
Molecular engineering of the bio/nano-interface for enzymatic electrocatalysis in fuel cells
Alan
Le Goff
 * and
Michael
Holzinger
* and
Michael
Holzinger
Univ. Grenoble Alpes, CNRS, DCM, 38000 Grenoble, France. E-mail: alan.le-goff@univ-grenoble-alpes.fr
First published on 7th September 2018
Abstract
The fascinating topic of converting chemical energy into electric power using biological catalysts, called enzymes, and sustainable fuels motivates a large community of scientists to develop enzymatic fuel cells. Enzymes provide the advantage of catalytic oxidation and reduction processes under ecologically friendly conditions and even in complex media due to their unique specificity. However, this specificity represents a constant challenge since every enzyme has its own distinguished structure and catalytic behaviour. In this context, great efforts have been invested to understand the operational modes of promising enzymes for the bioconversion of energy. The aim is to provide chemical functions and functionalities to enable or to facilitate an electron transfer between the enzymes and the electrode material to reach the maximum efficiency of the electrocatalytic process. Original and high performance examples are summarized here in a non-exhaustive manner focusing on the wiring strategy for a series of enzymes described in the literature.
Enzymatic fuel cells
Fuel cells are envisioned in the production of electrical power from renewable fuels in order to replace power production from fossil fuels or nuclear energy. A fuel cell is composed of two electrodes, which, respectively, achieve oxidation of the fuel at the anode and reduction of the oxidant at the cathode. Hydrogen is the mainly investigated fuel for fuel cells. However, for now, hydrogen is produced from steam reforming of fossil fuels. But, newly emerging technology will aim at combining clean hydrogen production from water electrolysis and energy harvesting from solar cells. Other important fuels such as ethanol, lactate or glucose are also highly investigated for harvesting energy from biomass. Enzymatic fuel cells (EFCs) are a subclass of fuel cells, relying on purified redox enzymes instead of metal catalysts to achieve the electrocatalytic oxidation of the fuel and the reduction of the oxidant (Fig. 1). The attractiveness of enzymes lies in (i) the high substrate specificity making them excellent catalysts operating in a complex medium or mixtures, (ii) their ability to achieve high catalytic turnovers at low temperatures and neutral pH, (iii) the low overpotential requirement for electrocatalytic reactions such as H2 oxidation or O2 reduction, and (iv) their ability to achieve these reactions without the need for noble or rare metals such as platinum. Among the catalysts for fuel cell applications, enzymes need specific handling techniques. It is important to note that the active site of enzymes is buried in the quasi-insulating protein shells. This leads to two major challenges. Firstly, enzymes are big catalysts as compared to metal atoms or molecular catalysts. One of the main challenges lies in the design of highly specific surfaces with the ability to immobilize a large amount of enzymes per surface unit. This is the reason why nanomaterials have been successfully used in the design of enzyme electrodes. Secondly, electrons have to be efficiently transferred to the active site. This review is devoted to describing the strategies that have been developed in order to study this aspect of enzyme wiring. Focusing on the molecular strategies to improve the bio/nano-interface is a response to the dual challenge of increasing the number of wired enzymes per surface unit while increasing the electron transfer rates during electrocatalysis. This review focuses on the different achievements of enzyme immobilization and wiring on specifically modified surfaces using molecular technological strategies. Furthermore, the most famous enzyme families used for enzymatic fuel cell applications will be focused. Exhaustive recent reviews on enzymatic fuel cells can be found in the literature.1–3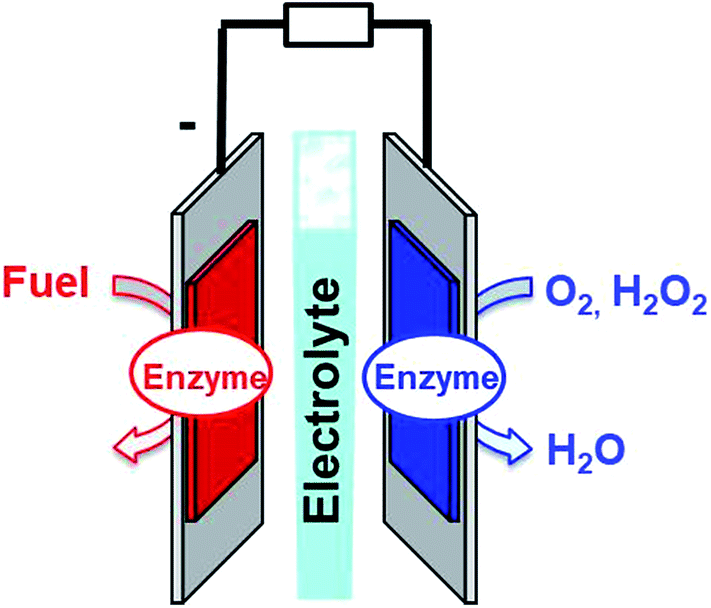 | ||
| Fig. 1 Schematic representation of an enzymatic fuel cell. | ||
Glucose fuel cells
The most widely investigated example of EFCs is glucose EFCs (GFCs). Owing to the presence of glucose and O2 in living organisms, blood or extracellular fluids, GFCs are envisioned to operate as an implanted source of energy. With respect to the high selectivity of enzymes in contrast to metal-based catalysts, enzymes are, for now, the sole type of catalysts which would be able to work in such an environment. This is the reason why many efforts have been put in to develop more powerful and more stable GFCs. These GFCs are mostly based on multicopper enzymes at the cathode for oxygen reduction.4–6 A recent alternative has been investigated using a bienzymatic system using glucose oxidase for the reduction of O2 into H2O2 and horseradish peroxidase for the high potential reduction of H2O2 at the electrode.7,8 At the anode, different types of glucose-oxidizing enzymes have been wired on the electrodes. These enzymes are mostly based on organic cofactors such as FAD for glucose oxidases and FAD-dehydrogenases, PQQ for PQQ dehydrogenases or NAD for NAD-dehydrogenases. Cellobiose dehydrogenase (CDH), which is also able to oxidize glucose, is composed of one domain containing a FAD cofactor linked to another domain containing a cytochrome β-type heme. Following the work of Heller and colleagues in the implantation of GFCs in living plants,9 GFCs have been recently integrated in fuel cells implanted in animal body. In 2010, the first example of a biofuel cell implanted in the retroperitoneal space of a rat was achieved.10 This has been followed by the implantation of GFCs in organisms such as snails,11 insects,12–14 clams,15 lobsters,16 rats,17–19 and rabbits.20 Several of these examples have produced sufficient power to run electronic devices.16,21 However, the stability of these devices is one of the main bottlenecks to overcome in order to make GFCs a reliable implanted source of energy. For now, in vitro studies show that GFCs can discontinuously run for over one year22 while it can only run for one week in vivo.10,21Hydrogen fuel cells
Another type of extensively studied enzymatic fuel cells is the hydrogen fuel cell (HFC). While the cathode is based on MCOs for the reduction of oxygen, the anode is based on hydrogen-oxidizing enzymes such as hydrogenases. Hydrogenases are metalloenzymes which catalyse the reversible oxidation of H2 into protons.1–3,23,24 The main families of hydrogenases are based on dinuclear active sites composed of a NiFe or a FeFe centre. Electrons are transferred to these active sites by a chain of iron–sulfur clusters. It has been demonstrated that NiFe exhibits exceptional electrocatalytic activity towards H2 oxidation with near-zero overpotential requirements. These enzymes have demonstrated their ability to be as efficient as Pt for the conversion of H2 into protons.24,25 However, most hydrogenases are sensitive to O2 inhibition. Therefore, several studies have been devoted to the increase of the enzyme oxygen resistance by (i) techniques such as site-directed mutagenesis,26 (ii) the use of protective viologen polymers27–30 or self-assembled redox protein nanowires,31 (iii) the study of oxygen-tolerant hydrogenases such as the membrane-bound hydrogenase from Aquifex aeolicus32–34 or NiFeSe hydrogenases35 and (iv) the design of specific fuel cell set-ups preventing the enzymes from being in contact with O2.36–38Other types of enzymatic fuel cells
While GFCs or HFCs are the main examples of the investigated EFCs, numerous efforts have also been put into the integration of a wide library of substrate-oxidizing dehydrogenases. This has especially led to the recent design of alcohol EFCs39 or lactate EFCs for harvesting energy from human sweat or tears.40,41 Furthermore, the combination of different dehydrogenases in association with an electrocatalyst of NADH oxidation allows the achievement of deep oxidation of fuels in enzyme cascade systems.42–46Challenges in enzyme wiring
Speeding up electron transfers
Among anodic electrocatalysts, redox enzymes are very peculiar catalysts, especially with respect to the so-called electrical “wiring” of these biocatalysts. Metals or molecular catalysts do not require highly specific electrode materials in order to control electron tunneling and electron flow between the electrode and catalytic centres. In contrast, enzyme active sites are located in a quasi-insulating protein shell. Furthermore, these biocatalysts are considered as big catalysts compared to metal atoms or molecular complexes. Therefore, careful attention must be paid to maximize a wired biocatalyst per surface unit. Different strategies are required to allow exchanges of electrons between the active site and the external circuit in order to optimize cell voltages and current outputs. Two main classes of electron transfer pathway have been described (Fig. 2).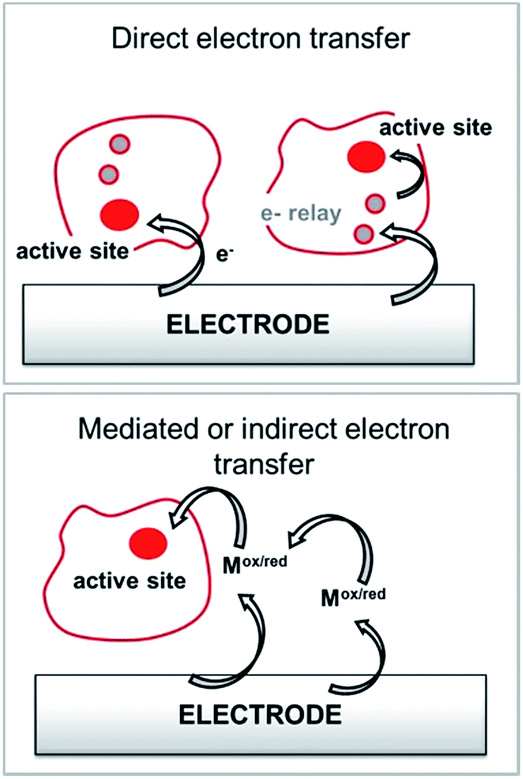 | ||
| Fig. 2 Principle for the transfer pathways of an immobilized redox enzyme. Direct electron transfer (DET) and indirect or mediated electron transfer (MET) via a small redox mediator (Mox/red). | ||
When the active site or the internal electron relays are located near the surface of the protein and the tunneling distance is short (below 2.5 nm), a direct electron transfer (DET) can occur. DET will intrinsically depend on the location of the active site inside the protein, the orientation of the protein and the shape of the protein lying on the electrode surface. When the distance between the catalytic centre or internal electron relays and the electrode surface is too high for efficient DET, a mediated electron transfer (MET) is required to ensure electron transfer. In this case, small redox molecules having the appropriate redox potential and reversible behaviour may be employed as electron shuttles, called redox mediators, to trigger electron transport. The efficiency of DET and MET will be influenced by different types of parameters. For MET, the immobilization strategy for the enzyme is not crucial, as long as a maximum enzyme concentration is ensured at a close distance to the electrode surface. For DET, the immobilization technique should take into account the orientation of the enzyme and a minimal distance between the electrode and enzyme active site. The presence of an internal electron relay, such as an iron–sulfur cluster or hemes, inside the protein often offers a facilitated ET pathway by allowing electrons to be transferred between a surface-located internal electron transfer relay and the active site.
