
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen production from formic acid decomposition in the liquid phase using Pd nanoparticles supported on CNFs with different surface properties†
Felipe
Sanchez
a,
Mohammad Hayal
Alotaibi
b,
Davide
Motta
a,
Carine Edith
Chan-Thaw
c,
Andrianelison
Rakotomahevitra
c,
Tommaso
Tabanelli
 d,
Alberto
Roldan
*a,
Ceri
Hammond
a,
Qian
He
a,
Tom
Davies
a,
Alberto
Villa
*e and
Nikolaos
Dimitratos
*a
d,
Alberto
Roldan
*a,
Ceri
Hammond
a,
Qian
He
a,
Tom
Davies
a,
Alberto
Villa
*e and
Nikolaos
Dimitratos
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: DimitratosN@cardiff.ac.uk; RoldanMartinezA@cardiff.ac.uk
bJoint Center of Excellence in Integrated Nano-Systems, King Abdulaziz City for Science and Technology, P. O. Box 6086, Riyadh 11442, Saudi Arabia
cInstitut pour la Maîtrise de l’Énergie, Université d’Antananarivo BP 566, 101 Antananarivo, Madagascar
dDipartimento di Chimica Industriale “Toso Montanari”, Alma Mater Studiorum Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy
eDipartimento di Chimica, Universitá degli studi di Milano, via Golgi 19, 20133, Milano, Italy. E-mail: Alberto.Villa@unimi.it
First published on 28th September 2018
Abstract
The development of safe and efficient H2 generation/storage materials toward a fuel-cell-based H2 economy as a long-term solution has recently received much attention. Herein we report the development of preformed Pd nanoparticles supported on carbon nanofibers (CNFs) via sol-immobilisation and impregnation techniques as efficient catalysts for the liquid phase decomposition of formic acid to H2. We used CNFs as the preferred choice of support and treated at three different temperatures for the deposition of Pd nanoparticles. They were thoroughly characterised using XRD, XPS, SEM-EDX, TEM, Raman spectroscopy and BET. We observed that the Pd particle size, metal exposure and CNF graphitisation grade play an important role in catalytic performance. We found that Pd/CNFs prepared by the sol-immobilisation method displayed higher catalytic performance than those prepared by the impregnation method, due to the smaller Pd particles and high Pd exposure of the catalysts prepared by the first method. Moreover, we have shown that the best results have been obtained using CNFs with a high graphitisation degree (HHT). DFT studies have been performed to gain insights into the reactivity and decomposition of formic acid along two-reaction pathways on Pd(111), Pd(011) and Pd(001) surfaces.
Dr Nikolaos Dimitratos (ND) is a Chancellor's Research Fellow at the Cardiff Catalysis Institute (CCI) and leads the Advanced Nanoparticulate Materials research group, which specialises in the development of nanoparticulate materials for a range of chemical applications. ND has extensive expertise in the synthesis, characterisation and catalytic testing of a range of materials for gas and liquid phase reactions. He has been involved in the development and management of several leading research projects (Auricat (ITN), Glycerol Challenge (TSB) and Dow Methane Challenge), having spent several years in the UK (at Liverpool, Cardiff and UCL) and Italy (Milan). He is currently a principal investigator in a range of projects at the UK Catalysis Hub (Nanoparticle Design) and a Co-Investigator on several EPSRC research grants totalling ∼£4.5 M. To date, ND has authored >100 research articles in top-tier journals including Science, Nature Chemistry and Angewandte Chemie, ACS Nano, as well as having four international patents, and an H-index of 38 with more than 5000 citations and has given >40 presentations (12 invited, two plenaries). |
Introduction
To meet the increasing energy demand without further damage to the environment, alternative energy sources have been considered. Amongst them, hydrogen is becoming one of the most promising energy sources for the future. Its versatility lies in the possibility to convert it to electricity or heat through electrochemical and catalytic processes.1 However, due to technical obstacles for controlling both the storage and release of hydrogen, general utilisation is limited. Therefore, research is being focused on exploring and finding storage materials that are able to fulfil these requirements.The physical storage of hydrogen has been demonstrated within porous networks such as carbon materials,2 metal–organic frameworks,3 zeolites,4 clathrate hydrates,5 and organic polymers.6 However, hydrogen can also be part of a compound i.e. it can be chemically stored, requiring a decomposition process to release it from compounds such as, ammonia borane,7 amines,8 sodium borohydride,9 alcohols,10 hydrous hydrazine11 and formic acid.12
Formic acid is a safe and convenient liquid storage material capable of releasing hydrogen under mild conditions. It may be produced during biomass upgrading, such as the hydration of 5-HMF, as well as by direct hydrogenation of CO2. Several properties such as its high stability in the absence of catalysts, being a liquid under ambient conditions, its high volumetric hydrogen content (4.4 wt%), its environmentally benign nature and nontoxicity, have promoted its importance in the field, becoming one of the most studied and promising liquid hydrogen carriers according to the U.S. Department of Energy. The decomposition of formic acid can proceed following two pathways, namely dehydrogenation (HCOOH → CO2 +H2, ΔG = −48.4 kJ mol−1) and dehydration (HCOOH → CO + H2O, ΔG = −28.5 kJ mol−1). Acidity or high temperature during the reaction usually promotes the dehydration pathway. Ultrapure hydrogen is necessary for the generation of energy with fuel cell devices, and since they are poisoned by CO, inlet CO concentration should remain below 20 ppm, otherwise a loss of long-term performance can occur. Mild conditions are also of key importance since these fuel cells are expected to supply energy to portable devices with a low heat management profile.
The homogeneous or heterogeneous catalytic decomposition of formic acid to hydrogen has thus been the topic of much investigation for the last ten years. However, issues such as the use of organic solvents, alongside separation problems, prevent the use of homogeneous catalysts in the device fabrication.13,14 Accordingly, heterogeneous catalysts have received much attention in the past few years. In this context, Pd, Au or Ag and their alloys have been widely studied.3,13,15–26 Numerous types of materials have been used as catalyst supports for formic acid dehydrogenation, i.e. activated carbon,23–25 zeolites,26 macroreticular resins,15 amines16 and MOFs.3,17,18 Carbon nanofibers (CNFs) and carbon nanotubes (CNTs) have also been successfully used as supports for the synthesis of supported metal nanoparticles for a wide range of important catalytic applications such as alcohol oxidation,27,28 nitrite reduction,29 oxygen reduction reactions,30,31 and hydrogen generation.32,33 When compared with other supports, CNFs present advantages such as the possibility to tailor the microstructures of CNFs by selecting growth techniques, the control of the surface chemistry by surface modification and the facile recovery of the metal by burning off the support.32 Moreover, carbon nanofibers present high mechanical strength, high thermal and electrical conductivity and good chemical stability in aggressive media.34 The carbon nanofibers used in this work are produced on a large scale and have applications in industry.35
In the present work, we report the effect of different types of CNFs as potential supports for depositing Pd nanoparticles and the catalytic performance in the case of the liquid phase decomposition of formic acid under mild reaction conditions. The CNFs used are: (1) pyrolytically stripped (PS-CNF), (2) low-heat treated (LHT-CNF) and (3) high-heat treated (HHT-CNF). The choice of the described CNFs allowed us to study the effect of the degree of CNF graphitisation. Furthermore, we studied the effect of these CNFs on the supported Pd particles' morphology prepared by a sol immobilisation method (using polyvinyl alcohol (PVA) as a stabiliser and NaBH4 as a reducing agent) and impregnation method – both commonly used preparative methodologies in academic and industrial groups. The characterisation of these catalysts was thoroughly performed by means of X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), scanning electron microscopy (SEM) with energy dispersive X-ray (EDX), temperature-programmed desorption of ammonia (NH3-TPD), Raman spectroscopy and BET (Brunauer–Emmett–Teller) surface area analysis. The catalyst performance study towards aqueous formic acid decomposition was carried out in a batch reactor. Finally, periodic density functional theory (DFT) calculations were employed to gain insights into the energy profiles along different decomposition pathways on Pd(001), Pd(011) and Pd(111) surfaces.
