
Amino-fulleropyrrolidines as electrotropic additives to enhance organic photovoltaics†
Supravat
Karak‡
a,
Zachariah A.
Page§
 *a,
Shaoguang
Li§
b,
Jonathan S.
Tinkham¶
c,
Paul M.
Lahti
c,
Volodimyr V.
Duzhko
a and
Todd
Emrick
*a
*a,
Shaoguang
Li§
b,
Jonathan S.
Tinkham¶
c,
Paul M.
Lahti
c,
Volodimyr V.
Duzhko
a and
Todd
Emrick
*a
aDepartment of Polymer Science and Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: zapage@mrl.ucsb.edu; tsemrick@mail.pse.umass.edu
bMaterials Research Lab, University of California, Santa Barbara, CA 93106, USA
cDepartment of Chemistry, University of Massachusetts, Amherst, Massachusetts 01003, USA
First published on 8th August 2018
Abstract
The synthesis, device fabrication, and electric poling of novel amino-functionalized fulleropyrrolidines in polymer solar cells is reported. Systematically varying the tether lengths between the fullerene cage and tertiary amines provides insight into enhanced device efficiency and stability after poling. DFT calculations in an external electric field result in alignment of molecular dipole moments, which corroborates our hypothesis that electrotropic polarization causes an increased built-in electrostatic potential difference across the device, and results in enhance performance.
Organic polymer solar cells (OPSCs) are regarded as inexpensive, flexible, and light-weight sources of energy, where ease of solution processability, abundance of raw starting materials, and mechanical integrity make them particularly attractive. Small amounts of additives to bulk-heterojunction (BHJ) active layers has led to significant advances in OPSC technology, by improving device performance and stability. For example, compatibilizers1–5 have been used to prevent phase-segregation to improve device lifetimes, while low vapor pressure solvents (e.g., 1,8-diiodooctane) modify the BHJ morphology and increase power conversion efficiency (PCE).6–8 Alternatively, dipolar additives that reorient in response to an applied electric field (i.e., electrotropic compounds) were recently shown to improve PCE values of standard-geometry solar cells (e.g., anode to cathode from bottom up) with small molecule-based BHJs, due to an increased electrostatic potential difference across the device.9 To better understand and utilize electrotropic materials in photovoltaic technology, a more systematic study between structure and function of dipolar additives in BHJ active layers is critical.
Dipolar materials have found utility as interlayers in OPSCs to elicit significant improvements in device performance, which, in-part, stems from the increase in built-in electrostatic potential across the device.10–22 Numerous examples of tertiary amine-functionalized materials have recently been developed and employed as efficient electrode work function modifiers through their use as interlayers. The proposed operational mechanisms for tertiary amines include the alignment of dipolar side-chains for p-type10 or insulating11 polymer backbones, doping of the acceptor component in the adjacent active layer,23 and self-doping of n-type polymers.12,17 Recently, we demonstrated that a tertiary amine-functionalized fulleropyrrolidine (C60-N-T3) is susceptible to ferroelectric polarization.9 Specifically, integration of C60-N-T3 into a small molecule BHJ active layer followed by electric field poling led to a significant improvement in device performance, which was largely attributed to the electric-field induced alignment of dipolar side groups, which increases the overall built-in potential difference across the device active layer. However, to-date electrotropism in OPSCs has only been shown with this one dipolar additive (C60-N-T3) and in one type of device configuration (standard geometry, small molecule BHJ).
To understand the versatility of electrotropism in solar cells, two new amine-functionalized fulleropyrrolidine derivatives, C60-N-T6 and C60-N-T11, with different tether lengths connecting the tertiary amines and fullerene core, were synthesized, and together with C60-N-T3, were studied as additives in inverted polymer-based solar cells. The effect of tether length on ferroelectric polarization was probed by testing device metrics over time. Additionally, density functional theory (DFT) calculations on the additives in the presence and absence of an external electric field was used to correlate chemical structure to device metrics.