MET is highly dependent on the redox potential, reversibility and structure of the redox mediator in order to maximize electron transfers. Furthermore, the electron transport through the redox mediator film through electron hopping should be maximized in order for it to not be the limiting factor of the electrocatalysis. All these crucial parameters, which we focus on in this review, have been taken into account in the design of molecular strategies for enzyme wiring at electrode surfaces.
Nanostructured bioelectrodes: maximizing wired enzymes per surface unit
Nanomaterial-based electrodes have become one major avenue for the development of bioelectrodes by the ability of nanoparticles to enhance the amount of immobilized active enzymes per surface or volume unit, to improve electron transfer kinetics and to optimize mass transport of biofuels.First, carbon nanotubes (CNTs) became the material of choice for bioelectrocatalytic applications. CNTs are nanowires composed of one or several graphene walls. CNTs can have many different diameters and lengths, and chirality that determine the electrical properties of single walled CNTs. Thin-diameter CNTs can have strong affinity to enzymes without leading to any protein denaturation or unfolding. This property allows the CNTs to closely approach the active site of redox enzymes, as compared to conventional graphite electrodes.47 Many examples of DET between CNTs and redox enzymes have been demonstrated, especially for metalloenzymes. Furthermore, one of the most powerful advantages of CNTs is the availability of many efficient functionalization methods in order to chemically modify their surface,48,49 owing to the reactivity of the sidewalls' pi-extended network. Several of these chemical modification reactions will be further described in the next section. Furthermore, CNT-based electrodes can be designed according to many different ways such as by direct growth on a metallic surface50 or starting from a homogeneous dispersion or solution which is further processed through transfer printing deposition,51 drop-casting,50 covalent binding of CNTs,52 CNT-doped polymers,50,53 or CNT-based paste coating.54 CNTs can furthermore be shaped55 forming free standing electrodes in the form of sheets (buckypapers),56 pellets,57 or fibres.58
Other types of carbon-based nanostructured materials such as carbon nanoparticles,59 graphene60 or carbon nanofibers33,61 have also shown enhancement of DET of metalloenzymes or improvement of enzyme surface coverages.
On the other side, the nanostructuring of gold electrodes, either by the use of mesoporous gold electrodes41,62 or gold nanoparticles63–67 or nanorods,68 has also provided high active surface area electrodes while also enhancing heterogeneous electron transfer rates. Several of these examples will be detailed in the next section of this review.
Covalent functionalization of electrodes for enzyme immobilization
In order to establish stable and controlled immobilization of enzymes on electrodes, several covalent modification techniques have been investigated in order to introduce functional groups on electrodes. Covalent techniques are especially well-suited for a controlled and stable functionalization of electrode surfaces for bringing novel physical–chemical properties to the electrode surface such as controlling the nanostructuring of the electrode or tuning the interface between the electrode and the enzyme by grafting negative or positive charges, hydrophilic or hydrophobic groups or reactive molecules. The functionalization strategy will also be strongly influenced by the nature of the electrode material.Orientation of enzymes on SAMs
Self-assembled monolayer (SAM) on gold is a functionalization technique which provides many advantages. SAMs form stable, highly homogeneous, highly organized thin functionalized films (1 to 3 nm thickness) on gold electrodes. While the terminal thiol provides stable chemisorption, the thickness of the film can be tuned according to the length of the alkyl chain. Furthermore, many terminal functions have been introduced in order to study the immobilization, orientation and electron transfer properties of metalloenzymes on gold (Fig. 3). L. Gorton and colleagues have investigated DET with metalloenzymes such as laccases and cellobiose dehydrogenase immobilized on thiol-modified gold electrodes.69–71 The use of negatively charged surfaces takes advantage of electrostatic interactions with the enzymes or a favorable dipolar moment to drive their immobilization on the electrodes.72 In a similar way, M. Tominaga and colleagues have investigated SAMs modified with carboxylic acid functions for the immobilization and orientation of bilirubin oxidases.73 Owing to the use of alkyl chains of different lengths, a tunneling distance of 17 Å could be estimated between the T1 copper centre and the gold electrode. A combined spectroscopic/electrochemical study revealed the orientation of laccases from Trametes hirsuta (ThLAC) on mixed aminophenyl-mercaptohexanol-modified gold electrodes.74 Gewirth and colleagues took advantage of the hydrophobic substrate cavity of laccase from Trametes versicolor (TvLAC) to immobilize and orientate the enzyme at the surface of an anthraquinone-based SAM.75 Its cavity has been shown earlier to strongly interact with polycyclic aromatics.76,77 Lojou and colleagues used hydrophilic and hydrophobic SAMs to influence the orientation of a NiFe hydrogenase, via a hydrophobic helix surrounded by detergents.78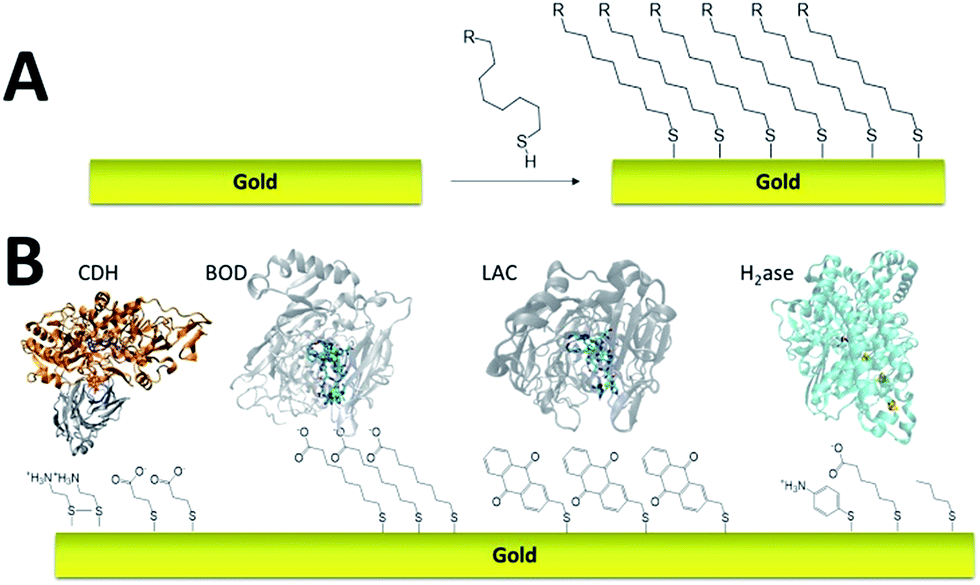 | ||
| Fig. 3 (A) Schematic representation of the formation of a SAM on gold. (B) Examples of thiols for the immobilization and direct wiring of different metalloenzymes. | ||
More recently, this technique has been successfully transferred to the surface of gold nanoparticles, playing the additional role of controlling and stabilizing the gold nanostructure. Gold nanoparticles (AuNPs) have also led to an increased number of wired enzymes per surface unit and excellent DET properties towards redox enzymes. While the immobilization of enzymes such as GOx on AuNPs did not show an efficient DET,64,65 this technique has shown a combination of highly efficient DET and high wired enzyme surface coverage with metalloenzymes such as galactose oxidases,66 laccases63,79 or hydrogenases.80 J. Abad and colleagues reported the modification of gold electrodes with a biphenyl dithiol monolayer, followed by the immobilization of thioctic acid-capped AuNPs (Fig. 4). The carboxylic acid groups were able to bind to the copper centre of galactose oxidase, triggering direct electroenzymatic oxygen reduction by the copper enzyme.66 We recently developed the functionalization of CNT-based electrodes with β-cyclodextrin-modified AuNPs79 (Fig. 4). Thanks to the supramolecular host–guest interactions, laccases specifically modified with pyrene were immobilized at these nanostructured electrodes. AuNPs increase both the enzyme surface coverage and electron transfer rates between the electrode and the T1 copper centre.