Experimental
Materials and chemicals
Formic acid (≥95%, Cat. W248703) and succinic acid (99%, Cat. S3674-100G) were obtained from Sigma Aldrich. Deionised water was used as the reaction solvent. CNFs PR24-PS, PR24-LHT and PR24-HHT were purchased from Applied Science Company. The CNFs consist of tubular fibres with an average diameter of 80 ± 30 nm and a specific surface area of around 50 m2 g−1. Schlögl and co-workers previously performed thorough characterisation of these materials.35 PS grade carbon nanofibers are produced by pyrolytically stripping the CNFs to remove polyaromatic hydrocarbons covering the outer CNF surface. LHT grade carbon nanofibers are produced by treating the CNFs at 1500 °C which carbonises any chemical vapour deposited carbon from the surface of the CNFs. HHT grade carbon nanofibers are produced by treating the CNFs at 3000 °C creating the most graphitic carbon nanofibers.Preparation of the Pd/CNF catalyst series
| pV = nRT. |
The initial rate is expressed as the volume produced within the first 5 minutes of reaction.
An approximation of the reaction order was achieved by representing the rate of gas formation versus the initial concentration of formic acid, and fitting the data to a power-law model equation:
| r = kCn |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 volumetric ratio with deionised water and analysed by using a HPLC model Agilent 1220 Infinity LC using a column MetaCarb 87H 250 × 4.6 mm, Agilent, at 60 °C and a flow rate of 0.4 mL min−1. The instrument is equipped with a Variable Wavelength (VW) Detector pre-set at 210 nm. The eluent was an aqueous solution of phosphoric acid (0.1 wt%). Succinic acid was used as an external standard for the quantification of formic acid.
:100 volumetric ratio with deionised water and analysed by using a HPLC model Agilent 1220 Infinity LC using a column MetaCarb 87H 250 × 4.6 mm, Agilent, at 60 °C and a flow rate of 0.4 mL min−1. The instrument is equipped with a Variable Wavelength (VW) Detector pre-set at 210 nm. The eluent was an aqueous solution of phosphoric acid (0.1 wt%). Succinic acid was used as an external standard for the quantification of formic acid.
The Pd bulk lattice parameter is 3.893 Å (ref. 53) which is in very good agreement with the one resulting from our cell optimisation (3.836 Å). We have modelled low-Miller index surfaces, where due to crystal symmetry, we have reduced the number of surfaces to the {111}, {011} (consisting of the equivalent (011), (101), and (110) faces), and {001} (which includes the equivalent (001), (010), and (100) faces). All have a coordination number of 12. We believe that this analysis will provide a deeper understanding since most of the literature focus on Pd(111) only.54,55 These surfaces are simulated by using a slab model containing 5 atomic layers; the two uppermost layers were relaxed without symmetry restrictions, and the bottom ones were frozen at the bulk lattice parameter. The slab contains 45 atoms per unit cell exposing an area of 66.217, 93.645 and 66.217 Å2 for (111), (011) and (001) respectively. We added a vacuum width of 15 Å between vertically repeated slabs, to avoid the interaction between them.
We defined the binding energy (EB) as the difference between the combined system and the isolated species, and the reaction energy (ER) of each step is calculated as the total energy difference between the final state (product(s)) and the initial state (reactant(s)). Thus, negative values of EB and ER indicate favourable adsorption and reaction respectively.
Results and discussion
Catalyst characterisation
The catalysts were initially characterised by X-ray diffraction (XRD), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) to determine the graphitisation degree and thus the surface properties of the material. The most visible feature in the X-ray diffraction patterns of the fresh series of catalysts is the diffraction peak at approximately 26°, assigned to the presence of the (002) plane of graphitic carbon (Fig. 1).35 The intensity of the corresponding diffraction peak increases during the heat treatment process since the utilisation of high-temperature post-treatment significantly enhances the graphitic character of carbon materials and therefore, the absence of amorphous carbon. The intensity of this characteristic diffraction peak is higher for the CNF-HHT samples.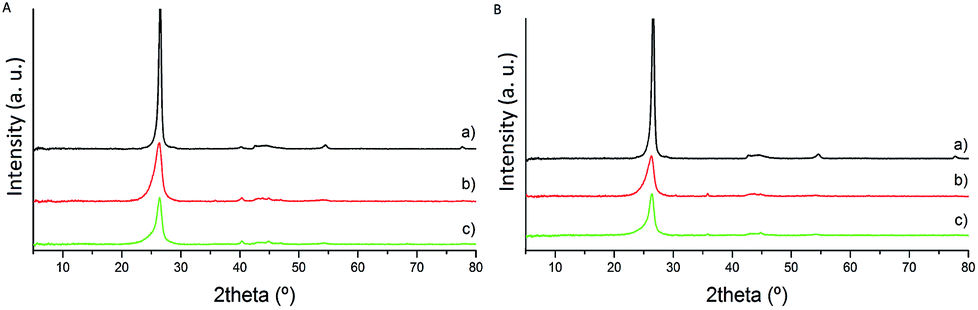 | ||
| Fig. 1 XRD patterns of fresh Pd/CNFs. (A) Catalysts synthesised by impregnation. (B) Catalysts synthesised by sol-immobilisation. (a) CNF-HHT, (b) CNF-LHT and (c) CNF-PS. | ||
In Fig. 2, the XRD patterns of the series of catalysts are presented in more detail. Between 42° and 46°, there is a broad diffraction peak attributed to either (100) or (101) planes of C. However since both hexagonal and rhombohedral graphite peaks are present in this region, it is difficult to relate specifically both species and diffraction peaks.35
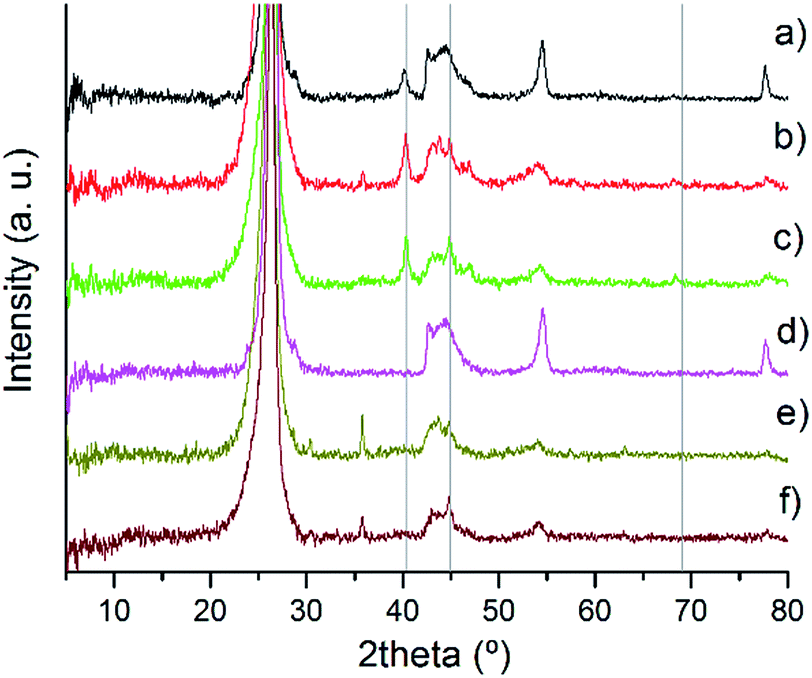 | ||
| Fig. 2 XRD patterns of fresh Pd/CNF: (a) PdIMP/CNF-HHT, (b) PdIMP/CNF-LHT, (c) PdIMP/CNF-PS, (d) PdSI/CNF-HHT, (e) PdSI/CNF-LHT, and (f) PdSI/CNF-PS. | ||
The two catalysts supported on CNF-HHT present intense and sharp diffraction peaks at 54° and 78°. They are also observed in the other catalysts albeit to a much lesser extent. The diffraction peaks at 78° correspond to the graphite (110) plane confirming the presence of rhombohedral graphite.35 The assignment of the diffraction peak at 54° is not straightforward since both graphite (004) and PdO (112) planes could be assigned to the same position.35,56 To clarify this argument, the XRD patterns of the bare supports were analysed, as shown in Fig. S1.† It confirms the presence of the graphite (004) plane; however, this peak shows a lower intensity compared with the XRD pattern of the Pd/CNFs. Therefore, we can suppose that the PdO(112) plane is overlaid by the graphite (004) plane and these results indicate the presence of PdO species. The characteristic planes (111), (200) and (220) of the face-centered cubic structure of Pd56,57 are assigned to the reflections at 2θ = 40.4°, 44.9° and 68.3° (grey lines in Fig. 2) and are present only in some catalysts as is evident from the XRD patterns. It is well known that one of the limitations of the XRD technique is its sensitivity to small crystallite sizes, with crystallite sizes lower than 5 nm being below the detection limit of the apparatus.33 Therefore, the absence of these patterns for the catalysts synthesised by sol-immobilisation could be attributed to a particle size below 5 mm. The crystallinity will be subsequently studied by means of TEM. On the other hand, for the catalysts synthesised by impregnation, it is easier to observe the presence of metallic Pd and PdO species, which means that the particles could be larger. The presence and percentage of metallic Pd and PdII species have been studied by XPS. Note that the diffraction peak at 36° only appears for the catalyst whose support has been treated at the lowest temperature; therefore, the most plausible reason is the presence of an unidentified contaminant. The XRD patterns of the used samples are displayed in Fig. S2.† The appearance and increment of the reflections at 2θ = 40.4° and 44.9° ascribed to the (111) and (200) planes of metallic Pd indicate (i) the progressive reduction of PdO to metallic Pd due to H2 generation and (ii) the increase in particle size due to agglomeration.
