.2:1 between the [Au(C6F5)(THT)] complex and the repeating unit of poly(4-vinylpyridine) blocks and then grew steadily in intensity (Fig. 2a–c). The corresponding titration curves revealed that the absorption values almost saturated at an [Au(C6F5)(THT)]/V molecular ratio of 1:1 (Fig. 2g). This was consistent with the two-coordinated mode of pentahalophenyl–gold(I) complexes. Therefore, it was concluded that the labile ligand of THT was replaced by the polymer pyridine groups. The resulting [Au(C6F5)(4-vinylpyridine)] units were arranged closely along with the Vm blocks. Accordingly, the absorption shoulders were related to aurophilic Au(I)⋯Au(I) interactions. Of note was the fact that the absorption intensity increased unexpectedly with increasing molecular weight of the noncoordinated Sn block (n = 106, 576, and 1114) even with the same degree of polymerization of poly(4-vinylpyridine) (m = 38, Fig. 2g). For example, at the stoichiometric mixing fraction, the absorption intensity ratio was calculated to be 1.0:3.6:5.0.
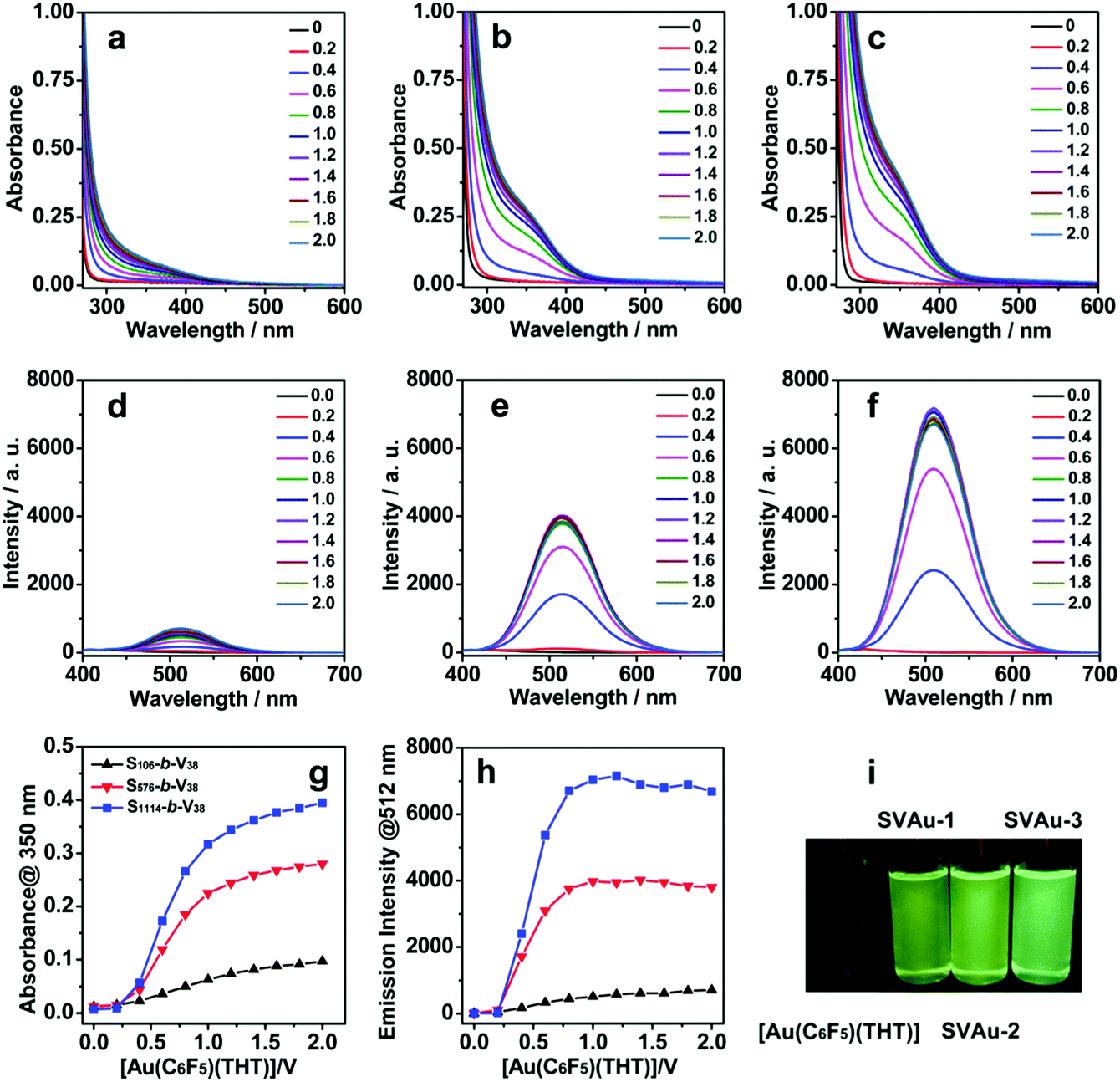 | ||
| Fig. 2 UV-vis absorption (a–c and g) and emission (d–f and h) spectral changes of Sn-b-Vm (0.4 mmol L−1 for the 4-vinylpyridine unit) upon dropwise addition of [Au(C6F5)(THT)]. The molar ratios between the [Au(C6F5)THT] complex and V repeating units were controlled at 0, 0.2, 0.4, 0.6, 0.8, 1.0 1.2, 1.4, 1.6, 1.8, and 2.0. The dichloromethane solutions of SVAu-1, SVAu-2, and SVAu-3 obtained at an [Au(C6F5)(THT)]/V molecular ratio of 1:1 showed a strong green luminescence under UV irradiation at 365 nm, while the dichloromethane solution of [Au(C6F5)(THT)] was nonemissive (i). | ||
On the other hand, both Sn-b-Vm and [Au(C6F5)(THT)] were nonemissive in their solution and solid states. However, the dropwise addition of [Au(C6F5)THT] to the solutions of Sn-b-Vm (0.4 mmol L−1) induced a steady increase in intensity of the phosphorescent emission bands at ca. 512 nm at an [Au(C6F5)(THT)]/V molecular ratio of ≥0.2:1 (Fig. 2d–f). The phosphorescence intensity reached the maximum value at an [Au(C6F5)(THT)]/V molecular ratio of close to 1 and showed a stepwise increase with increasing molecular weights of the polystyrene blocks (Fig. 2h). Under the stoichiometric mixing conditions, the phosphorescence emissions showed an intensity ratio of 1.0:7.7:13.7. These results were commendably consistent with