In an effort to increase the molecular dipole moment, two novel fulleropyrrolidine derivatives of C60-N-T3 were synthesized where the electron deficient fullerene core and electron rich tertiary amines were separated by longer tethers.13 All three derivatives were synthesized in two facile steps from commercial starting materials. First, Mitsunobu coupling of 2,3,4-trihydroxybenzaldehyde and an amino-alkyl-alcohol containing 3, 6 or 11 methylene units in the alkyl chain, provided the amino-benzaldehyde intermediates, as shown in Scheme 1. Second, a Prato reaction between the amino-benzaldehyde intermediates, sarcosine, and fullene-C60 yielded the desired fulleropyrrolidines, C60-N-T3, C60-N-T6 and C60-N-T11, corresponding to tethers with 3, 6, and 11 methylene units, respectively, separating the fulleropyrrolidine and tertiary amines (Scheme 1). Successful Prato reactions were indicated by a change in solution color from violet (C60) to brown, and confirmed using 1H nuclear magnetic resonance (NMR) spectroscopy. The appearance of doublets at ∼4.9 and 4.2 ppm are diagnostic of the geminal protons α to the cyclic amine of the pyrrolidine product. Moreover, matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry of the products confirmed the identity of the three fulleropyrrolidines, providing m/z values of 1157.60, 1284.45, and 1494.47 for [M + H+] molecular ions (details in the ESI†).
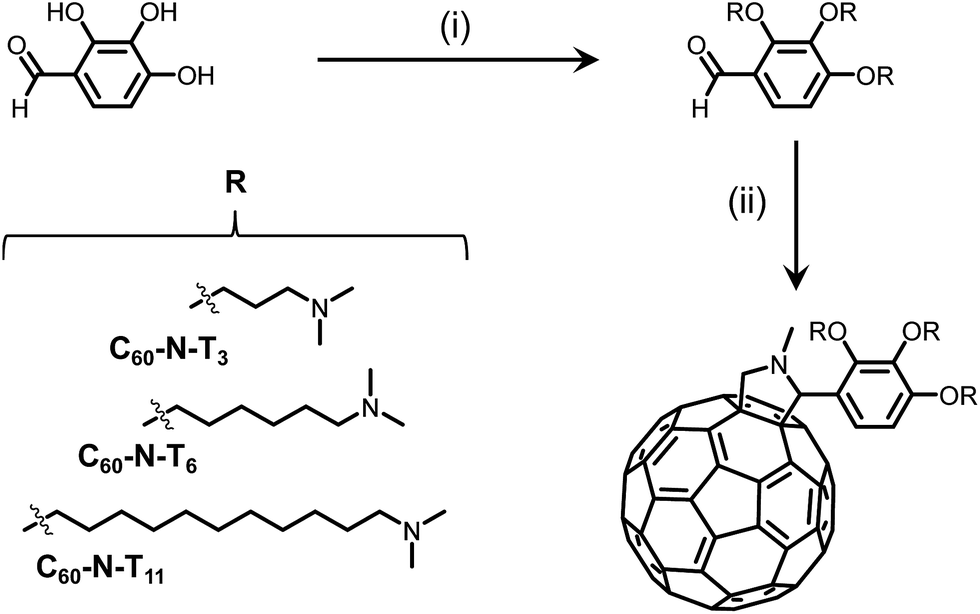 | ||
| Scheme 1 Synthesis of C60-N-T3, C60-N-T6, and C60-N-T11 additives. (i) Diisopropyl azodicarboxylate, triphenylphosphine, 3-(dimethylamino)X-1-ol, THF, 30–70%, where X corresponds to propan, hexan and undecan for C60-N-T3, C60-N-T6 and C60-N-T11, respectively; (ii) fullerene-C60, sarcosine, chlorobenzene, 33–54%. | ||
A schematic illustration of the inverted device configuration used in this study is provided in Fig. 1A. Poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5b′]-dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene)-2-carboxylate-2-6-diyl] (PTB7-Th) was the donor in the active layer and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) was the acceptor component (chemical structures shown in the ESI†). The top and bottom contacts were a transparent indium tin oxide (ITO)/poly[(9,9-bis(3′-(N,N-dimethylamino)propyl)-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)] (PFN) cathode and molybdenum trioxide (MoO3)/aluminum (Al) anode. These were selected for their favorable energy alignment for inverted device architectures, providing efficient extraction of photo-generated charge carriers. The amount of dipolar additives in the active layer was optimized with regard to the device performance for C60-N-T3 additives (0.5 volume percent (v%)), as shown in Fig. S1 and Table S1,† and matching quantities (0.5 v%) were used for devices containing C60-N-T6 and C60-N-T11. Fig. 1B provides the J–V characteristics under simulated solar irradiation for the reference device (without dipolar additives) and devices with 0.5 v% C60-N-T3, C60-N-T6, and C60-N-T11 additives prior to poling (open symbols) and after poling (−2 V, solid symbols).