XPS analysis of the as-synthesised and selected used samples was carried out to determine the electronic states of the samples (Tables 1 and 2). The XPS spectra of Pd 3d of the as-synthesised fresh catalysts are presented in Fig. 3 for impregnation and sol-immobilisation methods, respectively. Pd and O content and the percentage of Pd0 for both fresh and used series derived from XPS are shown in Table 1.
| Catalyst | Pd content (at%) | (%) Pd0 on the surface | O content (at%) | |||
|---|---|---|---|---|---|---|
| Fresh | Used | Fresh | Used | Fresh | Used | |
| PdIMP/CNF-HHT | 0.71 | 0.52 | 30.1 | 74.1 | 0.94 | 1.40 |
| PdIMP/CNF-LHT | 0.57 | 0.49 | 55.0 | 73.7 | 2.60 | 2.94 |
| PdIMP/CNF-PS | 0.40 | 0.23 | 45.1 | 61.2 | 2.94 | 6.34 |
| PdSI/CNF-HHT | 0.93 | 0.72 | 51.9 | 73.1 | 2.72 | 4.05 |
| PdSI/CNF-LHT | 1.44 | 0.98 | 53.1 | 69.8 | 4.49 | 3.49 |
| PdSI/CNF-PS | 0.90 | 0.04 | 53.7 | 84.9 | 5.07 | 18.23 |
| Catalyst | C sp2 (%) | C sp3 (%) | sp2/sp3 | I D/IG | |
|---|---|---|---|---|---|
| Fresh | Used | ||||
| CNF-HHT | — | — | — | 0.11 | — |
| CNF-LHT | — | — | — | 0.71 | — |
| CNF-PS | — | — | — | 0.75 | — |
| PdIMP/CNF-HHT | 82.10 | 7.50 | 10.95 | 0.26 | 0.21 |
| PdIMP/CNF-LHT | 72.01 | 17.43 | 4.13 | 0.78 | 0.73 |
| PdIMP/CNF-PS | 66.73 | 22.37 | 2.98 | 0.67 | 0.57 |
| PdSI/CNF-HHT | 81.87 | 8.11 | 10.09 | 0.08 | 0.23 |
| PdSI/CNF-LHT | 70.44 | 19.05 | 3.70 | 0.80 | 0.90 |
| PdSI/CNF-PS | 65.80 | 23.12 | 2.85 | 0.78 | 0.71 |
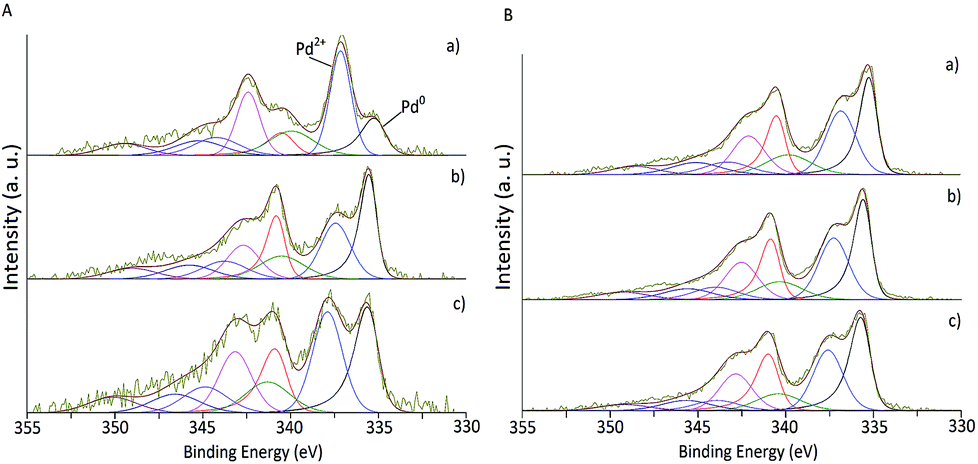 | ||
| Fig. 3 XPS spectra of fresh Pd/CNF. (A) Catalysts synthesised by impregnation. (B) Catalysts synthesised by sol-immobilisation. (a) CNF-HHT, (b) CNF-LHT and (c) CNF-PS. | ||
The Pd 3d5/2 component at 335 eV approximately was assigned to metallic Pd58 and the component at approximately 337 eV to PdII mainly present as PdO.59 As presented in Table 1, the impregnated samples display, in general, a lower Pd atomic percentage (0.40–0.71%) in contrast with the sol-immobilisation prepared samples (0.90–1.44%). From Fig. 3 and Table 1 it is evident that the majority of the catalysts prepared by the sol-immobilisation method display a higher content of Pd0 species indicating that the presence of the PVA ligand may inhibit the oxidation of the metallic Pd surface in ambient air. This feature has a significant impact on the catalyst activity as will be discussed later.
No apparent trend can be extracted for the variations in Pd surface content or the percentage of Pd0 according to modifications in the morphology through temperature treatment. XPS analyses of the used catalysts are presented in Fig. S3.†Table 1 shows a reduction in the atomic Pd content on the CNF surface. Since XPS is surface sensitive, the decrease of Pd content for the used catalysts could be explained by (i) Pd leaching during the reaction, (ii) migration of the nanoparticles to the inner wall of the nanofibers and (iii) increase of the Pd mean particle size. Table 1 displays the atomic percentage of Pd0 of the used catalysts, and it is clear that there is a significant increase of metallic Pd after the reaction, suggesting that the hydrogen released during the decomposition of HCOOH can facilitate the reduction of PdII to Pd0 species. These results are in agreement with the appearance or increase of the intensity of Pd0 diffraction peaks in the XRD patterns of the used catalysts (Fig. S2†).
XPS has been used to measure the relative concentration of sp3 and sp2 hybridisation types from the deconvolution of the C 1s region. The C 1s XPS spectra of the fresh and used series of catalysts are presented in Fig. S4.†Table 2 displays the concentration of sp3 and sp2 hybridisation and their ratio (sp2/sp3) since it determines the structural properties of carbon materials.35 The component appearing at approximately 284 eV is attributed to the presence of sp2-hybridised carbon species, whereas the component at 285 eV to the presence of sp3-hybridised carbon species.60 We observe that both the sp2 content and the ratio of sp2/sp3 increase with increasing annealing temperature as proof of surface graphitisation in agreement with previous reports.60 For instance, in the case of Pd/CNF-PS this ratio is 2.85–2.98; Pd/CNF-LHT, 3.70–4.13; and for Pd/CNF-HHT, 10.10–10.95, indicating that in CNF-HHT the sp2 bond is the most abundant. In agreement with this observation, the content of sp3-hybridised carbon species decreases as expected with increasing annealing temperature and the deposition of Pd metal nanoparticles has not significantly influenced the observed trend.
Oxygen has been analysed by the O 1s emission (Fig. S5†). O 1s peaks are well described by a Gaussian–Lorentzian curve after Shirley background subtraction. The assignment of the components in the O 1s spectrum is not straightforward. However, there is a certain degree of agreement in the position range of the peaks.