the aforementioned UV-vis spectral titration. The intense green phosphorescence was characteristic of the aurophilic Au(I)···Au(I) interactions that occurred between the [Au(C6F5)(4-vinylpyridine)] repeating units.3c,5 To verify the phosphorescence enhancements, the solutions formed at an [Au(C6F5)(THT)]/V molecular ratio of 1 were further examined by luminescence quantum yields (Φ). During the measurements, quinine sulfate was used as a reference in water (Φ = 0.56 in air). For the sake of convenience, the gold(I)-containing metallopolymers fabricated at the stoichiometric mixing fraction were named as SVAu-1, SVAu-2, and SVAu-3 for [Au(C6F5)(THT)]/S106-b-V38, [Au(C6F5)(THT)]/S576-b-V38, and [Au(C6F5)(THT)]/S1114-b-V38, respectively. The relative Φ values of SVAu-1, SVAu-2, and SVAu-3 were determined to be 3.0%, 6.7%, and 8.3%, respectively. This increasing order agreed well with the increasing tendency of both the UV-vis absorption (350 nm, Fig. 2a–c and g) and phosphorescence intensities (512 nm, Fig. 2d–f and h) with increasing molecular weight of the Sn block. Furthermore, the remarkable enhancements of the green phosphorescence were clearly observed under UV irradiation at 365 nm with the naked eye (Fig. 2i).
Transmission electron microscopy (TEM) images revealed that SVAu-1, SVAu-2, and SVAu-3 self-assembled in dichloromethane to form micelle-like aggregates (Fig. S1, ESI†), in which the insoluble [Au(C6F5)V38] core was encapsulated by a polystyrene corona (Fig. 1). This picture actually induced the aurophilic Au(I)⋯Au(I) interactions in the micellar core, leading to remarkable phosphorescence enhancements of the metallopolymers. To support the increasing spectral features with the increasing molecular weight of the noncoordinated Sn block, the dichloromethane solutions of SVAu-1, SVAu-2, and SVAu-3 were further examined by dynamic light scattering (DLS). The resulting hydrodynamic diameters (Dhs) were 79, 91, and 106 nm, respectively (Fig. 3a), leading to a decrease in mobility from SVAu-1 to SVAu-2 to SVAu-3 in solution. Therefore, the corresponding nonradiative decay of the excited states originating from both micelle–solvent interactions and micelle mobility would be suppressed in the order of SVAu-1 < SVAu-2 < SVAu-3. This coincided well with the increasing spectral and luminescence properties with increasing molecular weight of the Sn block.