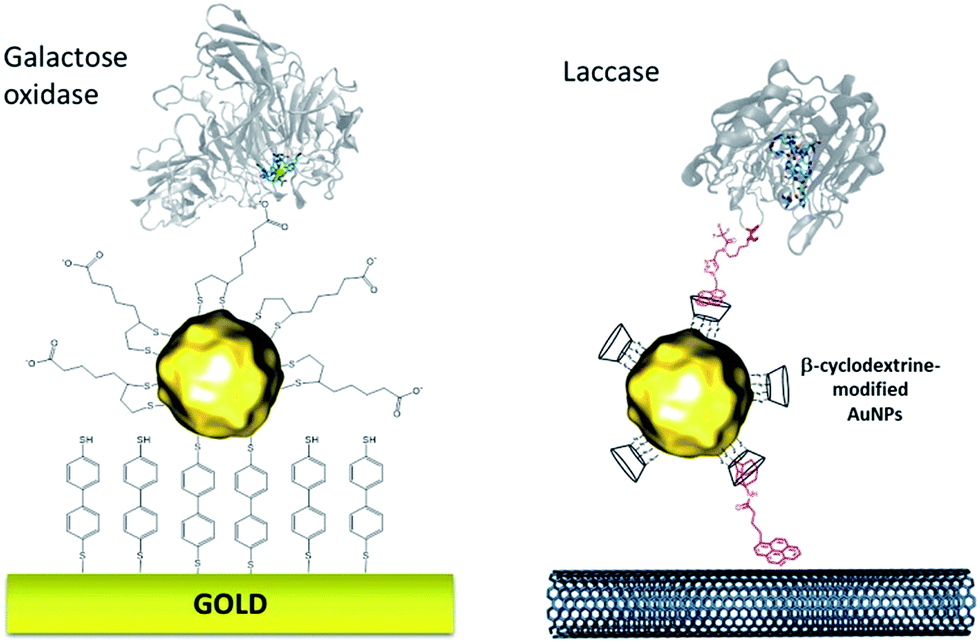 | ||
| Fig. 4 Example of thiol-modified gold nanoparticles for enzyme wiring of two copper enzymes. | ||
Electrografting
While SAMs are limited to metal surfaces such as gold or silver, electrografting has proven to be an efficient means for covalent modification of all types of conductive surfaces.81 Most electrografting processes can be controlled through electrochemical conditions and allow the formation of highly stable carbon–carbon or carbon–metal bonds. In particular, the irreversible electrochemical reduction of a phenyldiazonium derivative generates aryl radicals (driven by the formation of dinitrogen) at the vicinity of the electrode surface, triggering the grafting of aryl radicals on the surface and the subsequent growth of a polyphenylene layer (Fig. 5).82,83 A functional group in the para position of the diazonium affords the introduction of many different types of groups. Electrografting of aryldiazonium salts has proven to be highly efficient in the immobilization and wiring of redox enzymes.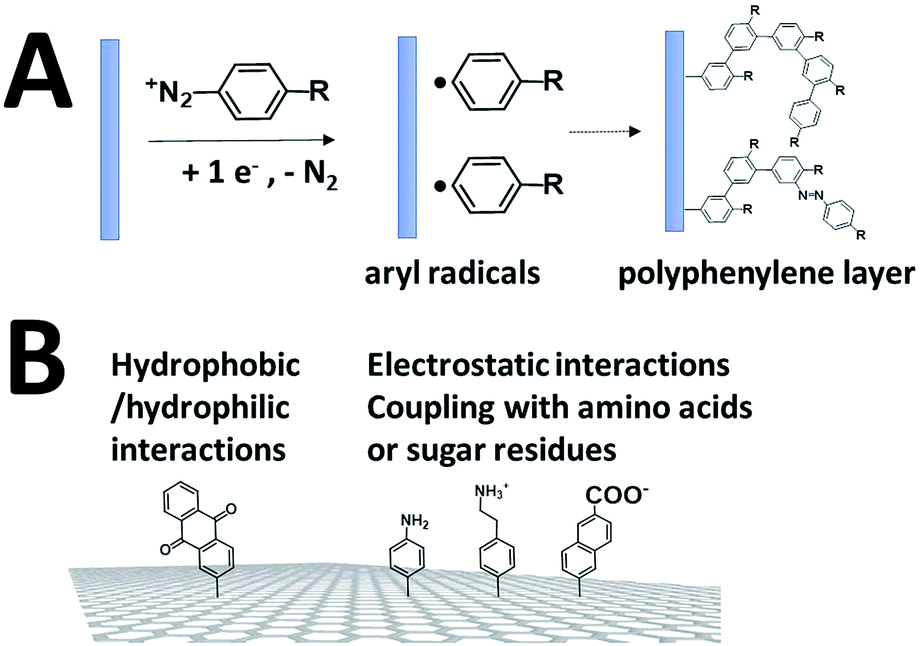 | ||
| Fig. 5 (A) Schematic representation of the grafting of aryldiazonium salts and the formation of a polyphenylene layer. (B) Diazonium functionalization of carbon-based nanomaterials for the wiring of enzymes. | ||
For instance, a set of hydrophobic amino acids forms a well-defined hydrophobic cavity at the surface of laccase from TvLAC. The Armstrong group took advantage of this hydrophobic patch to wire TvLAC at the surface of graphite electrodes previously modified by electrografting of aryldiazonium derivatives having pi-extended hydrophobic groups such as anthraquinone, anthracene, naphthalene or chrysene.76,77 A favorable orientation of the enzyme, allowing minimization of the distance between the T1 copper centre of the enzyme and the electrode, leads to efficient and stable DET for immobilized TvLAC (Fig. 5). A.L. De Lacey and colleagues extensively worked on the immobilization and orientation of hydrogenases and laccases on electrodes. The electrografting of 4-aminophenyl layers has taken advantage of the positive charge of ammonium groups to favor the orientation of a hydrogenase from Desulfovibrio gigas (via its favorable dipolar moment). The immobilization was then achieved via an amide coupling targeting carboxylic-acid residues at the surface of the enzyme. This technique was then successfully applied on CNT-based electrodes, leading to stable, high-current density direct electroenzymatic oxidation of H2.84 This technique has also been employed for the immobilization and orientation of ThLAC on low-density-graphite/AuNP composite electrodes or cellobiose dehydrogenase on covalently modified SWCNTs.85 C. Baffert and colleagues also used this technique to stabilize the direct electrochemistry of FeFe hydrogenases on an electrode.86 L. Stoica and colleagues have investigated different covalent routes to bind laccases from ThLAC on a carbon microfiber/CNT composite electrode.87 Diazonium functionalization of CNTs by aminophenyl groups was chosen to bind laccases via amide bonding with carboxylic and amino acids or via imido bonding via aldehyde residues formed by the oxidation of glycosylated enzyme residues. Inspired by the seminal work of Armstrong on the modification of planar electrodes with diazonium–anthraquinone derivatives for the direct wiring of TvLAC,76 R. Bilewicz and colleagues have transferred this technique to the surface of covalently functionalized CNTs.88,89 We have also investigated the covalent modification of reduced graphene oxide (RGO) with such anthraquinone–diazonium derivatives.60 The latter work has shown that RGO can be successfully used as a platform to bind laccases and improve its DET properties at CNT-based electrodes. As previously mentioned, the grafting of positive or negative charges can influence the orientation of the enzymes at the surface of the electrode by interactions with charged amino acids or by taking advantage of the dipolar moment of the enzyme. This strategy has successfully been adapted at the surface of CNTs for the oriented immobilization of NiFe hydrogenases or bilirubin oxidases.35,36,90 The modification of CNTs by electrografting naphthoate groups allows highly efficient DET with bilirubin oxidase from Myrothecium verrucaria (MvBOD) (Fig. 6).90 In contrast, positively charged groups such as aminoethylphenyl have a detrimental effect on the DET of MvBOD via the T1 centre. In this case, an alternative low-potential electrocatalytic pathway was evidenced.