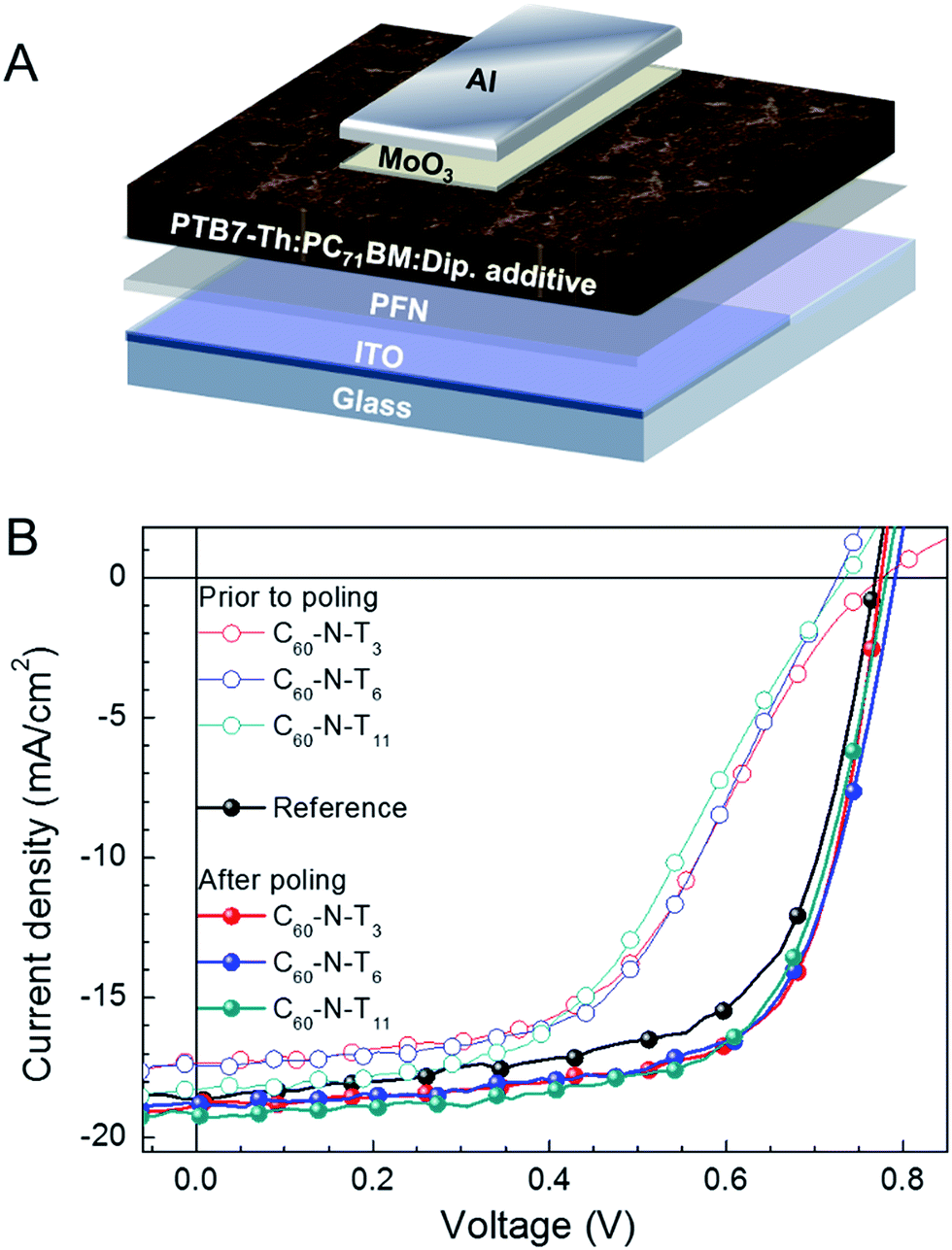 | ||
| Fig. 1 Solar cell device schematic and performance. (A) Inverted solar cell architecture. (B) Current density–voltage characteristics with and without dipolar additives before (open symbols) and after (solid symbols) −2 V poling. | ||
The key device performance parameters are summarized in Table 1. The reference devices show a maximum VOC = 0.77 V, JSC = 18.4 mA cm−2 and FF = 64.9%, providing a PCE (η) = 9.2%. However, the reference devices deteriorate with electric-field poling, reaching a VOC = 0.78 V, JSC = 16.0 mA cm−2, FF = 61.2% and η = 7.7% after 20 min at −2 V. Notably, devices with dipolar additives have a reduced performance relative to the reference devices prior to poling (metrics given in Fig. S2 and Table S3†). This result is similar to the observations made by Cao and coworkers for devices that contained amine-functionalized methanofullerenes (PC71BM-N) in the active layer, which was suggested to arise from hole trapping.12 However, after poling at −2 V (forward diode direction), the performance of devices containing dipolar additives improves by ∼10% compared to the reference devices (without dipolar additives), and >50% compared to as-made (non-poled) devices containing dipolar additives, providing PCE values of ∼10% for all three amine-functionalized fulleropyrrolidines. Specifically, devices with C60-N-T3 showed VOC = 0.78 V, JSC = 18.9 mA cm−2, FF = 69.1% and η = 10.1%, while devices with C60-N-T6 showed VOC = 0.79 V, JSC = 18.8 mA cm−2, FF = 67.8% and η = 10.1% and devices with C60-N-T11 showed VOC = 0.78 V, JSC = 19.1 mA cm−2, FF = 66.8% and η = 10.0% (Table 1). Notably the enhanced FF and JSC are largely responsible for the improvement of device performance, while the VOC remains comparable for poled devices with respect to reference devices. This result highlights the generality of the approach given that the improvement in device performance as a result of electric-field poling is similar to behavior observed for small-molecule-based solar cell devices having a regular (non-inverted) device architecture with C60-N-T3 additives.9
| Sample | Poling (min) | J SC (mA cm−2) | V OC (V) | FF (%) | η (%) (average) |
|---|---|---|---|---|---|
| Reference | 0 | 18.4 | 0.77 | 64.9 | 9.2 (9.0 ± 0.2) |
| Reference | 20 | 16.0 | 0.78 | 61.2 | 7.7 (7.5 ± 0.2) |
| C60-N-T3 | 0 | 17.3 | 0.78 | 50.2 | 6.8 (6.4 ± 0.3) |
| C60-N-T3 | 10 | 18.9 | 0.78 | 69.1 | 10.1 (9.7 ± 0.4) |
| C60-N-T6 | 0 | 17.4 | 0.72 | 55.0 | 6.9 (6.7 ± 0.3) |
| C60-N-T6 | 60 | 18.8 | 0.79 | 67.8 | 10.1 (9.8 ± 0.3) |
| C60-N-T11 | 0 | 18.3 | 0.73 | 49.2 | 6.6 (6.2 ± 0.4) |