The carbon–oxygen double bond is usually located in the range of 530.0–531.5 eV; regarding the assignment of C–O–H and C–O–C, in the literature several contrasting opinions have been expressed. However, since carbon is slightly more electronegative than hydrogen, carbon–oxygen single bonds in hydroxyl groups should appear at a slightly lower energy, in our case: 531.5–531.8 eV and carbon–oxygen ether-like single bonds at 533.0–533.2 eV; carboxylic groups are usually located in the range of 534.5–535.0 eV and even though the assignment is not clear, the peak at 536.7–537.1 eV might be assigned to adsorbed water.61–63
A clear trend when comparing the preparation method is observed for the intensity of the peak attributed to the carbon–oxygen single bonds in hydroxyl groups (C–O–H). The sol-immobilisation method leads to a higher presence of C–O–H compared to C–O–C due to the presence of PVA and its hydroxyl groups as observed in Fig. S5.† As expected, carbon–oxygen ether-like single bonds decrease when increasing the annealing temperature due to the graphitisation of the surface as previously presented.
In summary, XPS analyses have shown a higher concentration of sp2 hybridisation as a result of increasing the annealing temperature and consequently the graphitisation of the support. The XPS results also indicated that there are two types of Pd species present in the fresh samples. Catalysts prepared by the sol-immobilisation technique exhibit both a higher atomic Pd content and a higher percentage of Pd0, since the PVA ligand probably tends to inhibit the oxidation of the Pd surface.
Raman spectroscopy was performed to analyse the structure and graphitisation degree of the carbon nanofibers to a better extent. Raman spectra were measured in the range of 900–1900 cm−1. Fig. 4 and 5 display the Raman spectra of the bare supports and the catalysts subjected to different temperature treatments respectively and Table 2 shows the intensity of the peaks and the ID/IG ratio. The main peaks appear at 1348 and 1572 cm−1 and are attributed to the D and G bands of sp2 clusters respectively. The D band is caused by the presence of disorder in sp2-hybridised carbon and the G band is due to the stretching of the C–C bonds in sp2 systems leading to graphitisation. The relative intensity between these two bands (ID/IG) is related to the structural disorder and subsequently to the size of graphitic domains.64 All the Pd/CNFs studied in this work present both D and G Raman bands. For the bare supports, as expected, the ID/IG ratio decreases with increasing annealing temperature, presenting a more significant drop when the support is treated at the highest temperature. This should be the trend observed for the catalysts as well. However, we observe a lower than expected ID/IG ratio for the catalysts supported on CNF/PS (Fig. S6†). This fact gave us a hypothesis to consider: during the preparation method, the catalyst was washed to remove the remaining stabiliser or metal precursor impurities and this could have affected the amorphous phase of the carbon, being washed away to some extent and subsequently showing this low ID/IG ratio.
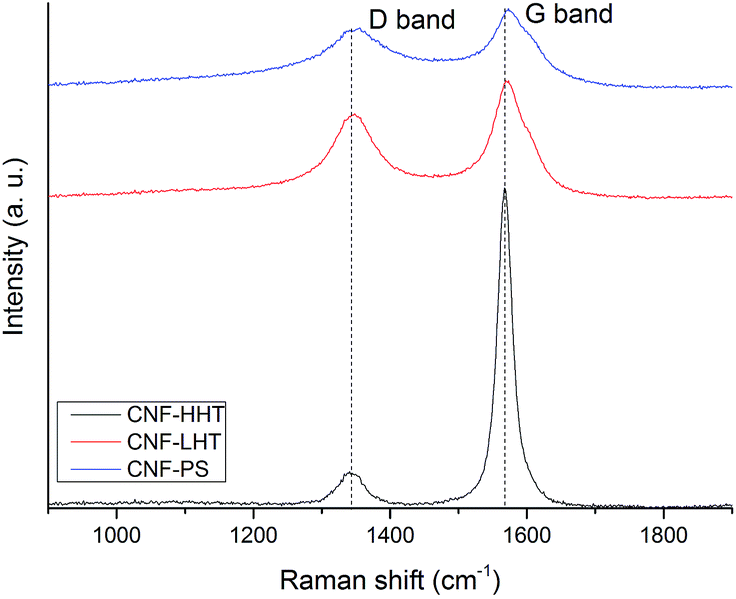 | ||
| Fig. 4 Raman spectra of the bare supports: CNF-HHT (black curve), CNF-LHT (red curve) and CNF-PS (blue curve). | ||
 | ||
| Fig. 5 Raman spectra of the fresh samples. (A) Catalysts synthesised by impregnation: PdIMP/CNF-HHT (black curve), PdIMP/CNF-LHT (red curve) and PdIMP/CNF-PS (blue curve). (B) Catalysts synthesised by sol-immobilisation. PdSI/CNF-HHT (black curve), PdSI/CNF-LHT (red curve) and PdSI/CNF-PS (blue curve). | ||