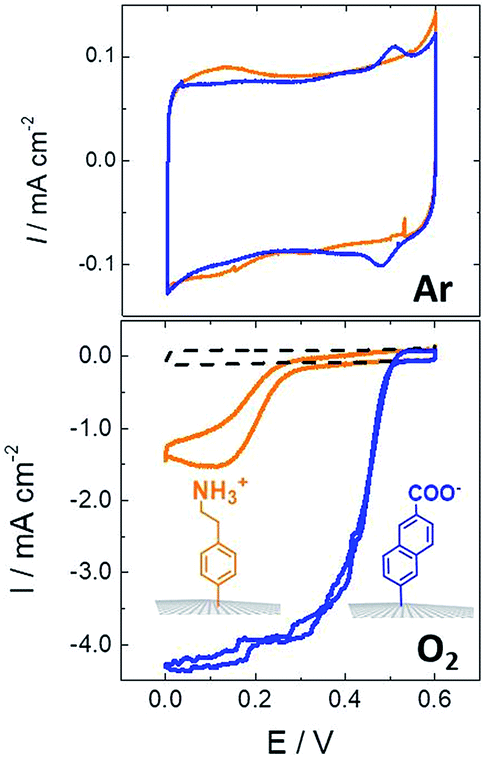 | ||
| Fig. 6 Direct electrochemistry of MvBOD immobilized on MWCNT electrodes modified with naphthoate and aminoethylphenyl diazonium salts under argon and O2 (scan rate = 10 mV s−1, McIlvaine buffer pH 7, ref: SCE).90 | ||
In a different work, we demonstrated that naphthoate-modified MWCNTs were also able to favour the direct wiring of BOD from Magnaporthe oryzae (MoBOD).91 A reductive activation mechanism was evidenced and a high potential/high current density air-breathing biocathode was designed with a maximum current density of 7 mA cm−2. Furthermore, this type of electrostatic interaction cannot be extended to BOD from other organisms. For instance, BOD from Bacillus pumilus (BpBOD) is not favourably oriented on napthoate-modified MWCNTs,58 but favourably oriented on both pristine and positively charged surfaces.36,72 We also performed a similar study for the immobilization of oxygen-tolerant hydrogenases, NiFe from Aquifex aeolicus (Aa[NiFe]H2ase)36 and NiFeSe from Desulfomicrobium baculatum (Db[NiFeSe]H2ase).35 While both enzymes show an enhanced DET at negatively charged surfaces, (Db[NiFeSe]H2ase) also exhibits enhanced DET at anthraquinone-modified MWCNTs, as confirmed by QCM-D and DFT calculations. Using these covalently modified MWCNTs, hydrogen/air fuel cells were designed. Gas-diffusion bioelectrodes were integrated in a fuel cell system, delivering a power of 0.7 mW cm−2 (ref. 36) for Aa[NiFe]H2ase/BpBOD (Fig. 7) and 0.9 mW cm−2 for the Db[NiFeSe]H2ase/MvBOD system.35
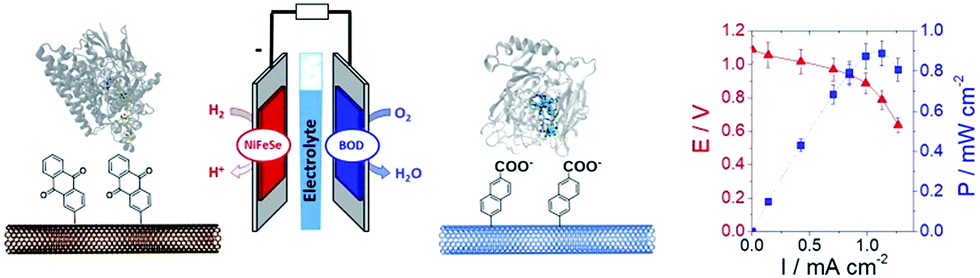 | ||
| Fig. 7 Schematic representation of an H2/air enzymatic fuel cell based on the immobilization of NiFeSe hydrogenase at the anode and MvBOD at the cathode. Polarization and power curve of the H2/air enzymatic fuel cell (phosphate buffer, pH 7.6, 25 °C). | ||
The electro-oxidation of amines has also proven to be an efficient means for the grafting of functional groups on surfaces92,93 (Fig. 8). P. Bartlett and colleagues used this strategy to covalently modify CNTs with anthraquinone groups.94,95 This leads to the efficient immobilization and orientation of laccases from ThLAC on hierarchically structured CNT/carbon microfiber/graphite electrodes. The electrocatalytic oxygen reduction exhibits high current densities with oxygen mass transport limitations. K. Kano and colleagues grafted aryl amines and alkyl amines to study the influence of negative or positive charges on the orientation of BODs.93 They concluded that the negatively charged surface in association with hydrophobic interactions promotes the orientation of BOD on Ketjen black electrodes. Other types of covalent modification of surfaces have also proven to be efficient in the immobilization and wiring of enzymes. In particular, the CNT sidewalls have demonstrated many efficient strategies to introduce functional groups at their surface, apart from electrografting techniques. The presence of oxidized sites, such as carboxylic acids at CNT defects, can also be used to graft enzymes or DET promoters. S. Minteer and colleagues have used this strategy to graft anthracene groups at CNT defects and induce the immobilization and direct wiring of laccases.96
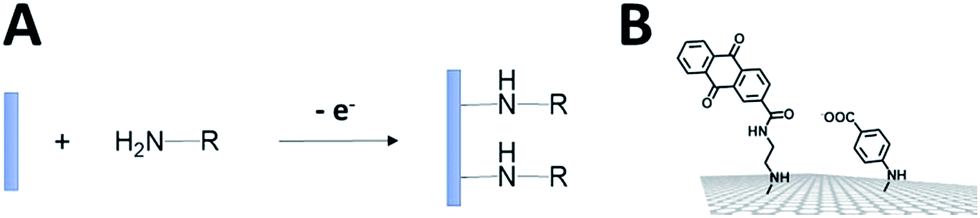 | ||
| Fig. 8 (A) Schematic representation of the electrochemical oxidation of amines on the electrode. (B) Example of amine derivatives grafted on electrodes for the direct wiring of enzymes. | ||
All these covalent strategies have also been used to graft redox mediators at the surface of nanostructured materials. In particular, ferrocene or ABTS redox mediators have been grafted for the indirect wiring of GOx97–99 and laccases,99 respectively. However, both current densities and overpotentials of MET-based electrocatalysis have shown much lower performances as compared to DET in these cases.
Non-covalent functionalization of electrodes
Pi-stacking of polyaromatics
Non-covalent strategies represent a soft and facile technique to modify the surface of nanostructured materials. This is especially the case with CNT sidewalls or graphene sheets100 where the pi-extended network strongly interacts with polycyclic aromatics by pi-stacking interactions. In particular, many types of pyrene molecules have been synthesized and employed to favour the interactions between the enzyme and CNTs (Fig. 9). Pyrene can form strong interactions with CNT sidewalls, especially in water. For instance, H. D. Abruña and colleagues characterized the strong interactions between a Co(II) bis-terpyridyl complex bearing three pyrene groups and a single-layer graphene electrode, with a ΔGads of −38.8 ± 0.2 kJ mol−1.101 The immobilisation of NADH-oxidation catalysts modified by the pyrene molecule has been studied on CNT electrodes. The control over functionalization could be assessed by using Langmuir isotherm models, giving association constants in the range of 200–2000 L mol−1 and underlining the strong and stable interaction of pyrene molecules with CNTs. The binding constant was also dependent on the nature of the pyrene molecule, the number of pyrene groups, etc.102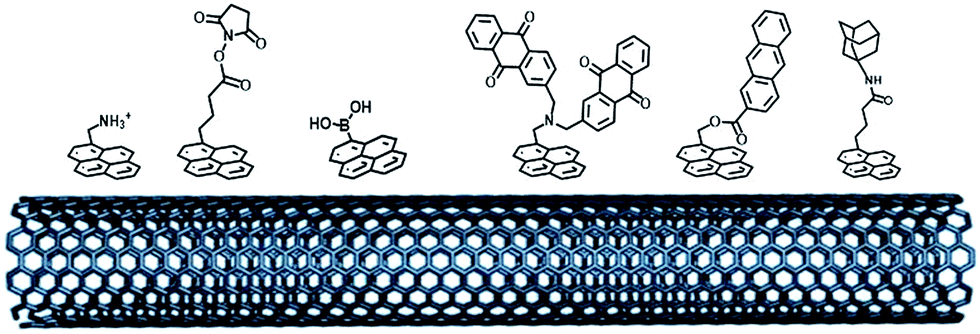 | ||
| Fig. 9 Examples of pyrene molecules for the immobilization of redox enzymes by electrostatic or hydrophobic interactions and covalent bonding. | ||
As previously mentioned, different groups have taken advantage of the hydrophobic cavity of TvLAC in order to immobilize and wire TvLAC at the surface of CNTs. We and others have developed noncovalent functionalization of CNTs by pyrene molecules to modify CNTs with anthraquinone,103 anthracene104 or adamantane groups.105 In particular, the use of pyrene–adamantane molecules led to a stable immobilization of TvLAC, owing to the strong interaction between the substrate cavity and adamantane groups (Fig. 10). The highly efficient ORR catalysis reached 2.5 mA cm−2 under oxygen.94,105
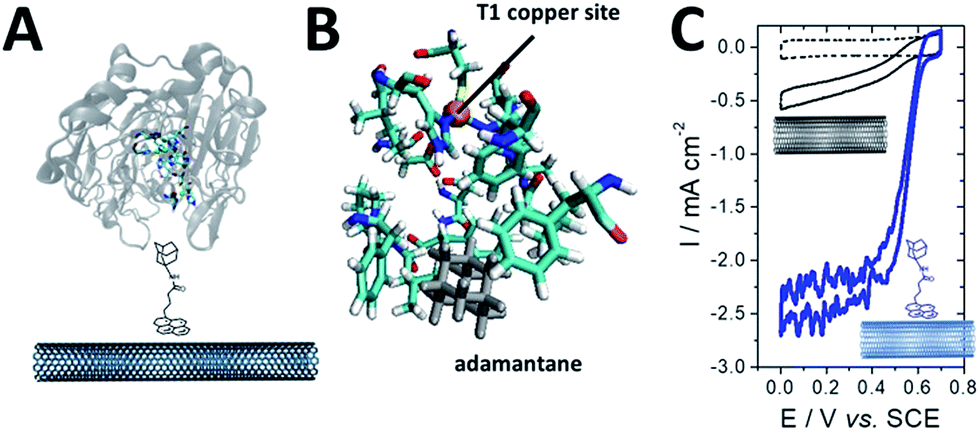 | ||
| Fig. 10 (A) Schematic representation of the immobilization of a pyrene–adamantane-modified CNT. (B) Characterization of the adamantane–enzyme interactions by molecular dynamic simulations. (C) Direct electrochemistry (CV) of MvBOD immobilized on MWCNT electrodes modified with TvLAC and MWCNT electrodes modified with pyrene-adamantane and TvLAC105 (scan rate = 10 mV s−1, McIlvaine buffer pH 5, under O2, ref: SCE). | ||
Pyrene bearing positive or negative charges, such as pyrene methylammonium or pyrene carboxylates, has been successfully used to introduce negative and positive charges to the surface of CNTs and promote DET with hydrogenases or bilirubin oxidases.72,106 Using this strategy, high power enzymatic hydrogen fuel cells delivering a maximum power density of 1.7 mW cm−2 were designed using carbon-nanofiber-based electrodes.106
The formation of a covalent bond between functional groups and amino acids located at the surface of the enzyme has also proven to be an efficient and stable route for the immobilization and orientation of metalloenzymes. Pyrene modified with an activated ester group was first designed by the H. Dai group for enzyme immobilization,107 and then widely employed on CNTs for the binding of enzymes. Katz and coworkers have modified buckypaper electrodes with this activated-ester pyrene derivative to achieve immobilization of laccases. These laccase-modified buckypaper electrodes have been integrated in a GFC using a GOx-based buckypaper anode and finally implanted in living organisms such as clams,108 snails109 and lobsters16 delivering maximum power outputs up to 160 μW cm−2 and OCVs of 0.54 V. P. Atanassov and colleagues have employed pyrene or perylene derivatives as linkers for the binding of BODs integrated in air-diffusion electrodes.110 In a different work, the same group additionally used syringaldazine in order to control the orientation of both laccases and BODs on CNT sidewalls.111 In this respect, we investigated the pi–pi-stacking of porphyrins at MWCNT sidewalls. In a substrate-mimicking strategy, we have demonstrated that protoporphyrin IX and hemin, precursors of bilirubin, were able to favor the orientation of MvBOD towards the T1 copper centre on MWCNT electrodes. High potential/high current density towards direct oxygen reduction was observed with 4 mA cm−2 maximum current density, accompanied by quasi-ideal sigmoidal CV wave shapes. These porphyrin-modified MWCNT/BOD cathodes were integrated in different types of GFCs based on buckypaper electrodes using a FAD-GDH-based bioanode112 or in a flow-through system.113
Taking advantage of the glycosylated proteins, pyrene–boronic acid has been used to modify CNT sidewalls while achieving covalent bonding with sugar residues of horseradish peroxidases. Enhanced DET, as well as high current density of 0.2 mA cm−2 towards H2O2 reduction, was observed at these bioelectrodes.114
The pi-stacking strategy has also been employed for the immobilization of redox mediators or NADH catalysts at CNT-based electrodes (Fig. 11). Pyrene-modified ferrocene,98 ABTS,115 naphthoquinone116 and polypyridyl metal complexes100,117 have been immobilized on CNTs for indirect wiring of enzymes such as GOx or laccases. Pyrene-modified (phenanthrolinequinone)RuII complexes have also been used in a dual combination with NAD-dependent dehydrogenases, achieving high current densities of 0.9 mA cm−2 towards the oxidation of glucose102,118 However, in this particular field, these redox mediators have not reached the performances of redox polymers and hydrogels yet.