| C60-N-T11 | 120 | 19.1 | 0.78 | 66.8 | 10.0 (9.7 ± 0.3) |
Interestingly, by systematically varying the tether length, temporal characteristics of poled electrotropic additives are significantly altered. Fig. 2A shows the normalized PCE of devices with different additives for different durations of electric-field poling. The individual J–V characteristics of the devices are shown in Fig. S2† and device performance parameters are summarized in Tables S2–S5.† As stated previously, the PCE values for the reference devices decreased from 9.2 to 7.7% over the course of poling at −2 V for 20 min, while in contrast, all devices containing dipolar additives show improved performance as a result of poling over time scales on the same order of magnitude. Prior to poling, devices with C60-N-T3 had a PCE of 6.8% (6.4 ± 0.4%), which steadily improved over 10 min of poling at −2 V, reaching a maximum PCE of 10.1% (9.7 ± 0.4%) (Fig. S2b and Table S3†). Additional poling (+10 min), resulted in a PCE decrease to 9.3% (9.1 ± 0.2%) primarily due to a lower JSC = 17.9 mA cm−2. The peak performance highlights the balance between dipole orientation to improve performance and device degradation under continuous electrical stress. Similarly, devices with C60-N-T6 (Fig. S2c and Table S4†) and C60-N-T11 (Fig. S2d and Table S5†) initially benefited from poling, although on different time scales; η = 6.9% (6.7 ± 0.3%) to 10.1% (9.8 ± 0.3%) after 60 min of poling for devices with C60-N-T6 and η = 6.2% (6.7 ± 0.4%) to 10.0% (9.7 ± 0.3%) after 120 min of poling for devices with C60-N-T11. The devices degraded with extended poling, likely due to the trade-off between performance-enhancing orientation of dipoles and degradation under electrical stress. Therefore, it is anticipated that photovoltaic devices with enhanced stability could benefit further from the use of dipolar additives, since deleterious background degradation would be eliminated. External quantum efficiency (EQE) spectra of the corresponding devices are shown in Fig. S3.† For all devices, high and uniform EQE values were observed between 400–800 nm, suggesting efficient conversion of photons to free charge carriers throughout the visible region of the solar spectrum. For reference devices the EQE peaks at ∼76% around 700 nm, while nearly ∼90% EQE is observed for electrotropic devices at the same wavelength, which corresponds to the peak absorption of PTB7-Th. The improved EQE for devices containing electrotropic additives as compared to the reference devices is consistent with the improved J–V characteristics.