 | ||
| Fig. 3 DLS plots of gold(I)-containing metallopolymers (SVAu-1–SVAu-6) dispersed in dichloromethane. The solution concentration was 0.4 mmol L−1 for the 4-vinylpyridine unit. | ||
In addition, SVAu-1, SVAu-2, and SVAu-3 could be collected as white solids by removing dichloromethane and THT under reduced pressure. Furthermore, the samples were further precipitated in a minimal amount of dichloromethane with copious volumes of diethylether. The combination of elemental and thermogravimetric analyses (30–700 °C)3c demonstrated the compositions of these gold(I)-containing metallopolymers (Table S2, ESI†). The gold weight contents of SVAu-1, SVAu-2, and SVAu-3 were 16.82%, 8.10%, and 5.26%, corresponding to the 50%, 84%, and 92% coordination rates of 4-vinylpyridine units with [Au(C6F5)THT], respectively. Their 1H NMR spectra showed that the characteristic resonances of the Sn blocks emerged clearly, while the resonance signals of the Vm block were not observed (Fig. S2, ESI†). This was consistent with the spherical micelles: the insoluble [Au(C6F5)V38] cores were closely enwrapped by polystyrene coronas (Fig. 1). Accordingly, the Vm block was completely immobilized, leading to the invisible proton signals. The metallopolymers showed strong green emissions and were further subjected to emission spectral measurements (Fig. S3, ESI†). In the emission spectra of SVAu-1 and SVAu-2, bands appeared at λmax values of 530 and 528 nm, respectively. In the case of SVAu-3, the emission band was blue-shifted toward a higher energy centered at 519 nm. Such a blue shift was presumably due to a longer Au(I)⋯Au(I) separation with a higher molecular weight of the Sn block in SVAu-3. The excitation spectra showed broad bands centered at 345 nm, consistent with the absorption shoulder bands at 350 nm observed in the UV-vis absorption spectra (Fig. 2a–c). The Φ values of SVAu-1, SVAu-2, and SVAu-3 in the solid state were determined to be 5.2%, 17%, and 16% by the absolute method using an integrating sphere. The lower Φ value for SVAu-1 was consistent with the lower phosphorescence intensity and the lower Φ value found in the solution of the gold(I)-containing metallopolymer formed by S106-b-V38 and [Au(C6F5)(THT)]. Their long emission lifetimes in the microsecond range were indicative of a phosphorescence origin (Table S3, ESI†).
Interestingly, with casual grinding of SVAu-1, the original green luminescence changed to bright yellow-green luminescence under UV irradiation at 365 nm (Fig. 4). Upon successive fuming with dichloromethane within 5 min, the yellow-green luminescence was changed back to the original green luminescence (Fig. 4b–d). Of note was the fact that the phosphorescent emission band associated with aurophilic Au(I)⋯Au(I) interactions only showed a slight red shift (Fig. 4a). Therefore, mechanical grinding probably induced anisotropic arrangements of the repeating unit of [Au(C6F5)V], leading to a recognizable emission color change. A similar situation was also captured in small molecular gold(I)–isocyanide complexes.6 The powder X-ray diffraction pattern of the ground sample was gentler than that of the original one, in good agreement with their different emission colors (Fig. S4, ESI†). However, such a reversible mechanochromic luminescence was not captured in the cases of SVAu-2 and SVAu-3, probably due to their higher molecular weights of the Sn blocks leading to inaccessible micellar [Au(C6F5)V38] cores by mechanical grinding.