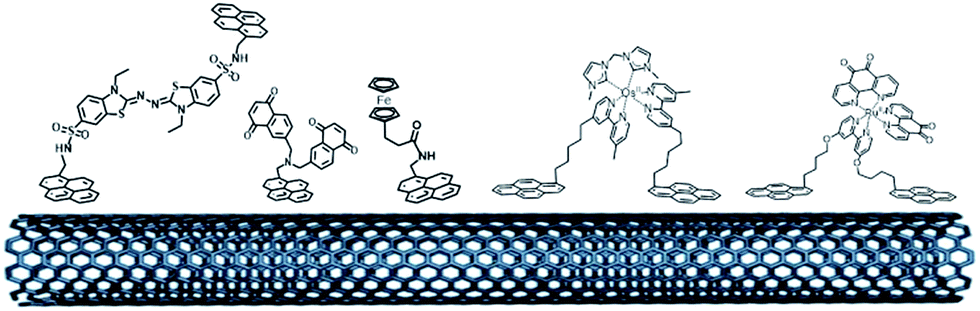 | ||
| Fig. 11 Organic and inorganic redox molecules modified with pyrene groups for the modification of CNTs and the indirect wiring of enzymes. | ||
Conducting and redox polymers (redox hydrogels, polypyrroles)
The entrapment of enzymes in a redox polymer matrix is one major technique in enzyme wiring on electrodes. These techniques avoid two major issues in enzyme wiring: the stable immobilization of the enzyme on the electrode and the electron transfer from the enzyme to the active site. Furthermore, the design of redox polymers offers many advantages by the fine control over the structure of the polymer via polymerization process conditions and the intrinsic structure of the monomer. Polymer backbones, linkers and redox molecules can be assembled during the synthesis to form a library of flexible monomers. Last but not least, polymers can also provide a biocompatible microenvironment which allows a controllable access of the substrate or inhibitors and a protective matrix towards inhibition, deactivation or denaturation. The combination of redox polymers and nanostructured materials drastically increases the number of wired enzymes per surface unit thanks to both the high specific surface of nanomaterials and the 3D-matrix effect of redox polymers.Conjugated or conducting polymers such as polypyrroles have been extensively studied in the field of enzyme electrodes for biosensors and biofuel cell applications. Furthermore, the recent combination of pyrrole oxidative electropolymerisation and carbon-based nanomaterials affords the design of stable and nanostructured bioelectrodes with a large number of wired enzymes per surface unit.119 Pyrrole monomers bearing pyrene groups have been studied at MWCNT electrodes for the supramolecular orientation of laccases via hydrophobic interactions.120 These electrogenerated polymers were compared with polymers bearing activated ester groups, which create covalent amide bonding with laccases without favoured orientation. Supramolecular immobilization of TvLAC showed more than two times higher electrocatalytic current densities as compared to amide bonds. This was also accompanied by an excellent stability over one month. Pyrrole monomers modified with methylviologen groups have been combined at SWCNT and MWCNT electrodes.28 This electrogenerated redox polymer is able to entrap NiFe hydrogenases from Desulfovibrio fructosovorans during the oxidative electropolymerisation. Thanks to MET via the methylviologen matrix, MET-based electrocatalysis exhibited a current density of 0.3 mA cm−2 for H2 oxidation. Early studies of immobilisation of GOx at electrodes have also involved the use of ferrocene-modified pyrroles.121
One of the most extensively studied redox polymers for enzymatic fuel cell applications is from the osmium-based hydrogel family (Fig. 12). This family is based on a polymer backbone which is often a polyvinyl chain and a redox coordination complex based on osmium(II) bearing N-heterocyclic ligands such as imidazole or pyridine. Owing to the rich library of available ligands, the osmium(III)/osmium(II) redox potential can be tuned to cover the whole required redox potentials to enable MET for many redox enzymes by a proper match between the redox potential of the enzyme active site and the osmium complex. In the design of enzymatic fuel cells, the A. Heller group has made a major contribution in the design of osmium hydrogels for wiring enzymes such as laccases or glucose oxidases.122,123
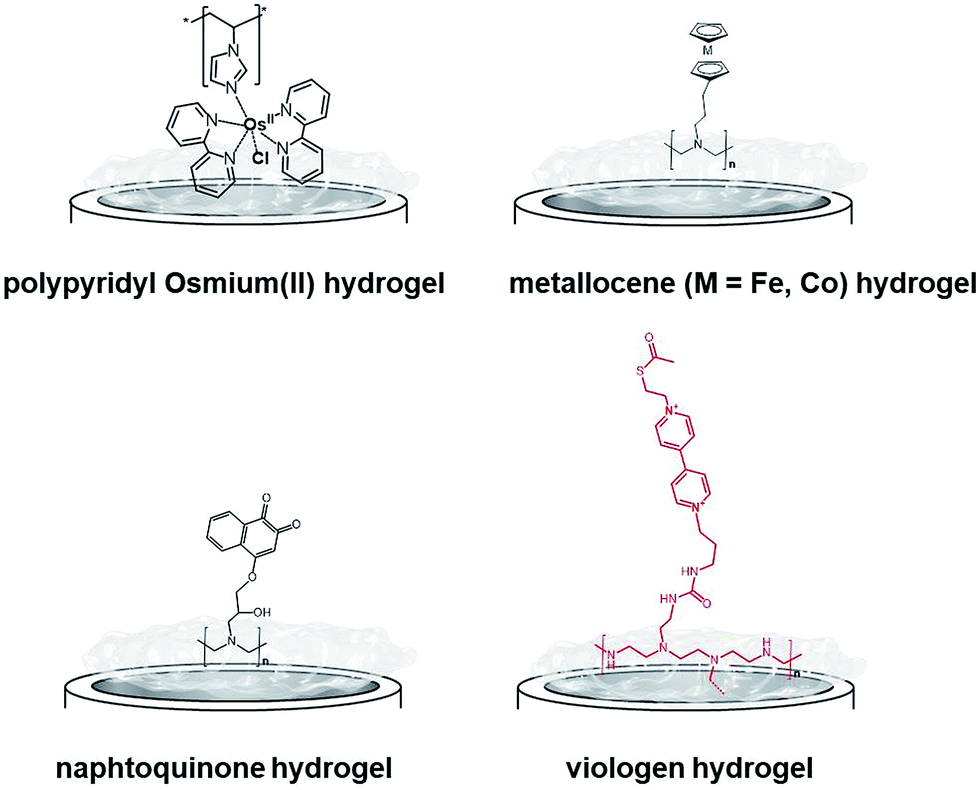 | ||
| Fig. 12 Main examples of redox hydrogels based on inorganic and organic redox mediators for the entrapment, wiring and protection of redox enzymes. | ||
However, at this time, these biofuel cells were mostly active below neutral pH. In this respect, bilirubin oxidases and laccases from different organisms124–127 were entrapped in osmium hydrogels to reduce O2 at physiological pH. Osmium polymers have been recently used with macroporous gold electrodes or CNT-based electrodes. D. Leech and colleagues combined laccase and an osmium polymer on highly ordered macroporous gold electrodes, previously obtained by nanosphere lithography using 500 nm-diameter polystyrene beads.128 N. Mano and coworkers combined osmium hydrogels with CNT fibres. A GOx-based anode and a BOD-based cathode were connected and the biofuel cell delivered 0.75 mW cm−2.129 Kwon and colleagues have designed biscrolled CNT yarns and combined osmium-based hydrogels and enzymes at these nanostructured electrodes to design a high-power glucose fuel, delivering a power density of 2.2 mW cm−2.130
While early immobilisation of glucose-oxidising enzymes in redox hydrogels has involved the use of metallocenes,121 S. Minteer and colleagues have recently modified polyethyleneimine with ferrocene for the wiring of glucose oxidases,131 PQQ-dependent alcohol and aldehyde dehydrogenases,132,133 lactate oxidases,134,135 and FAD-GDH136 on CNT-based electrodes. Cobaltocene-modified poly(allylamine) has also been recently used for the immobilization and wiring of molybdenum-dependent formate dehydrogenase for the electroenzymatic CO2 reduction at a carbon electrode.137 As an alternative to inorganic transition-metal-based hydrogels, Minteer's group have designed different naphthoquinone-based hydrogels (Fig. 12).138 Owing to the synthesis of different naphthoquinone derivatives and redox polymers, FAD-GDH was efficiently entrapped in these organic polymers and delivered low-potential/high current electrocatalytic oxidation of glucose with a maximum current density of 5.4 mA cm−2. These bioanodes were associated with DET-type cathodes relying on the orientation of BODs on functionalized CNTs. The glucose/O2 fuel cell delivered a high power density of 2.3 mW cm−2 in the presence of 100 mM glucose.