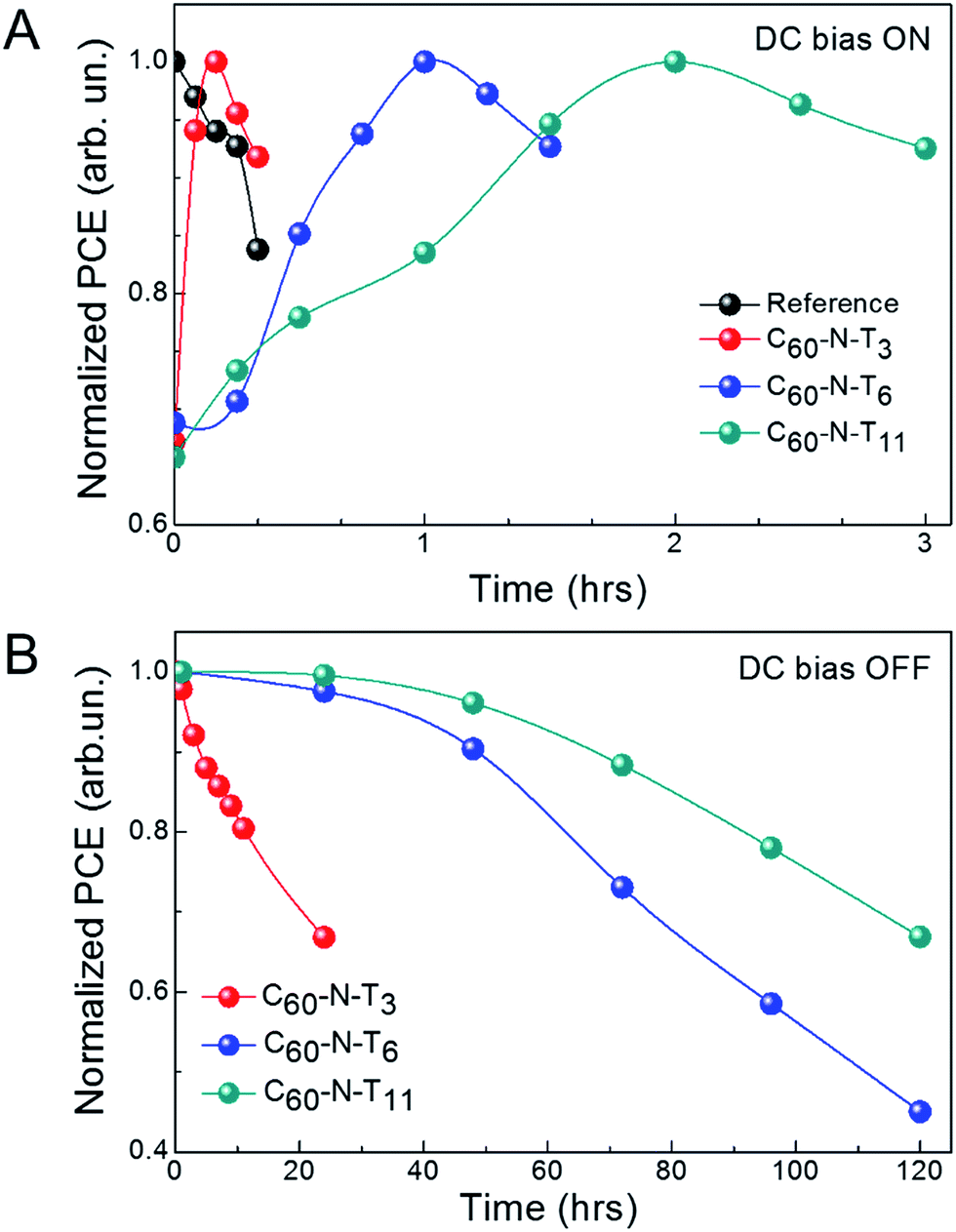 | ||
| Fig. 2 Temporal characteristics of ferroelectric polarization. (A) Normalized power conversion efficiency of devices as a function of poling time, (B) normalized power conversion efficiency of devices after poling (e.g., DC bias off). | ||
Additional device improvement observed upon poling in the presence of electrotropic additives was the retention time for the ferroelectric polarization. Fig. 2B shows the normalized efficiency of devices with fulleropyrrolidine additives as a function of time after poling (e.g., DC bias off). C60-N-T3 based devices showed a quick dissolution of the effect, losing 30% efficiency within 24 h. Alternatively, C60-N-T6 and C60-N-T11 based devices showed prolonged stability, losing 30% of their initial efficiency after 72 and 120 h, respectively. It is interesting to note that the poling time required for achieving the highest efficiency of the current devices and their prolonged stability vary systematically with the additive molecules. To better understand the origin of the effect and explain temporal variations, density functional theory (DFT) calculations were undertaken.
The DFT calculations24,25 probed molecular geometries, dipole moments, and electrostatic potential maps for the ground states of the fulleropyrrolidine additives, as shown in Fig. 3A. The computed dipole moment for C60-N-T3 is 4.97 D, which originates from the lone pair electrons of the tertiary amines, as suggested by the electrostatic potential maps. The antiparallel position of the two amine groups may compensate each other, which results in a lower permanent dipole moment in the optimized structure. In contrast, the optimized structures of C60-N-T6 and C60-N-T11 show distinct interactions between the