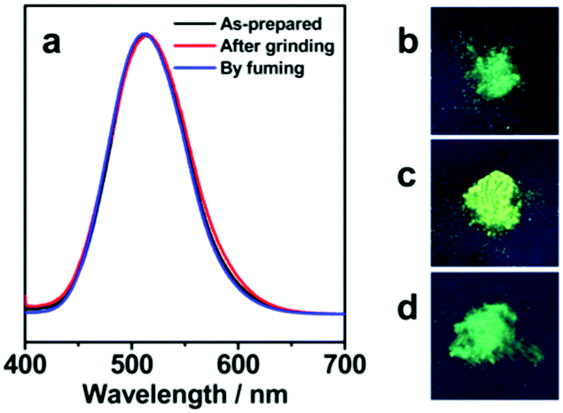 | ||
| Fig. 4 Emission spectra of SVAu-1 in the as-prepared, after-grinding, and by-fuming states (a). The inset photographs show SVAu-1 before (b) and after grinding (c), and after treatment with CH2Cl2 (d) under UV irradiation at 365 nm. | ||
To confirm the coordination mode of [Au(C6F5)(4-vinylpyridine)], SVAu-1, SVAu-2, and SVAu-3 were subjected to Fourier transform infrared (FT-IR) spectroscopy measurements. In the resulting FT-IR spectra (Fig. S5, ESI†), the pyridine band at 1414 cm−1 disappeared and shifted to 1615 cm−1. On the other hand, bands associated with the Au(C6F5) group emerged at 1505, 1063, 956, and 807 cm−1.3c These features demonstrated that the labile THT ligand was substituted by the much stronger pyridine-based ligands of Sn-b-Vm to form highly luminescent gold(I)-containing metallopolymers.
Similarly, the block copolymers of Sn-b-Vm were also titrated with [AuCl(THT)] in dichloromethane. The absorption intensity at 350 nm increased gradually (Fig. 5a–c). Similar to the situation found in the series of [Au(C6F5)(THT)]/Sn-b-Vm, this absorption band was associated with aurophilic Au(I)⋯Au(I) interactions and showed a gradual increase in intensity with increasing molecular weight of the polystyrene blocks (Fig. 5d). The DLS measurements for SVAu-4, SVAu-5, and SVAu-6 formed by [AuCl(THT)]/Sn-b-Vm exhibited Dhs of 85, 120, and 142 nm, respectively (Fig. 3b). This increasing order agrees well with the increasing absorption intensity with increasing molecular weight of the Sn block. As shown in the TEM images, they also self-assembled into micellar aggregates with a [Au(V38)Cl] core and a polystyrene corona (Fig. S1, ESI†). This was also consistent with the 1H NMR spectral result (Fig. S6, ESI†). However, the [Au(THT)Cl]/Sn-b-Vm metallopolymers were totally nonemissive in both the solution and solid states. This was presumably due to the weak field Cl− ligand leading to efficient radiationless decay via a low-lying triplet ligand field state. In their solid samples, the 4-vinylpyridine units were coordinated with [Au(THT)Cl], and the coordination rates were 45%, 66%, and 100%, respectively (Table S2, ESI†). This increasing tendency was consistent with that occurring in the case of [Au(C6F5)(THT)]/Sn-b-Vm (vide supra).
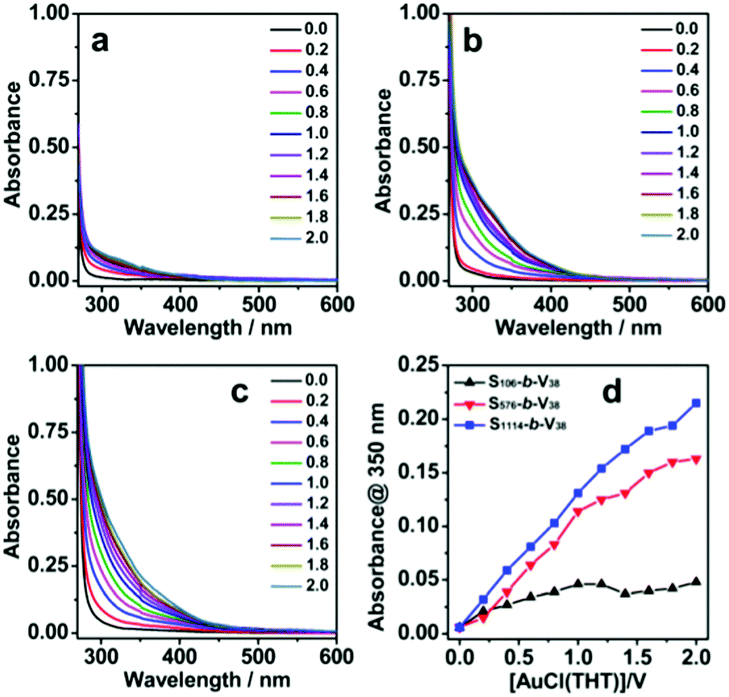 | ||
| Fig. 5 UV-vis absorption (a–d) spectral changes of Sn-b-Vm (0.4 mmol L−1 for the 4-vinylpyridine unit) upon titration with [Au(THT)Cl]. | ||
In summary, we have prepared two types of gold(I)-containing metallopolymers of [Au(C6F5)(THT)]/Sn-b-Vm and [AuCl(THT)]/Sn-b-Vm by harnessing coordination-driven self-assembly. Both of them form spherical micelles with a gold(I)-based core and a polystyrene corona. The former metallopolymers exhibit intense green phosphorescence, which is associated with aurophilic Au(I)⋯Au(I) interactions. With the increasing weight fraction of the polystyrene block, the metallopolymers show an unexpected increase in both the phosphorescence intensities and quantum yields. This is a characteristic feature of aggregation-induced