In order to prove this hypothesis, the bare support was treated with water for 1 hour under stirring and dried after filtration under the same conditions as the previous samples. The Raman spectrum of the dried sample showed a further decrease in ID/IG: 0.61 for PdIMP/CNF-PS and 0.67 for PdSI/CNF-PS, confirming the fact that the amorphous carbon was removed to a certain extent from the surface when preparing the catalyst. The difference observable between the concentrations of sp2 and sp3 from XPS for the catalysts is in agreement with the results obtained from Raman; the catalysts annealed at 3000 °C present the highest graphitisation and a big difference with LHT samples (annealed at 1500 °C) as observable in the sp2/sp3 ratio. Between LHT and PS samples there is a small difference confirmed by both the concentrations of sp2 and sp3 and the ID/IG ratio.
The particle size distributions of the catalyst series for both impregnated and sol-immobilised samples were assessed from the analyses of bright field TEM micrographs. The samples synthesised via the impregnation route (Fig. 6) present a particle size distribution in the 2.5–10 nm range, with an average particle size of 5.9 nm, and those catalysts prepared by the sol-immobilisation method present a narrower particle size distribution of 2–8 nm (Fig. 7) and a smaller average particle size of 4.2 nm.
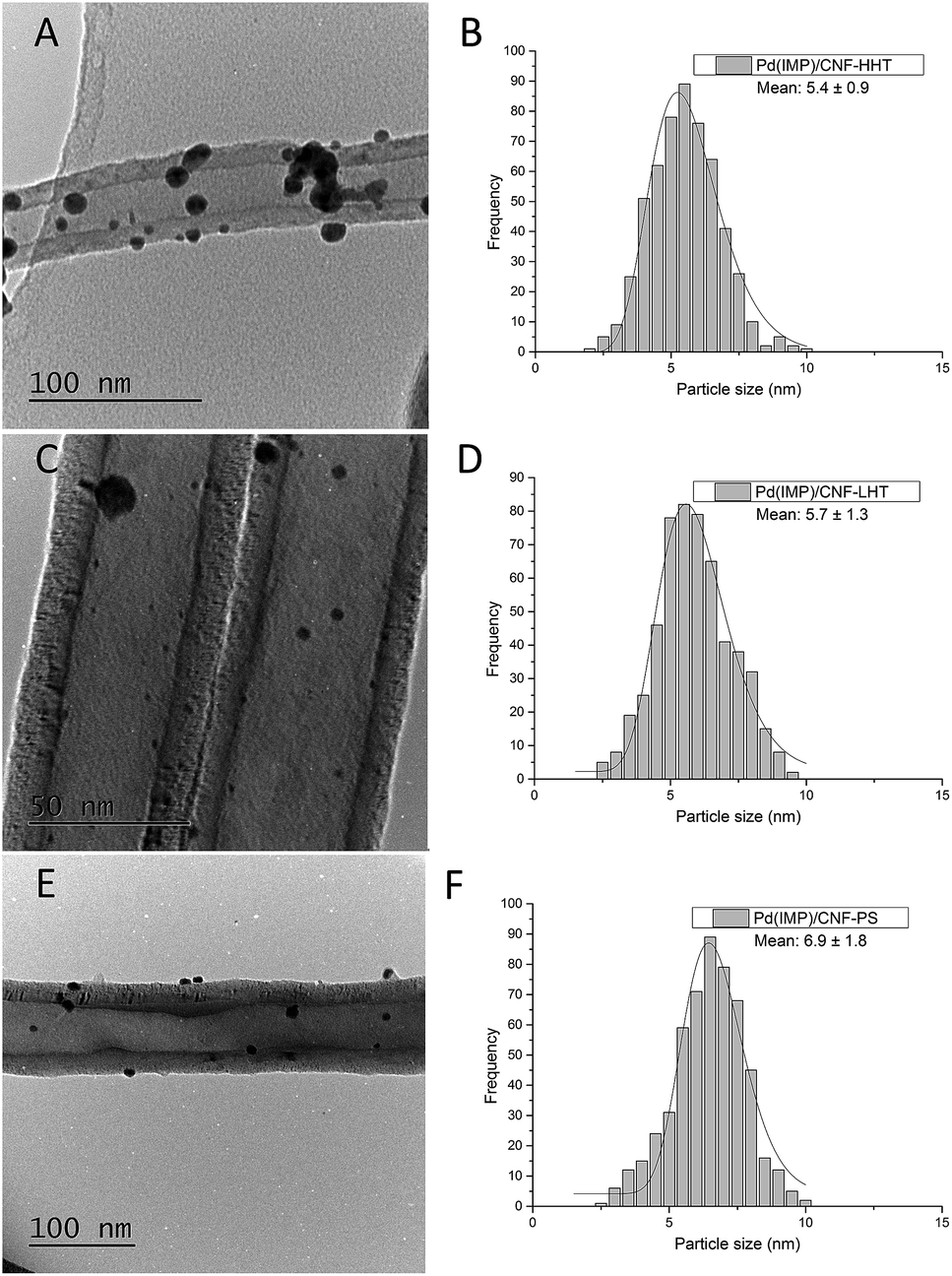 | ||
| Fig. 6 Bright field TEM micrographs and corresponding histograms of the particle size distributions for the fresh catalysts prepared by impregnation. (A and B) PdIMP/CNF-HHT, (C and D) PdIMP/CNF-LHT, and (E and F) PdIMP/CNF-PS. | ||
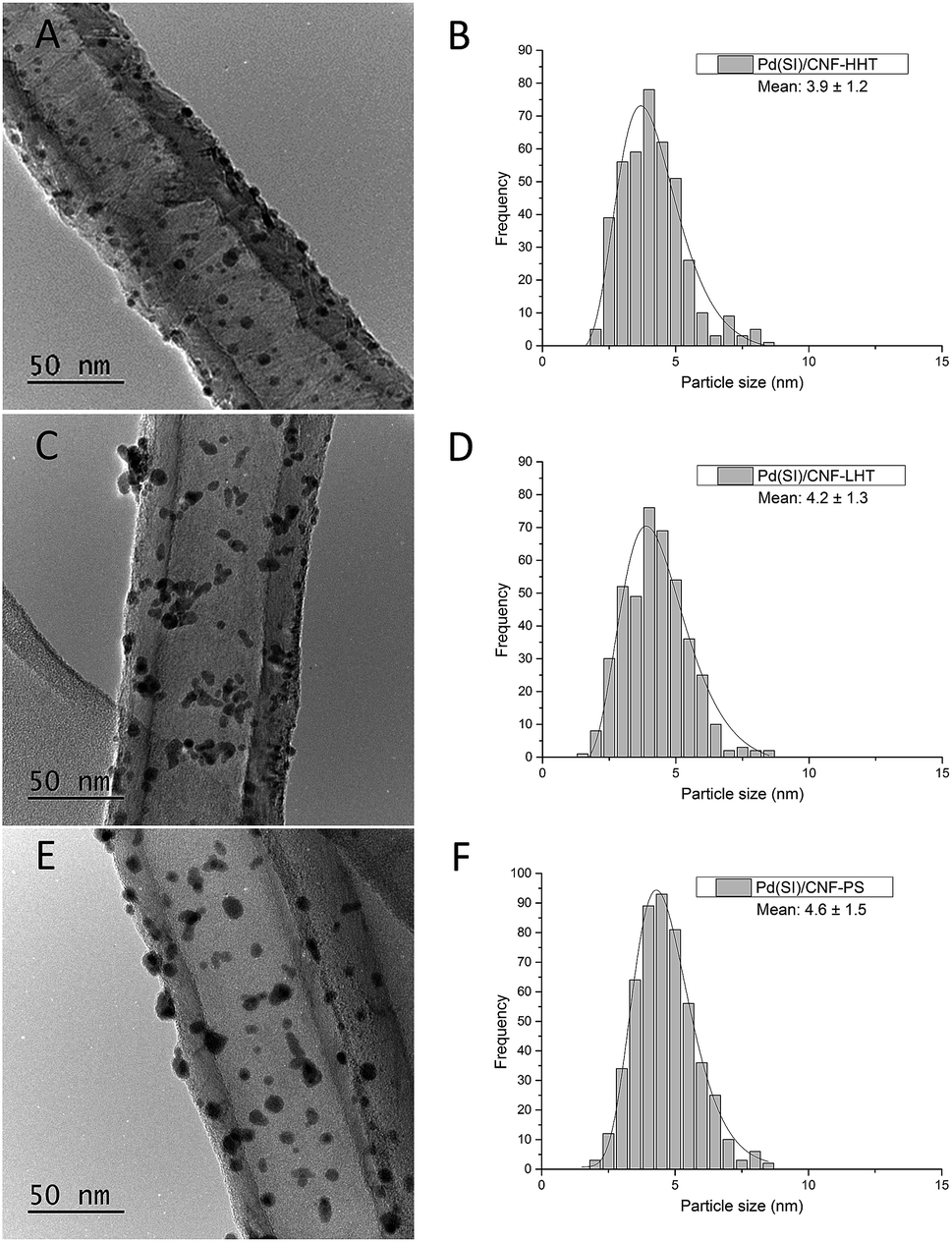 | ||
| Fig. 7 Bright field TEM micrographs and corresponding histograms of the particle size distributions for the fresh catalysts prepared by sol-immobilisation. (A and B) PdSI/CNF-HHT, (C and D) PdSI/CNF-LHT, and (E and F) PdSI/CNF-PS. | ||
Table 3 shows the mean particle size of the as-synthesised fresh and used catalysts. Fig. S7 and S8† present the representative TEM micrographs and particle size distribution for the used catalysts. No remarkable increment in the particle size has been observed for the used catalysts. TEM analyses provide evidence that the Pd nanoparticles were more evenly dispersed on the catalysts prepared by sol-immobilisation in comparison with the impregnated samples. Regarding graphitisation, it is remarkable how the particle size tends to decrease for the catalysts whose support has been treated at increasing annealing temperature. The concentration of sp2 could explain this behaviour and sp3 carbon previously calculated. Carbon with sp2 hybridisation is less reactive than carbon with sp3. The catalysts supported on CNF-HHT present a high percentage of sp2, which means that less reactive sites are present on the carbon surface. During the preparation method, sol-immobilisation or impregnation methods, the nanoparticles bind to the nanofiber preferentially by the most reactive sites of carbon (sp3). This could facilitate a smaller particle size for HHT-CNF since the concentration of sp3 sites is lower and supposedly, these sites will be distributed more spatially within the nanofiber.
| Catalyst | Fresh | Used | ||
|---|---|---|---|---|
| Mean particle size (nm) | Standard deviation | Mean particle size (nm) | Standard deviation | |
| PdIMP/CNF-HHT | 5.4 | 0.9 | 5.5 | 0.4 |
| PdIMP/CNF-LHT | 5.7 | 1.3 | 7.3 | 0.4 |
| PdIMP/CNF-PS | 6.9 | 1.8 | 6.8 | 0.3 |
| PdSI/CNF-HHT | 3.9 | 1.2 | 4.5 | 0.3 |
| PdSI/CNF-LHT | 4.2 | 1.3 | 6.2 | 0.4 |
| PdSI/CNF-PS | 4.6 | 1.5 | 5.4 | 0.4 |
The distribution and dispersion of Pd within the Pd/CNF catalysts were evaluated with SEM-EDX. Fig. 8 displays a typical SEM image of the fresh and used PdIMP/CNF-HHT. No significant variation is appreciable between the fresh and the used samples. To deeply characterise the catalysts using this technique, EDX analysis on a large area during SEM observation was performed confirming the presence of Pd. Total metal loading derived from the EDX analysis for both fresh and used catalysts series is presented in Table 4. The total metal loading of the as-synthesised catalysts approaches the theoretical value of 1 wt%, and it is not considerably affected by the preparation method used in this work.