tertiary amine groups and the electron deficient fullerene core, allowed by the increased flexibility of the longer hydrocarbon-tethers, which reduces the dipole moments to 3.98 and 4.21 D, respectively. While the amine–fullerene interaction is apparent in the optimized structures, the electrostatic maps show no ground-state electron transfer (e.g., n-doping of fullerene), in agreement with the photoexcitation shown to induce electron transfer in PC71BM in the presence of amines.26,27 To investigate the effect of electric field on the structure and electrostatic properties of the molecules, an external field, with strength on the same order of magnitude as device poling (∼106–107 V m−1), was applied to the calculation. The optimized structures and their corresponding electrostatic maps are shown in Fig. 3B, revealing dramatic structural changes as the tertiary amines extend away from the fullerene core. This extension causes an increase of dipole moments for C60-N-T6 and C60-N-T11 to approximately 4.8 D, which is comparable with C60-N-T3 and is consistent with the nearly identical peak PCE values obtained for all poled devices. Moreover, larger conformational changes for derivatives with longer side chains correlate with the requirement for increased poling and retention times. It is speculated that local Joule heating that occurs in response to the flow of electrical current during poling can be used to explain the relatively rapid chain alignment during poling in comparison to the observed relaxation post-poling, where the side-chains are effectively “frozen” in the surrounding matrix. Therefore, synthesizing additive molecules with large dipole moments that are distributed over long side chains may further improve device efficiency and stability.