While osmium- or naphthoquinone-based hydrogels have been essentially investigated for the wiring of glucose-oxidizing enzymes or oxygen reducing copper enzymes, hydrogenases have been mostly entrapped in redox polymers based on viologen (Fig. 12). Viologen has a low reversible redox system which matches well with the redox potential of most hydrogenases. A recent study on viologen-based hydrogels has also shown their ability to achieve MET for an entrapped oxygen sensitive [NiFe] hydrogenase and to protect them from high potential deactivation and oxidative deactivation by oxygen.27,139 This anode was integrated in a biofuel cell in front of a biocathode modified with MvBOD. The biofuel cell delivered 0.2 mW cm−2 at 40 °C and pH 7.27 This strategy was further extended for the entrapment of [NiFeSe] hydrogenases29 and for O2-sensitive [FeFe] hydrogenases which can be irreversibly deactivated by O2.30 In a novel type of MET-based redox polymer, we have recently proposed the use of redox protein nanowires.31,140 A chimeric protein has been designed with a prion domain which is responsible for the formation of amyloid fibrils through self-assembly and a rubredoxin domain which is responsible for the redox activity (Fig. 13). Owing to the self-assembly, a redox protein hydrogel is formed and can be used to entrap redox enzymes and achieve MET. These protein nanowires have been successfully employed for the immobilization and wiring of TvLAC140 and Aa[NiFe]H2ases.31 The redox potential of rubredoxin is well-suited as a redox partner for the wiring of hydrogenases, achieving low-potential oxidation of H2via interprotein electron transfers. Depending on the immobilization process of the hydrogenases in the nanofiber matrix, a competition exists between DET to the electrode and MET through the nanofibers.31 However, charge transport diffusion coefficient (DCT) values are in the range of 4 × 10−12 cm2 s−1 and might limit the mediated electrocatalysis. For comparison, viologen-based hydrogel has a higher DCT value of 4.7 10−9 cm2 s−1 (ref. 139) while osmium(II)-based hydrogels have a DCT value of 5.8 10−6 cm2 s−1.141 This arises from the larger size of rubredoxin redox centres as compared to viologen and the superior mobility of viologen or osmium complexes attached to the respective polyethyleneimine or polyvinylpyridine polymer backbone.
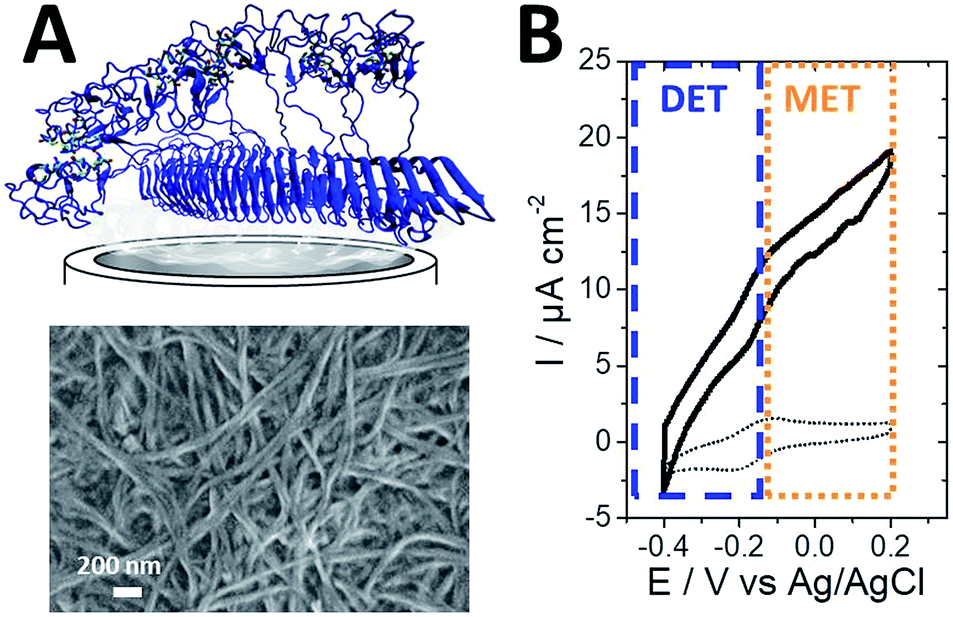 | ||
| Fig. 13 (A) Schematic representation and SEM image of the protein nanofiber electrode. (B) CV of AaH2ase entrapped in the redox protein nanofiber hydrogel, exhibiting H2 oxidation electrocatalysis with a mixed contribution of DET and MET (scan rate = 10 mV s−1, 0.1 M PBS pH 7, 60 °C, ref: Ag/AgCl). | ||
Conclusions
Many original examples of molecularly engineered systems have been developed leading to improved performances of EFCs. The power outputs and generated energy of such devices start to reach the necessary requirements to replace the currently used batteries to power some mobile devices with low power consumption. However, the operational lifetime is still insufficient for realistic applications but, this issue is more and more faced by the scientific community. One strategy relies on the confinement of enzymes in matrices with controlled pore sizes from meso- to macroporous structures to protect, to a certain extent, the protein structure thus increasing their operational lifetime and controlling substrate diffusion. These matrices will not only improve the wired enzyme loadings but also the stability of such fragile catalysts towards deactivation, inhibition or denaturation. Furthermore, the access and study of novel enzymes via mutagenesis or novel sources, accompanied by the study of structure–function relationships, will target the improvement of the enzyme–electrode interface and bioelectrocatalysis. These improvements will target different aspects of enzyme catalysis such as intrinsic stability, orientation towards the electrode, proton and electron transfer steps during the electroenzymatic reaction. There will certainly be other promising strategies in the near future which will apply this green power generation technology to commercialized products. These strategies will most likely involve the molecular engineering of the interface between these novel matrices and enzymes.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- S. Cosnier, A. J. Gross, A. Le Goff and M. Holzinger, J. Power Sources, 2016, 325, 252–263 CrossRef CAS.
- I. Mazurenko, X. Wang, A. de Poulpiquet and E. Lojou, Sustainable Energy Fuels, 2017, 1, 1475–1501 RSC.
- N. Mano and A. de Poulpiquet, Chem. Rev., 2017, 118, 2392–2468 CrossRef PubMed.
- N. Mano and L. Edembe, Biosens. Bioelectron., 2013, 50, 478–485 CrossRef CAS PubMed.
- A. Le Goff, M. Holzinger and S. Cosnier, Cell. Mol. Life Sci., 2015, 72, 941–952 CrossRef CAS PubMed.
- C. Agnès, B. Reuillard, A. Le Goff, M. Holzinger and S. Cosnier, Electrochem. Commun., 2013, 105–108 CrossRef.
- K. Elouarzaki, M. Bourourou, M. Holzinger, A. Le Goff, R. S. Marks and S. Cosnier, Energy Environ. Sci., 2015, 8, 2069–2074 RSC.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS PubMed.
- P. Cinquin, C. Gondran, F. Giroud, S. Mazabrard, A. Pellissier, F. Boucher, J.-P. Alcaraz, K. Gorgy, F. Lenouvel, S. Mathé, P. Porcu and S. Cosnier, PLoS One, 2010, e10476 CrossRef PubMed.
- L. Halámková, J. Halámek, V. Bocharova, A. Szczupak, L. Alfonta and E. Katz, J. Am. Chem. Soc., 2012, 134, 5040–5043 CrossRef PubMed.
- M. Rasmussen, R. E. Ritzmann, I. Lee, A. J. Pollack and D. Scherson, J. Am. Chem. Soc., 2012, 134, 1458–1460 CrossRef CAS PubMed.
- K. Shoji, Y. Akiyama, M. Suzuki, T. Hoshino, N. Nakamura, H. Ohno and K. Morishima, Biomed. Microdevices, 2012, 14, 1063–1068 CrossRef CAS PubMed.
- K. Shoji, Y. Akiyama, M. Suzuki, N. Nakamura, H. Ohno and K. Morishima, Biosens. Bioelectron., 2016, 78, 390–395 CrossRef CAS PubMed.
- A. Szczupak, J. Halámek, L. Halámková, V. Bocharova, L. Alfonta and E. Katz, Energy Environ. Sci., 2012, 5, 8891–8895 RSC.
- K. MacVittie, J. Halamek, L. Halámková, M. Southcott, W. D. Jemison, R. Lobel and E. Katz, Energy Environ. Sci., 2012, 6, 81–86 RSC.
- J. A. Castorena-Gonzalez, C. Foote, K. MacVittie, J. Halámek, L. Halámková, L. A. Martinez-Lemus and E. Katz, Electroanalysis, 2013, 25, 1579–1584 CrossRef CAS.
- H. Cheng, P. Yu, X. Lu, Y. Lin, T. Ohsaka and L. Mao, Analyst, 2012, 138, 179–185 RSC.
- F. C. P. F. Sales, R. M. Iost, M. V. A. Martins, M. C. Almeida and F. N. Crespilho, Lab Chip, 2013, 13, 468–474 RSC.
- T. Miyake, K. Haneda, N. Nagai, Y. Yatagawa, H. Onami, S. Yoshino, T. Abe and M. Nishizawa, Energy Environ. Sci., 2011, 4, 5008–5012 RSC.
- A. Zebda, S. Cosnier, J.-P. Alcaraz, M. Holzinger, A. Le Goff, C. Gondran, F. Boucher, F. Giroud, K. Gorgy, H. Lamraoui and P. Cinquin, Sci. Rep., 2013, 3, 1516 CrossRef CAS PubMed.
- B. Reuillard, C. Abreu, N. Lalaoui, A. Le Goff, M. Holzinger, O. Ondel, F. Buret and S. Cosnier, Bioelectrochemistry, 2015, 106, 73–76 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, Y. S. Chaudhary and F. A. Armstrong, Energy Environ. Sci., 2012, 7470–7490 RSC.
- P.-P. Liebgott, A. L. de Lacey, B. Burlat, L. Cournac, P. Richaud, M. Brugna, V. M. Fernandez, B. Guigliarelli, M. Rousset, C. Léger and S. Dementin, J. Am. Chem. Soc., 2010, 986–997 Search PubMed.
- N. Plumere, O. Ruediger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Poeller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef CAS PubMed.
- J. Baur, A. Le Goff, S. Dementin, M. Holzinger, M. Rousset and S. Cosnier, Int. J. Hydrogen Energy, 2011, 12096–12101 CrossRef CAS.