 | ||
| Fig. 8 (A) SEM image of fresh PdIMP/CNF-HHT, (B) SEM image of used PdIMP/CNF-HHT, (C) mapping images of fresh PdIMP/CNF-HHT, and (D) mapping images of used PdIMP/CNF-HHT. | ||
| Catalyst | Pd loading EDX (wt%) | Pd loading AAS (wt%) | |
|---|---|---|---|
| Fresh | Used | Fresh | |
| PdIMP/CNF-HHT | 1.03 | 1.01 | 0.99 |
| PdIMP/CNF-LHT | 1.03 | 0.99 | 1.01 |
| PdIMP/CNF-PS | 1.04 | 0.95 | 1.02 |
| PdSI/CNF-HHT | 0.91 | 1.01 | 0.94 |
| PdSI/CNF-LHT | 1.09 | 0.96 | 1.06 |
| PdSI/CNF-PS | 1.05 | 1.05 | 1.03 |
A comparison of fresh and used catalysts does not reveal significant variation in the metal loading during the reaction; however, SEM-EDX mapping in Fig. 8 exposes that whereas for the fresh sample Pd is homogeneously distributed in the catalyst, areas with some extent of particle agglomeration and higher Pd particle density are observed in the used catalysts. EDX is a bulk-sensitive technique and we observed a similar metal loading for all the catalyst series, whereas, from the XPS data in Table 1, we obtained a higher Pd atomic percentage on the CNF surface, in the case of the sol-immobilised samples. Therefore, we can hypothesise that, with the sol-immobilisation method, Pd nanoparticles were preferentially distributed on the surface of the CNFs, whereas with the impregnation method, a portion of the formed Pd nanoparticles was located and distributed in the inner walls of CNFs, besides being deposited on the external surface.
Table 5 presents the total surface area determined from the BET equation. It ranges from approximately 36 m2 g−1 to 52 m2 g−1. This low surface area compared with that of carbon nanotubes (up to 1200 m2 g−1) is caused by the thickness of the walls (ca. 45 nm). Since BET surface area instruments have a certain error, in some cases up to 10%, no conclusion can be extracted for the apparent variations in the surface area of these samples.
| Catalyst | Support (m2 g−1) | Impregnation (m2 g−1) | Sol-immobilisation (m2 g−1) |
|---|---|---|---|
| Pd/CNF-HHT | 34 | 37 | 36 |
| Pd/CNF-LHT | 32 | 41 | 36 |
| Pd/CNF-PS | 43 | 50 | 47 |
Catalytic activity of Pd/CNF catalysts for HCOOH decomposition
Fig. 9 displays a comparison of the effect of the preparation methods on the catalytic performance of the Pd/CNFs with different morphologies (A to C). TOFs obtained are presented in Table 6 and compared with previously reported data.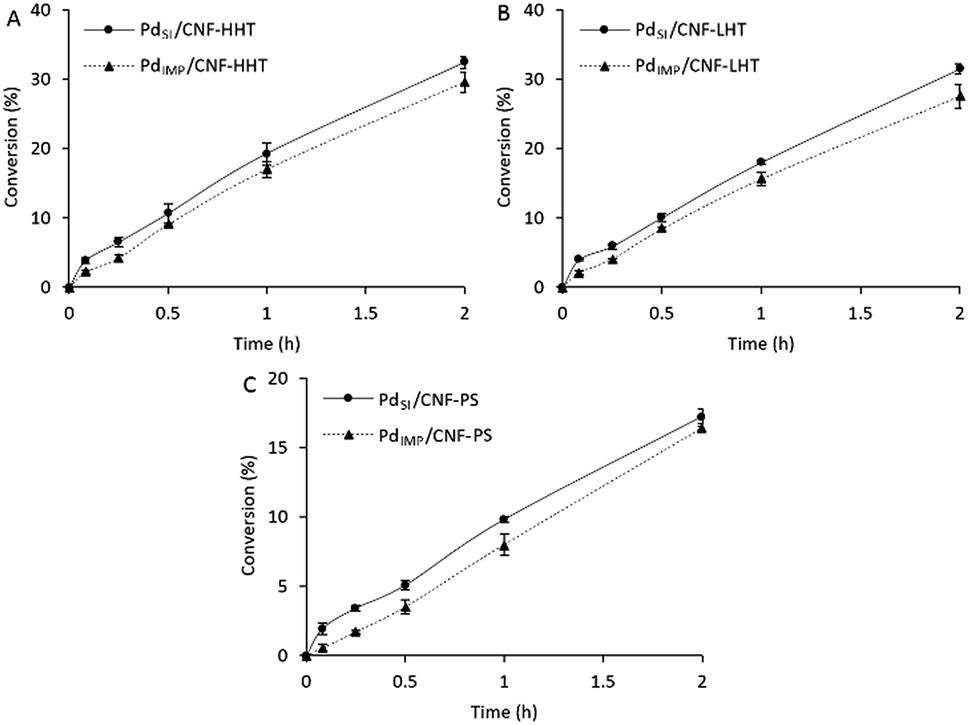 | ||
| Fig. 9 Formic acid dehydrogenation reaction on (A) Pd/CNF-HHT, (B) Pd/CNF-LHT, and (C) Pd/CNF-PS. Comparison of preparation methods. | ||
| Catalyst | T (°C) | Reagent | TOF (h−1) | Activation energy (kJ mol−1) | Ref. | |
|---|---|---|---|---|---|---|
| Initial | 2 h | |||||
| a TOF calculated after 50 min. b TOF calculated after 160 min. c TOF calculated based on the surface metal sites. | ||||||
| PdIMP/CNF-HHT | 30 | Formic acid (0.5 M) | 563.2 | 27.5 | This work | |
| PdIMP/CNF-LHT | 30 | 527.5 | ||||
| PdIMP/CNF-PS | 30 | 136.3 | ||||
| PdSI/CNF-HHT | 30 | 979.1 | 26.2 | |||
| PdSI/CNF-LHT | 30 | 965.2 | ||||
| PdSI/CNF-PS | 30 | 484.4 | ||||
| Pd/C | 21 | Formic acid (1.33 M) | 18 | 15a | 53.7 | 25 |
| 30 | 48 | 28a | ||||
| Pd/C (citric acid) | 25 | Formic acid | 64b | 57 | ||
| Pd/C | 30 | Formic acid/sodium formate 1:9 |
228.3 | 24 | ||
| Au41Pd59/C | 50 | Formic acid (1 M) | 230 | 28 ± 2 | 13 | |
| Ag@Pd (1:1) |
35 | Formic acid | 156c | 30 | 19 | |
| 50 | 252c | |||||
| Ag/Pd alloy (1:1) |
20 | 144c | ||||
| Ag42Pd58 | 50 | Formic acid (1 M) | 382 | 22 ± 1 | 65 | |
| Pd–MnOx/SiO2–NH2 | 20 | Formic acid (0.265 M) | 140 | 61.9 | 66 | |
| 50 | 1300 | |||||
| Ag0.1Pd0.9/rGO | 25 | Formic acid | 105 | 67 | ||
As observed from Table 6, the Pd/CNFs prepared by the sol-immobilisation method are more active for the formic acid liquid phase decomposition compared with the Pd/CNFs prepared by the impregnation procedure. This higher activity can be partially explained by XPS data; for the colloidal method used there is a higher atomic Pd percentage on the surface and a higher content of metallic Pd. These two facts can enhance catalyst activity. Furthermore, the particles' size plays an important role regarding the catalytic activity. Analyses by TEM confirmed a lower mean particle size for the sol-immobilisation samples. Therefore, the higher catalytic activity observed could be attributed to the smaller Pd mean particle size and as a consequence the increasing proportion of Pd surface atoms. However, after 2 hours of reaction, as observed in Fig. 9, the three pairs of catalysts led to similar final conversion even though the TOF values differ significantly during the initial period. As previously commented, PVA used during the sol-immobilisation catalyst preparation leads to a higher presence of C–O–H that might be occupying active sites and leading to a slightly faster deactivation than catalysts prepared by the impregnation method.
Table S1† presents the H2, CO2 and CO concentrations evolved from formic acid decomposition by the most active catalysts studied in this work. As observed, very low CO concentration is produced by both preparation methods, with the lowest level of CO production being observed for catalysts prepared by the colloidal method.
DFT results
The characterisation data of the supported Pd nanoparticles indicate that the majority of the Pd species are metallic and there is a variation in the Pd mean particle size in the range of 4–7 nm. Moreover, the catalytic activity showed a clear dependence on the Pd mean particle size. Therefore, in order to provide insights into the decomposition of formic acid on different sites that are expected to be present on Pd/CNFs with different particle sizes, we focused our attention to carry out DFT calculations. They provided insights into the energetics of the pathways of formic acid decomposition (dehydrogenation versus dehydration) on the most usual Pd(001), Pd(011) and Pd(111) surfaces.The most common (111), (011) and (001) surfaces have been used to simulate each step of formic acid decomposition. The energy profile (Fig. 10) presents the energy requirements of two different paths initiated by O–H (1: HCOO formation) and C–H (2: COOH formation) dissociation.