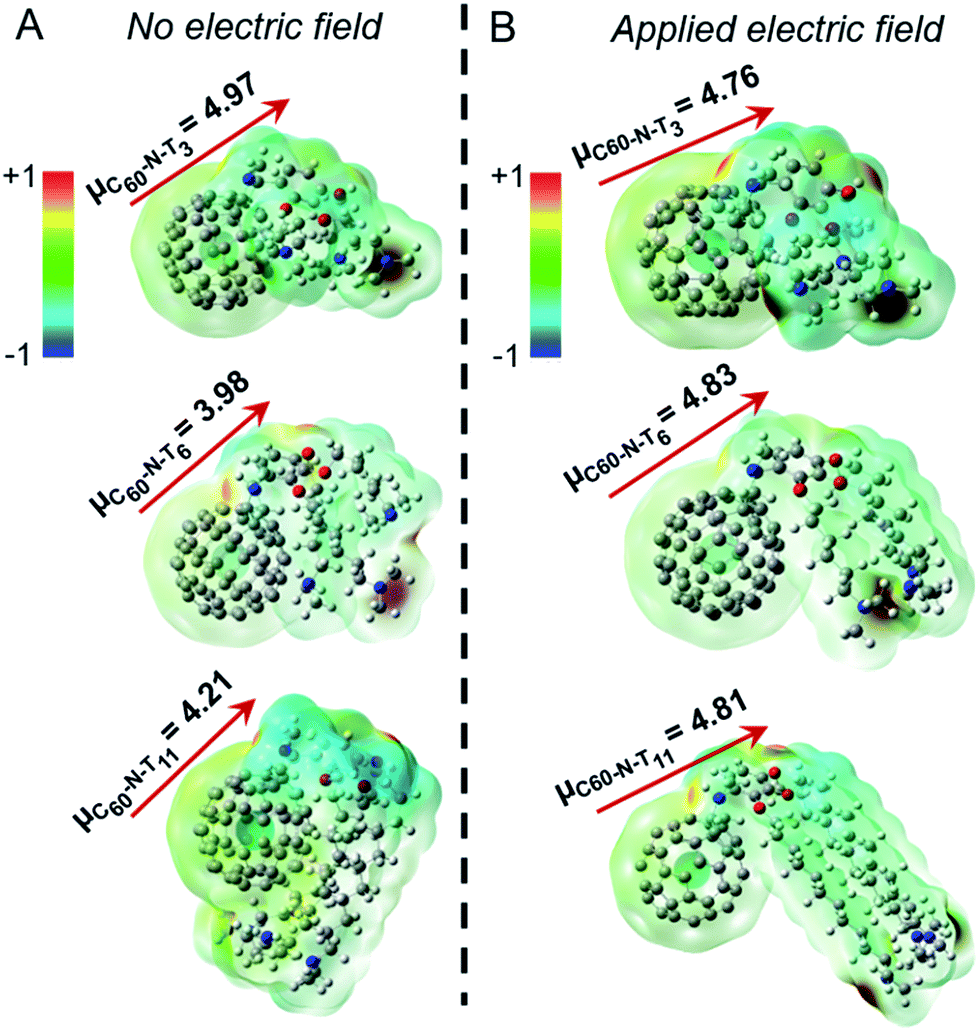 | ||
| Fig. 3 Molecular geometries with dipole moment vectors (left) and electrostatic potential maps (right) for electrotropic additives under no electric field (A) and under an external electric field (B). | ||
Conclusions
In summary, the synthesis, device integration and computational analysis of three amine-functionalized fulleropyrrolidine derivatives, C60-T-N3, C60-T-N6 and C60-T-N11, were reported. Extending the tether length between the electron rich amines and deficient fullerene core led to systematic variations in poling times necessary to reach peak device power conversion efficiencies and ferroelectric polarization retention times. Combining photovoltaic device performance analysis with DFT calculations revealed underlying structure–property relationships between the newly developed electrotropic molecules and photophysics associated with electrical field poling. Of significance, the longer tether groups led to significantly improved retention times of ferroelectric polarization in the inverted photovoltaic devices, reaching PCE values in excess of 10%. This study opens new prospects for energy harvesting by developing high-efficiency solution processable OPV devices using dipolar additives and electric field poling to enhance performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Supported by the Polymer-Based Materials for Harvesting Solar Energy (PHaSE), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, under award DE-SC0001087. We thank PHaSE and the NSF-MRSEC on Polymers at UMass (DMR-0820506) for facilities support.Notes and references
- J. U. Lee, J. W. Jung, T. Emrick, T. P. Russell and W. H. Jo, J. Mater. Chem., 2010, 20, 3287–3294 RSC.
- B. K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Fréchet, Adv. Mater., 2006, 18, 206–210 CrossRef.
- C. Yang, K. Lee, J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416–5423 RSC.
- H. Fujita, T. Michinobu, S. Fukuta, T. Koganezawa and T. Higashihara, ACS Appl. Mater. Interfaces, 2016, 8, 5484–5492 CrossRef PubMed.
- F. Lombeck, A. Sepe, R. Thomann, R. H. Friend and M. Sommer, ACS Nano, 2016, 10, 8087–8096 CrossRef PubMed.