emission (AIE). The AIE phenomenon has been observed typically in tetrahydrofuran/water mixture solutions of propeller-shaped (macro)molecules.7 However, the underlying mechanism is totally different from the present case and due to the effective restriction of intramolecular rotation when the AIE-active (macro)molecules aggregate in solution. Therefore, the gold(I)-containing metallopolymers can be regarded as a novel class of AIE materials, but with a different luminescence enhancement mechanism. Furthermore, the metallopolymer formed by [Au(C6F5)(THT)]/S106-b-V38 shows a typical mechanochromic feature from green to yellow-green emissions, which can be reversed back to the original state by fuming with dichloromethane. These emission-enhanced and multiple chromic metallopolymers have great potential for applications in optoelectronic devices and luminescence sensing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21474044, 21674044, and 21504036), the Fundamental Research Funds for the Central Universities (lzujbky-2017-k08 and lzujbky-2016-42) and the Open Project of State Key Laboratory of Supramolecular Structure and Materials of Jilin University (sklssm201701). This project was supported by the Open Research Fund of the State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (2017-08).Notes and references
- (a) U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892 CrossRef CAS; (b) G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS PubMed; (c) C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2011, 255, 2469 CrossRef CAS.
- (a) R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714 CrossRef CAS PubMed; (b) M. Baron, S. Bellemin-Laponnaz, C. Tubaro, M. Basato, S. Bogialli and A. Dolmella, J. Inorg. Biochem., 2014, 141, 94 CrossRef CAS PubMed; (c) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed; (d) H. Schmidbaur, Gold Bull., 2000, 33, 3 CrossRef CAS.
- (a) T. Moriuchi, K. Yoshii, C. Katano and T. Hirao, Tetrahedron Lett., 2010, 51, 4030 CrossRef CAS; (b) T. Moriuchi, M. Yamada, K. Yoshii, C. Katano and T. Hirao, Macromol. Symp., 2012, 317-318, 206 CrossRef CAS; (c) M. A. Rawashdeh-Omary, J. M. López-de-Luzuriaga, M. D. Rashdan, O. Elbjeirami, M. Monge, M. Rodríguez-Castillo and A. Laguna, J. Am. Chem. Soc., 2009, 131, 3824 CrossRef CAS PubMed; (d) M.-C. Brandys, M. C. Jennings, R. J. Puddephatt, R. E. Prud’homme and C. Géraldine Bazuin, J. Inorg. Organomet. Polym., 2010, 20, 519 CrossRef CAS; (e) K. J. T. Noonan, B. H. Gillon, V. Cappello and D. P. Gates, J. Am. Chem. Soc., 2008, 130, 12876 CrossRef CAS PubMed.
- (a) R. Usón, A. Laguna and J. Vicente, J. Chem. Soc., Chem. Commun., 1976, 353 RSC; (b) R. Usón, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, Inorg. Synth., 1989, 26, 85 CrossRef.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos, J. Pérez and M. Rodríguez-Castillo, Gold Bull., 2000, 40, 172 CrossRef.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instruments, TEM images, solid emission spectra, and FT-IR spectra. See DOI: 10.1039/c7sm02148h |
| This journal is © The Royal Society of Chemistry 2018 |