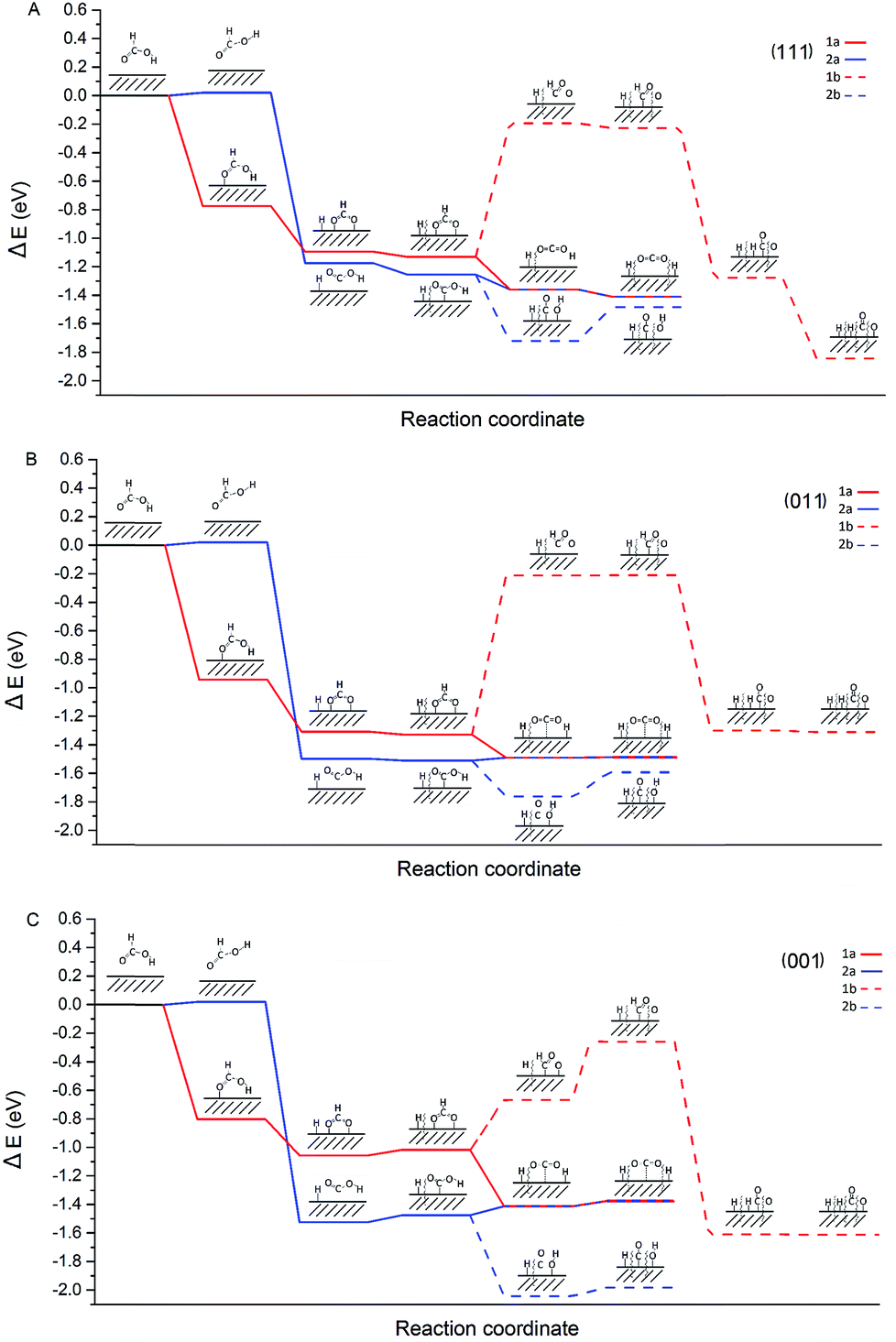 | ||
| Fig. 10 Potential energy surface for formic acid decomposition on (A) Pd(111), (B) Pd(011) and (C) Pd(001) surfaces. Red and blue lines indicate HCOO and COOH paths respectively. The solid lines lead to CO2 whereas the dashed line to CO. | ||
Since 1a (via HCOO) is the main pathway followed by formic acid decomposition, according to Fig. 10, it is clear that for both (111) and (011) surfaces, this pathway is exothermic. On the other hand, for the (001) surface, pathway 1a finds two steps in which absorption energies increase, having a negative impact on the catalyst activity. In fact, the last step in pathway 1a, the H spillover, on the (001) has a potential energy of ER = −1.38 eV, while on the (111) surface ER = −1.41 eV and on (011) ER = −1.49 eV. Hence, the resulting energy of the system is higher for the (001) surface and therefore, we can conclude that very small nanoparticles with a large extensive area of the (001) surface would be detrimental to both catalyst activity and CO evolution. In our investigation, we have presented the maximum activity for the catalysts with a mean particle size of approximately 4 nm. This diameter seems to be large enough to avoid the (001) surface to a large extent.
Taking into account the observed catalytic and potential energy surface trends, the catalytic activity increases with the increase of (111) and (011) surfaces, and furthermore, it is evident that the cis configuration is the pathway (2a) that leads to CO formation even at low temperature is particularly enhanced in the case of the (001) surface. Therefore, it is crucial to design experimental methodologies to develop supported Pd nanoparticles exposed with a higher degree of (111) and (011) surfaces.
Conclusions
A series of monometallic Pd nanoparticles supported on several carbon nanofibers (CNFs) were synthesised. Three CNFs with different graphitisation grades were used. Sol-immobilisation and impregnation techniques were selected as model preparation methods widely used for the deposition of Pd nanoparticles.Pd/CNFs prepared by the sol-immobilisation method exhibit higher catalytic activity when compared with catalysts prepared by the impregnation method due to (i) the higher surface atomic Pd percentage, (ii) higher percentage of Pd0 and (iii) smaller Pd particle size. Probably, the sol-immobilisation method tends to distribute Pd metal nanoparticles on the surface of the nanofibers, whereas the impregnation method leads to a partial filling of the nanofibers and distribution of Pd nanoparticles in the inner wall, besides the distribution on the outer CNF surface.
The heat treatment on CNFs affects catalyst activity. The catalytic performance of the samples significantly increases with the increase of annealing temperature. A heat treatment at 1500 °C did not significantly modify the surface properties of CNFs. In contrast, treating CNFs at 3000 °C rearranges the structure improving the order and the degree of graphitisation. This increase of activity with annealing temperature has been attributed to the presence of smaller Pd nanoparticles formed and improved dispersion. A TOF of 979 h−1 was achieved by PdSI/CNF-HHT, the most active catalyst in this series, with high selectivity for H2 (>99%) under mild reaction conditions, e.g. 30 °C. Finally, DFT studies provide valuable insights into the role of under-coordinated sites in terms of activity and preference of reaction pathways toward CO2 and H2 evolution against CO formation and provide answers to the important question of how to avoid/minimise the formation of CO. (001) was found to be the surface in which CO would evolve at faster rates compared with (011) and (111). Therefore, a higher degree of (111) and (011) surfaces could in our opinion be a possible solution in order to minimise CO formation.
Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
The authors would like to thank the EPSRC for the financial support. Felipe Sanchez thanks Cardiff University for the PhD studentship. Davide Motta wishes to thank the EPSRC Catalysis CDT (EP/L016443/1) for the PhD scholarship. Ceri Hammond is grateful to The Royal Society for provision of a University Research Fellowship (UF140207) and additional research grant funding (CHG\R1\170092). This work has also used the computational facilities of the Advanced Research Computing at Cardiff (ARCCA) Division, Cardiff University, and HPC Wales. All data created during this research are openly available in the University of Cardiff Research Portal. Information about the data underpinning the results here, including how to access them, can be found in the Cardiff University data catalogue: https://doi.org/10.17035/d.2018.0057542883.Notes and references
- T. N. Veziroǧlu and S. Şahin, Energy Convers. Manage., 2008, 49, 1820–1831 CrossRef.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS PubMed.