- K. R. Graham, P. M. Wieruszewski, R. Stalder, M. J. Hartel, J. Mei, F. So and J. R. Reynolds, Adv. Funct. Mater., 2012, 22, 4801–4813 CrossRef.
- H. Jhuo, S. Liao, Y. Li, P. Yeh, S. Chen, W. Wu, C. Su, J. Lee, N. L. Yamada and U. Jeng, Adv. Funct. Mater., 2016, 26, 3094–3104 CrossRef.
- J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619–3623 CrossRef PubMed.
- S. Karak, Z. A. Page, J. S. Tinkham, P. M. Lahti, T. Emrick and V. V. Duzhko, Appl. Phys. Lett., 2015, 106, 103303 CrossRef.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 CrossRef.
- Y. Zhou, C. Fuentes-Hernandez, J. Shim, J. Meyer, A. J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T. M. Khan, H. Sojoudi, S. Barlow, S. Graham, J.-L. Brédas, S. R. Marder, A. Kahn and B. Kippelen, Science, 2012, 336, 327–332 CrossRef PubMed.
- C. Duan, W. Cai, B. B. Y. Hsu, C. Zhong, K. Zhang, C. Liu, Z. Hu, F. Huang, G. C. Bazan, A. J. Heeger and Y. Cao, Energy Environ. Sci., 2013, 6, 3022–3034 RSC.
- Z. A. Page, Y. Liu, V. V. Duzhko, T. P. Russell and T. Emrick, Science, 2014, 346, 441 CrossRef PubMed.
- Y. Liu, Z. A. Page, T. P. Russell and T. Emrick, Angew. Chem. Int. Ed., 2015, 54, 11485–11489 CrossRef PubMed.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Adv. Mater., 2014, 26, 3473–3477 CrossRef PubMed.
- F. Liu, Z. A. Page, V. V. Duzhko, T. P. Russell and T. Emrick, Adv. Mater., 2013, 25, 6868–6873 CrossRef PubMed.
- C. Sun, Z. Wu, H. L. Yip, H. Zhang, X. F. Jiang, Q. Xue, Z. Hu, Z. Hu, Y. Shen, M. Wang, F. Huang and Y. Cao, Adv. Energy Mater., 2016, 6, 1501534 CrossRef.
- T. Y. Juang, J. C. Kao, J. C. Wang, S. Y. Hsu and C. P. Chen, Adv. Mater. Interfaces, 2018, 5(10), 1800031 CrossRef.
- B. H. Jiang, Y. J. Peng and C. P. Chen, J. Mater. Chem. A, 2017, 5, 10424 RSC.
- Z. Xiao, Q. Dong, P. Sharma, Y. Yuan, B. Mao, W. Tian, A. Gruverman and J. Huang, Adv. Energy Mater., 2013, 3, 1581 CrossRef.
- K. S. Nalwa, J. A. Carr, R. C. Mahadevapuram, H. K. Kodali, S. Bose, Y. Chen, J. W. Petrich, B. Ganapathysubramanian and S. Chaudhary, Energy Environ. Sci., 2012, 5, 7042 RSC.
- Z. Xiao, Q. Dong, Q. Wang, W. Tian, H. Huang and J. Huang, J. Photovolt., 2015, 5(5), 1408 Search PubMed.
- S. Fabiano, S. Braun, X. Liu, E. Weverberghs, P. Gerbaux, M. Fahlman, M. Berggren and X. Crispin, Adv. Mater., 2014, 26, 6000–6006 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef.
- L. Nian, W. Zhang, N. Zhu, L. Liu, Z. Xie, H. Wu, F. Würthner and Y. Ma, J. Am. Chem. Soc., 2015, 137, 6995–6998 CrossRef PubMed.
- Z. Wu, C. Sun, S. Dong, X. F. Jiang, S. Wu, H. Wu, H. L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and additional device testing. See DOI: 10.1039/c8se00294k |
| ‡ Present address: Centre for Energy Studies, Indian Institute of Technology, Delhi, Hauz Khas, New Delhi – 110016, India. |
| § Present address: Materials Research Lab, University of California, Santa Barbara, CA 93106, USA. |
| ¶ Present address: Department of Chemistry and Geochemistry, Colorado School of Mines, Golden, CO 80401, USA. |
| This journal is © The Royal Society of Chemistry 2018 |