- A. Ruff, J. Szczesny, S. Zacarias, I. A. C. Pereira, N. Plumeré and W. Schuhmann, ACS Energy Lett., 2017, 2, 964–968 CrossRef.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- S. Rengaraj, R. Haddad, E. Lojou, N. Duraffourg, M. Holzinger, A. LeGoff and V. Forge, Angew. Chem., Int. Ed., 2017, 56, 7774–7778 CrossRef CAS PubMed.
- A. Ciaccafava, A. De Poulpiquet, V. Techer, M. T. Giudici-Orticoni, S. Tingry, C. Innocent and E. Lojou, Electrochem. Commun., 2012, 23, 25–28 CrossRef CAS.
- A. de Poulpiquet, A. Ciaccafava, R. Gadiou, S. Gounel, M. T. Giudici-Orticoni, N. Mano and E. Lojou, Electrochem. Commun., 2014, 42, 72–74 CrossRef CAS.
- K. Monsalve, I. Mazurenko, N. Lalaoui, A. Le Goff, M. Holzinger, P. Infossi, S. Nitsche, J. Y. Lojou, M. T. Giudici-Orticoni, S. Cosnier and others, Electrochem. Commun., 2015, 60, 216–220 CrossRef CAS.
- S. Gentil, S. M. Che Mansor, H. Jamet, S. Cosnier, C. Cavazza and A. Le Goff, ACS Catal., 2018, 3957–3964 CrossRef CAS.
- N. Lalaoui, A. de Poulpiquet, R. Haddad, A. Le Goff, M. Holzinger, S. Gounel, M. Mermoux, P. Infossi, N. Mano, E. Lojou and S. Cosnier, Chem. Commun., 2015, 51, 7447–7450 RSC.
- K. So, Y. Kitazumi, O. Shirai, K. Nishikawa, Y. Higuchi and K. Kano, J. Mater. Chem. A, 2016, 4, 8742–8749 RSC.
- H. Xia, K. So, Y. Kitazumi, O. Shirai, K. Nishikawa, Y. Higuchi and K. Kano, J. Power Sources, 2016, 335, 105–112 CrossRef CAS.
- P. K. Addo, R. L. Arechederra and S. D. Minteer, J. Power Sources, 2011, 196, 3448–3451 CrossRef CAS.
- W. Jia, G. Valdés-Ramírez, A. J. Bandodkar, J. R. Windmiller and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 7233–7236 CrossRef CAS PubMed.
- X. Xiao, T. Siepenkoetter, P. O. Conghaile, D. Leech and E. Magner, ACS Appl. Mater. Interfaces, 2018, 10, 7107–7116 CrossRef CAS PubMed.
- G. T. R. Palmore, H. Bertschy, S. H. Bergens and G. M. Whitesides, J. Electroanal. Chem., 1998, 443, 155–161 CrossRef CAS.
- D. Sokic-Lazic, A. R. de Andrade and S. D. Minteer, Electrochim. Acta, 2011, 56, 10772–10775 CrossRef CAS.
- D. Sokic-Lazic and S. D. Minteer, Biosens. Bioelectron., 2008, 24, 939–944 CrossRef CAS PubMed.
- D. Sokic-Lazic and S. D. Minteer, Electrochem. Solid-State Lett., 2009, 12, F26–F28 CrossRef CAS.
- Y. H. Kim, E. Campbell, J. Yu, S. D. Minteer and S. Banta, Angew. Chem., Int. Ed., 2013, 52, 1437–1440 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Electrochim. Acta, 2012, 82, 179–190 CrossRef CAS.
- P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214–2230 RSC.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef CAS PubMed.
- L. Hu, D. S. Hecht and G. Grüner, Chem. Rev., 2010, 110, 5790–5844 CrossRef CAS PubMed.
- K. Fuchsberger, A. Le Goff, L. Gambazzi, F. M. Toma, A. Goldoni, M. Giugliano, M. Stelzle and M. Prato, Small, 2011, 7, 524–530 CrossRef CAS PubMed.
- J. J. Gooding, Electrochim. Acta, 2005, 50, 3049–3060 CrossRef CAS.
- M. Hughes, G. Z. Chen, M. S. P. Shaffer, D. J. Fray and A. H. Windle, Chem. Mater., 2002, 14, 1610–1613 CrossRef CAS.
- P. J. Britto, K. S. V. Santhanam and P. M. Ajayan, Bioelectrochem. Bioenerg., 1996, 41, 121–125 CrossRef CAS.
- N. Li, X. Chen, L. Stoica, W. Xia, J. Qian, J. Aßmann, W. Schuhmann and M. Muhler, Adv. Mater., 2007, 19, 2957–2960 CrossRef CAS.
- A. J. Gross, M. Holzinger and S. Cosnier, Energy Environ. Sci., 2018, 11, 1670 RSC.
- B. Reuillard, A. Le Goff, C. Agnès, M. Holzinger, A. Zebda, C. Gondran, K. Elouarzaki and S. Cosnier, Phys. Chem. Chem. Phys., 2013, 4892–4896 RSC.
- M. Bourourou, M. Holzinger, K. Elouarzaki, A. Le Goff, F. Bossard, C. Rossignol, E. Djurado, V. Martin, D. Curtil, D. Chaussy, A. Maaref and S. Cosnier, Chem. Commun., 2015, 51, 14574–14577 RSC.
- K. Szot, W. Nogala, J. Niedziolka-Jönsson, M. Jönsson-Niedziolka, F. Marken, J. Rogalski, C. N. Kirchner, G. Wittstock and M. Opallo, Electrochim. Acta, 2009, 54, 4620–4625 CrossRef CAS.
- N. Lalaoui, A. LeGoff, M. Holzinger, M. Mermoux and S. Cosnier, Chem.–Eur. J., 2015, 21, 3198–3201 CrossRef CAS PubMed.
- A. de Poulpiquet, H. Marques-Knopf, V. Wernert, M. T. Giudici-Orticoni, R. Gadiou and E. Lojou, Phys. Chem. Chem. Phys., 2013, 16, 1366–1378 RSC.
- U. Salaj-Kosla, S. Pöller, W. Schuhmann, S. Shleev and E. Magner, Bioelectrochemistry, 2013, 91, 15–20 CrossRef CAS PubMed.
- C. Gutiérrez-Sánchez, M. Pita, C. Vaz-Domínguez, S. Shleev and A. L. De Lacey, J. Am. Chem. Soc., 2012, 134, 17212–17220 CrossRef PubMed.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- J. T. Holland, C. Lau, S. Brozik, P. Atanassov and S. Banta, J. Am. Chem. Soc., 2011, 133, 19262–19265 CrossRef CAS PubMed.
- J. M. Abad, M. Gass, A. Bleloch and D. J. Schiffrin, J. Am. Chem. Soc., 2009, 131, 10229–10236 CrossRef CAS PubMed.
- M. Falk, V. Andoralov, Z. Blum, J. Sotres, D. B. Suyatin, T. Ruzgas, T. Arnebrant and S. Shleev, Biosens. Bioelectron., 2012, 37, 38–45 CrossRef CAS PubMed.
- C. Di Bari, S. Shleev, A. L. De Lacey and M. Pita, Bioelectrochemistry, 2016, 107, 30–36 CrossRef CAS PubMed.
- A. Lindgren, T. Larsson, T. Ruzgas and L. Gorton, J. Electroanal. Chem., 2000, 494, 105–113 CrossRef CAS.
- M. Pita, S. Shleev, T. Ruzgas, V. M. Fernández, A. I. Yaropolov and L. Gorton, Electrochem. Commun., 2006, 8, 747–753 CrossRef CAS.
- P. Ramírez, N. Mano, R. Andreu, T. Ruzgas, A. Heller, L. Gorton and S. Shleev, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1364–1369 CrossRef PubMed.
- I. Mazurenko, K. Monsalve, J. Rouhana, P. Parent, C. Laffon, A. Le Goff, S. Szunerits, R. Boukherroub, M.-T. Giudici-Orticoni, N. Mano and E. Lojou, ACS Appl. Mater. Interfaces, 2016, 8, 23074–23085 CrossRef CAS PubMed.
- M. Tominaga, M. Ohtani and I. Taniguchi, Phys. Chem. Chem. Phys., 2008, 10, 6928–6934 RSC.
- C. Vaz-Domínguez, M. Pita, A. L. de Lacey, S. Shleev and A. Cuesta, J. Phys. Chem. C, 2012, 16532–16540 CrossRef.
- M. S. Thorum, C. A. Anderson, J. J. Hatch, A. S. Campbell, N. M. Marshall, S. C. Zimmerman, Y. Lu and A. A. Gewirth, J. Phys. Chem. Lett., 2010, 1, 2251–2254 CrossRef CAS PubMed.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710 RSC.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2008, 140, 319–335 RSC.
- A. Ciaccafava, P. Infossi, M. Ilbert, M. Guiral, S. Lecomte, M. T. Giudici-Orticoni and E. Lojou, Angew. Chem., Int. Ed., 2012, 51, 953–956 CrossRef CAS PubMed.
- N. Lalaoui, P. Rousselot-Pailley, V. Robert, Y. Mekmouche, R. Villalonga, M. Holzinger, S. Cosnier, T. Tron and A. Le Goff, ACS Catal., 2016, 6, 1894–1900 CrossRef CAS.
- K. Monsalve, M. Roger, C. Gutierrez-Sanchez, M. Ilbert, S. Nitsche, D. Byrne-Kodjabachian, V. Marchi and E. Lojou, Bioelectrochemistry, 2015, 106, 47–55 CrossRef CAS PubMed.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1997, 119, 201–207 CrossRef CAS.
- M. Delamar, R. Hitmi, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS.
- M. A. Alonso-Lomillo, O. Rüdiger, A. Maroto-Valiente, M. Velez, I. Rodríguez-Ramos, F. J. Muñoz, V. M. Fernández and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- F. Tasca, W. Harreither, R. Ludwig, J. J. Gooding and L. Gorton, Anal. Chem., 2011, 83, 3042–3049 CrossRef CAS PubMed.
- C. Baffert, K. Sybirna, P. Ezanno, T. Lautier, V. Hajj, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Anal. Chem., 2012, 84, 7999–8005 CrossRef CAS PubMed.