- H. Dai, B. Xia, L. Wen, C. Du, J. Su, W. Luo and G. Cheng, Appl. Catal., B, 2015, 165, 57–62 CrossRef CAS.
- D. Bae, H. Park, J. S. Kim, J. bok Lee, O. Y. Kwon, K. Y. Kim, M. K. Song and K. T. No, J. Phys. Chem. Solids, 2008, 69, 1152–1154 CrossRef CAS.
- H. P. Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2014, 122, 112–132 CrossRef CAS.
- J. Germain, J. M. J. Fréchet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- J. Li, Q.-L. Zhu and Q. Xu, Catal. Sci. Technol., 2015, 5, 525–530 RSC.
- J. Shen, L. Yang, K. Hu, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2015, 40, 1062–1070 CrossRef CAS.
- J. Manna, B. Roy and P. Sharma, J. Power Sources, 2015, 275, 727–733 CrossRef CAS.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- M. Zheng, R. Cheng, X. Chen, N. Li, L. Li, X. Wang and T. Zhang, Int. J. Hydrogen Energy, 2005, 30, 1081–1089 CrossRef CAS.
- F. Sanchez, D. Motta, A. Roldan, C. Hammond, A. Villa and N. Dimitratos, Top. Catal., 2018, 61, 254–266 CrossRef CAS.
- Ö. Metin, X. Sun and S. Sun, Nanoscale, 2013, 5, 910–912 RSC.
- Q.-L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 RSC.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114–1119 CrossRef CAS.
- P. Sponholz, D. Mellmann, H. Junge and M. Beller, ChemSusChem, 2013, 6, 1172–1176 CrossRef CAS PubMed.
- H. Dai, N. Cao, L. Yang, J. Su, W. Luo and G. Cheng, J. Mater. Chem. A, 2014, 2, 11060–11064 CAS.
- S.-T. Gao, W. Liu, C. Feng, N.-Z. Shang and C. Wang, Catal. Sci. Technol., 2016, 6, 869–874 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. a J. Bagot, E. a Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed.
- X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 3540–3542 RSC.
- L. Yang, X. Hua, J. Su, W. Luo, S. Chen and G. Cheng, Appl. Catal., B, 2015, 168–169, 423–428 CrossRef CAS.
- Q. Lv, L. Feng, C. Hu, C. Liu, W. Xing, H. Wang, Y. Liu, M. Li, H. Huang, H. M. Xu, R. J. Hong and H. Shen, Catal. Sci. Technol., 2015, 5, 2581–2584 RSC.
- X. Wang, G. W. Qi, C. H. Tan, Y. P. Li, J. Guo, X. J. Pang and S. Y. Zhang, Int. J. Hydrogen Energy, 2014, 39, 837–843 CrossRef CAS.
- C. Hu, J. K. Pulleri, S. W. Ting and K. Y. Chan, Int. J. Hydrogen Energy, 2014, 39, 381–390 CrossRef CAS.
- M. Navlani-García, M. Martis, D. Lozano-Castelló, D. Cazorla-Amorós, K. Mori and H. Yamashita, Catal. Sci. Technol., 2015, 5, 364–371 RSC.
- A. Villa, D. Wang, N. Dimitratos, D. Su, V. Trevisan and L. Prati, Catal. Today, 2010, 150, 8–15 CrossRef CAS.
- A. Villa, D. Wang, P. Spontoni, R. Arrigo, D. Su and L. Prati, Catal. Today, 2010, 157, 89–93 CrossRef CAS.
- J. K. Chinthaginjala, A. Villa, D. S. Su, B. L. Mojet and L. Lefferts, Catal. Today, 2012, 183, 119–123 CrossRef CAS.
- B. Huang, Y. Liu and Z. Xie, J. Mater. Chem. A, 2017, 5, 23481–23488 RSC.
- B. Huang, L. Peng, F. Yang, Y. Liu and Z. Xie, J. Energy Chem., 2017, 26, 712–718 CrossRef.
- P. Li, Y. L. Huang, D. Chen, J. Zhu, T. J. Zhao and X. G. Zhou, Catal. Commun., 2009, 10, 815–818 CrossRef CAS.
- M. P. Lazaro, E. Garcia-Bordeje, D. Sebastian, M. J. Lazaro and R. Moliner, Catal. Today, 2008, 138, 203–209 CrossRef CAS.
- I. Kang, Y. Y. Heung, J. H. Kim, J. W. Lee, R. Gollapudi, S. Subramaniam, S. Narasimhadevara, D. Hurd, G. R. Kirikera, V. Shanov, M. J. Schulz, D. Shi, J. Boerio, S. Mall and M. Ruggles-Wren, Composites, Part B, 2006, 37, 382–394 CrossRef.
- J.-P. Tessonnier, D. Rosenthal, T. W. Hansen, C. Hess, M. E. Schuster, R. Blume, F. Girgsdies, N. Pfänder, O. Timpe, D. S. Su and R. Schlögl, Carbon N. Y., 2009, 47, 1779–1798 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 5, 4709–4714 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. Perdew, A. Ruzsinszky, G. Csonka, O. Vydrov, G. Scuseria, L. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- N. D. Mermin, Phys. Rev., 1965, 137, 1441–1443 CrossRef.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Irrera, A. Roldan, G. Portalone, N. H. De Leeuw and N. H. De Leeuw, J. Phys. Chem. C, 2013, 117, 3949–3957 CrossRef CAS.
- N. Y. Dzade, A. Roldan and N. H. De Leeuw, J. Chem. Phys., 2013, 139, 124708 CrossRef CAS PubMed.
- S. S. Tafreshi, A. Roldan, N. Y. Dzade and N. H. De Leeuw, Surf. Sci., 2014, 622, 1–8 CrossRef CAS.
- S. Haider, A. Roldan and N. H. De Leeuw, J. Phys. Chem. C, 2014, 118, 1958–1967 CrossRef CAS.
- N. Dzade, A. Roldan and N. de Leeuw, Minerals, 2014, 4, 89–115 CrossRef CAS.
- F. Zhang, J. D. Gale, B. P. Uberuaga, C. R. Stanek and N. A. Marks, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 1–7 Search PubMed.
- K. Mathew and R. G. Hennig, 2016, eprint arXiv: 1601.03346, 1–6.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 1–9 CrossRef PubMed.
- J. D. Pack and H. J. Monkhorst, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- H. W. King and F. D. Manchester, J. Phys. F: Met. Phys., 1978, 8, 15–26 CrossRef CAS.
- J. Cho, S. Lee, S. P. Yoon, J. Han, S. W. Nam, K. Y. Lee and H. C. Ham, ACS Catal., 2017, 7, 2553–2562 CrossRef CAS.
- J. Cho, S. Lee, J. Han, S. P. Yoon, S. W. Nam, S. H. Choi, K. Y. Lee and H. C. Ham, J. Phys. Chem. C, 2014, 118, 22553–22560 CrossRef CAS.
- C. J. Huang, F. M. Pan, T. C. Tzeng, C. Li and J. T. Sheu, J. Electrochem. Soc., 2009, 156, J28–J31 CrossRef CAS.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang, Y. Ping and Q. Jiang, Sci. Rep., 2012, 2, 598–604 CrossRef PubMed.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 387–394 CrossRef CAS.
- M. C. Militello and S. J. Simko, Surf. Sci. Spectra, 1994, 3, 395–401 CrossRef CAS.
- F. Y. Xie, W. G. Xie, L. Gong, W. H. Zhang, S. H. Chen, Q. Z. Zhang and J. Chen, Surf. Interface Anal., 2010, 42, 1514–1518 CrossRef CAS.
- R. Arrigo, M. Havecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlogl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- O. Baghriche, S. Rtimi, C. Pulgarin, C. Roussel and J. Kiwi, Appl. Catal., B, 2013, 130–131, 65–72 CrossRef CAS.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2009, 175, 55–65 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- S. Zhang, O. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed.
- A. Bulut, M. Yurderi, Y. Karatas, M. Zahmakiran, H. Kivrak, M. Gulcan and M. Kaya, Appl. Catal., B, 2015, 164, 324–333 CrossRef CAS.
- S. F. Ho, A. Mendoza-Garcia, S. Guo, K. He, D. Su, S. Liu, O. Metin and S. Sun, Nano Lett., 2012, 12, 1102–1106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00338f |
| This journal is © The Royal Society of Chemistry 2018 |