- C. Gutiérrez-Sánchez, W. Jia, Y. Beyl, M. Pita, W. Schuhmann, A. L. De Lacey and L. Stoica, Electrochim. Acta, 2012, 82, 218–223 CrossRef.
- K. Stolarczyk, D. Lyp, K. Zelechowska, J. F. Biernat, J. Rogalski and R. Bilewicz, Electrochim. Acta, 2012, 79, 74–81 CrossRef CAS.
- M. Karaśkiewicz, E. Nazaruk, K. Żelechowska, J. F. Biernat, J. Rogalski and R. Bilewicz, Electrochem. Commun., 2012, 20, 124–127 CrossRef.
- N. Lalaoui, M. Holzinger, A. Le Goff and S. Cosnier, Chem.–Eur. J., 2016, 22, 10494–10500 CrossRef CAS PubMed.
- S. Gentil, M. Carrière, S. Cosnier, S. Gounel, N. Mano and A. LeGoff, Chem.–Eur. J., 2018, 24, 8404 CrossRef CAS PubMed.
- A. Adenier, M. M. Chehimi, I. Gallardo, J. Pinson and N. Vilà, Langmuir, 2004, 20, 8243–8253 CrossRef CAS PubMed.
- H. Xia, Y. Kitazumi, O. Shirai and K. Kano, J. Electroanal. Chem., 2016, 763, 104–109 CrossRef CAS.
- M. Sosna, L. Stoica, E. Wright, J. D. Kilburn, W. Schuhmann and P. N. Bartlett, Phys. Chem. Chem. Phys., 2012, 14, 11882 RSC.
- M. Sosna, J.-M. Chrétien, J. D. Kilburn and P. N. Bartlett, Phys. Chem. Chem. Phys., 2010, 12, 10018–10026 RSC.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683–1690 CrossRef CAS.
- A. Callegari, S. Cosnier, M. Marcaccio, D. Paolucci, F. Paolucci, V. Georgakilas, N. Tagmatarchis, E. Vazquez and M. Prato, J. Mater. Chem., 2004, 807–810 RSC.
- A. Le Goff, F. Moggia, N. Debou, P. Jegou, V. Artero, M. Fontecave, B. Jousselme and S. Palacin, J. Electroanal. Chem., 2010, 57–63 CrossRef CAS.
- E. Nazaruk, K. Sadowska, J. F. Biernat, J. Rogalski, G. Ginalska and R. Bilewicz, Anal. Bioanal. Chem., 2010, 398, 1651–1660 CrossRef CAS PubMed.
- A. Le Goff, B. Reuillard and S. Cosnier, Langmuir, 2013, 8736–8742 CrossRef CAS PubMed.
- J. A. Mann, J. Rodríguez-López, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 17614–17617 CrossRef CAS PubMed.
- B. Reuillard, A. Le Goff and S. Cosnier, Chem. Commun., 2014, 50, 11731–11734 RSC.
- M. Bourourou, K. Elouarzaki, N. Lalaoui, C. Agnès, A. Le Goff, M. Holzinger, A. Maaref and S. Cosnier, Chem.–Eur. J., 2013, 19, 9371–9375 CrossRef CAS PubMed.
- F. Giroud and S. D. Minteer, Electrochem. Commun., 2013, 34, 157–160 CrossRef CAS.
- N. Lalaoui, R. David, H. Jamet, M. Holzinger, A. Le Goff and S. Cosnier, ACS Catal., 2016, 4259–4264 CrossRef CAS.
- I. Mazurenko, K. Monsalve, P. Infossi, M.-T. Giudici-Orticoni, F. Topin, N. Mano and E. Lojou, Energy Environ. Sci., 2017, 10, 1966–1982 RSC.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS PubMed.
- A. Szczupak, J. Halámek, L. Halámková, V. Bocharova, L. Alfonta and E. Katz, Energy Environ. Sci., 2012, 5, 8891–8895 RSC.
- L. Halámková, J. Halámek, V. Bocharova, A. Szczupak, L. Alfonta and E. Katz, J. Am. Chem. Soc., 2012, 134, 5040–5043 CrossRef PubMed.
- C. Lau, E. R. Adkins, R. P. Ramasamy, H. R. Luckarift, G. R. Johnson and P. Atanassov, Adv. Energy Mater., 2012, 2, 162–168 CrossRef CAS.
- Y. Ulyanova, S. Babanova, E. Pinchon, I. Matanovic, S. Singhal and P. Atanassov, Phys. Chem. Chem. Phys., 2014, 16, 13367–13375 RSC.
- A. J. Gross, X. Chen, F. Giroud, C. Abreu, A. Le Goff, M. Holzinger and S. Cosnier, ACS Catal., 2017, 7, 4408–4416 CrossRef CAS.
- C. Abreu, Y. Nedellec, A. J. Gross, O. Ondel, F. Buret, A. Le Goff, M. Holzinger and S. Cosnier, ACS Appl. Mater. Interfaces, 2017, 9, 23836–23842 CrossRef CAS PubMed.
- B. Reuillard, A. Le Goff, M. Holzinger and S. Cosnier, J. Mater. Chem. B, 2014, 2, 2228–2232 RSC.
- M. Bourourou, K. Elouarzaki, M. Holzinger, C. Agnès, A. Le Goff, N. Reverdy-Bruas, D. Chaussy, M. Party, A. Maaref and S. Cosnier, Chem. Sci., 2014, 5, 2885–2888 RSC.
- F. Giroud, R. D. Milton, B.-X. Tan and S. D. Minteer, ACS Catal., 2015, 5, 1240–1244 CrossRef CAS.
- N. Lalaoui, B. Reuillard, C. Philouze, M. Holzinger, S. Cosnier and A. Le Goff, Organometallics, 2016, 35, 2987–2992 CrossRef CAS.
- N. Lalaoui, N. Means, C. Walgama, A. LeGoff, M. Holzinger, S. Krishnan and S. Cosnier, ChemElectroChem, 2016, 3, 2058–2062 CrossRef CAS.
- A. Le Goff, M. Holzinger and S. Cosnier, Analyst, 2011, 1279–1287 RSC.
- N. Lalaoui, K. Elouarzaki, A. Le Goff, M. Holzinger and S. Cosnier, Chem. Commun., 2013, 49, 9281–9283 RSC.
- J. M. Dicks, S. Hattori, I. Karube, A. P. Turner and T. Yokozawa, Ann. Biol. Clin. (Paris), 1989, 47, 607–619 CAS.
- T. Chen, S. C. Barton, G. Binyamin, Z. Gao, Y. Zhang, H.-H. Kim and A. Heller, J. Am. Chem. Soc., 2001, 123, 8630–8631 CrossRef CAS PubMed.
- V. Soukharev, N. Mano and A. Heller, J. Am. Chem. Soc., 2004, 126, 8368–8369 CrossRef CAS PubMed.
- S. C. Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Phys. Chem. B, 2001, 105, 11917–11921 CrossRef CAS.
- S. C. Barton, M. Pickard, R. Vazquez-Duhalt and A. Heller, Biosens. Bioelectron., 2002, 17, 1071–1074 CrossRef CAS PubMed.
- Y. Ackermann, D. A. Guschin, K. Eckhard, S. Shleev and W. Schuhmann, Electrochem. Commun., 2010, 12, 640–643 CrossRef CAS.
- L.-L. Kiiskinen, L. Viikari and K. Kruus, Appl. Microbiol. Biotechnol., 2002, 59, 198–204 CrossRef CAS PubMed.
- S. Boland and D. Leech, Analyst, 2012, 137, 113 RSC.
- F. Gao, L. Viry, M. Maugey, P. Poulin and N. Mano, Nat. Commun., 2010, 1, 2 Search PubMed.
- C. H. Kwon, S.-H. Lee, Y.-B. Choi, J. A. Lee, S. H. Kim, H.-H. Kim, G. M. Spinks, G. G. Wallace, M. D. Lima, M. E. Kozlov, R. H. Baughman and S. J. Kim, Nat. Commun., 2014, 5, 3928 CrossRef CAS PubMed.
- S. Aquino Neto, R. D. Milton, L. B. Crepaldi, D. P. Hickey, A. R. de Andrade and S. D. Minteer, J. Power Sources, 2015, 285, 493–498 CrossRef CAS.
- S. Aquino Neto, D. P. Hickey, R. D. Milton, A. R. De Andrade and S. D. Minteer, Biosens. Bioelectron., 2015, 72, 247–254 CrossRef CAS PubMed.
- S. Aquino Neto, R. D. Milton, D. P. Hickey, A. R. De Andrade and S. D. Minteer, J. Power Sources, 2016, 324, 208–214 CrossRef CAS.
- D. P. Hickey, R. C. Reid, R. D. Milton and S. D. Minteer, Biosens. Bioelectron., 2016, 77, 26–31 CrossRef CAS PubMed.
- R. A. Escalona-Villalpando, R. C. Reid, R. D. Milton, L. G. Arriaga, S. D. Minteer and J. Ledesma-Garcia, J. Power Sources, 2017, 342, 546–552 CrossRef CAS.
- K. L. Knoche, D. P. Hickey, R. D. Milton, C. L. Curchoe and S. D. Minteer, ACS Energy Lett., 2016, 1, 380–385 CrossRef CAS.
- M. Yuan, S. Sahin, R. Cai, S. Abdellaoui, D. P. Hickey, S. D. Minteer and R. D. Milton, Angew. Chem., Int. Ed., 2018, 57, 6582–6586 CrossRef CAS PubMed.
- R. D. Milton, D. P. Hickey, S. Abdellaoui, K. Lim, F. Wu, B. Tan and S. D. Minteer, Chem. Sci., 2015, 6, 4867–4875 RSC.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
- L. Altamura, C. Horvath, S. Rengaraj, A. Rongier, K. Elouarzaki, C. Gondran, A. L. B. Maçon, C. Vendrely, V. Bouchiat, M. Fontecave, D. Mariolle, P. Rannou, A. Le Goff, N. Duraffourg, M. Holzinger and V. Forge, Nat. Chem., 2017, 9, 157–163 CrossRef CAS PubMed.
- F. Mao, N. Mano and A. Heller, J. Am. Chem. Soc., 2003, 125, 4951–4957 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |