
Nickel–copper supported on a carbon black hydrogen oxidation catalyst integrated into an anion-exchange membrane fuel cell†
Aaron
Roy
a,
Morteza R.
Talarposhti
a,
Stanley J.
Normile
 b,
Iryna V.
Zenyuk
b,
Vincent
De Andrade
c,
Kateryna
Artyushkova
a,
Alexey
Serov
a and
Plamen
Atanassov
*a
b,
Iryna V.
Zenyuk
b,
Vincent
De Andrade
c,
Kateryna
Artyushkova
a,
Alexey
Serov
a and
Plamen
Atanassov
*a
aDepartment of Chemical & Biological Engineering, Center for Micro-Engineered Materials, University of New Mexico, Albuquerque, NM 87131, USA. E-mail: plamen@unm.edu
bDepartment of Mechanical Engineering, Tufts University, Medford, MA 02155, USA
cAdvanced Photon Source, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 20th July 2018
Abstract
This work introduces the first practical platinum group metal-free (PGM-free) electrocatalyst for the hydrogen oxidation reaction (HOR) in alkaline membrane fuel cells (AMFCs), based on nickel-rich Ni95Cu5-alloy nanoparticles supported on Ketjenblack (KB) family carbon blacks. The catalyst synthesis is scalable and results in an expected true alloy of NiCu, which is thoroughly characterized by XRD, microscopy and XPS. The reactivity of the catalyst towards the HOR is studied by cyclic voltammetry and explained in view of its composition and structure. This catalyst showed the highest specific activity compared to previously reported NiCu electrocatalysts and was successfully integrated into an AMFC membrane electrode assembly (MEA) using a commercially available state-of-the-art membrane and an ionomer. Single MEA fuel cell tests have demonstrated a power density of 350 mW cm−2 at 80 °C, which sets a technical record for a PGM-free anode in realistic operating conditions. The MEA with the NiCu/KB anode catalyst layer was evaluated by in situ nano- and in operando micro-X-ray computed tomography (CT) and the results suggest that the nickel state in NiCu is hydrophobic in nature, where the NiCu surface may be isostructural with β-Ni(OH)2. The hydrophobic nature of the electrocatalyst allows for improved water distribution in the MEA and overall fuel cell as observed by in operando micro-X-ray CT.
Introduction
Growing concerns regarding CO2 emissions from the burning of fossil fuels, the finite abundance of fossil fuels, and the environmental impacts of the use of fossil fuels by industries are driving researchers to develop technologies which will enable hydrogen energy and hydrogen energy storage.1 Hydrogen fuel cells for energy conversion are therefore of considerable interest. The reduction of fuel cell costs by transitioning from proton exchange membrane fuel cells (PEMFCs) to anion exchange membrane fuel cells (AEMFCs) can be achieved due to (i) a lower overpotential required for the oxygen reduction reaction (ORR) at the cathode of AEMFCs, (ii) the ability to use Earth-abundant, platinum group metal-free (PGM-free) catalyst materials for the ORR at the cathode and also for the hydrogen oxidation reaction (HOR) at the anode in AEMFCs, and (iii) a wide range of polymers which can be employed to achieve OH− conductivity in the polymer membranes of AEMFCs.2–4The past few decades have seen a significant improvement in AEMFC performance, reaching power densities as high as 0.8–1.4 W cm−2, achieved by utilizing polymer membranes with a hydroxide conductivity ranging from 60 to 132 mS cm−1, usually derived from quaternary ammonia (QA) sites on a polymer backbone.5–7 Common factors among these results leading to high AEMFC performance are (i) the improved OH− conductivity of the ionomer and membrane, (ii) the use of highly active PtRu ORR/HOR catalysts, and (iii) the implementation of water management strategies.2,8 Although significant progress has been made in the advancement of OH− conductivity of anion exchange ionomers and membranes, as well as the development of PGM-free ORR cathode materials,9–11 less progress has been made in the advancement of the PGM-free HOR in alkaline media. The main challenge in developing PGM-free HOR catalysts for AEMFCs lies in the different kinetic mechanism of the HOR at high pH versus low pH, where the former is limited by the Volmer step of the hydrogen oxidation reaction.12,13 Recent understanding of the HOR mechanism in alkaline media suggests a bifunctional mechanism for the HOR on bimetallic catalysts, involving the formation of Had species on a primary active site followed by the 1 e− reduction of water forming OHad clusters at a secondary site,14,15 which can facilitate the Volmer step of the HOR and improve the AEMFC performance using PGM-free anode materials. These findings are consistent with the reports in the literature of highly active HOR catalysts in alkaline media derived from Earth-abundant materials such as carbon supported Ni, NiMo, and NiCu.16–18
Bimetallic systems based on Ni alloyed with base metals such as Mo, Cu, and W, have shown so far the most promise as PGM-free HOR catalysts for AEMFCs with the highest cell power density reported in the literature of ∼120 mW cm−2 for carbon-supported Ni95Mo5.17 However, NiMo presents challenges related to the performance loss due to flooding at high relative humidity, likely due to the hydrophilic nature of the catalyst pre-determined by the abundance of surface hydroxide species.19–21 In the present study, we report the superior activity of a NiCu nano-alloyed HOR catalyst and its AEMFC performance. We identified a nickel-rich Ni95Cu5 nanoparticle catalyst, supported on Ketjenblack, with a true alloy structure as a candidate for a thorough in operando micro- and nano-CT study of the catalyst layer and water management under various humidified conditions (50% and 100% RH). Alloying of nickel with copper and its effects on catalyst electrochemical activity and performance in a single membrane-electrode assembly (MEA) AEMFC are being discussed.
Experimental
NiCu/KB catalyst synthesis
NiCu/Ketjenblack was synthesized by the incipient wetness method followed by the thermal reduction of nickel and copper precursors. The atomic ratio of nickel to copper was selected as 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 with a metal loading on Ketjenblack EC600J of 50 wt%. Necessary amounts of nickel nitrate and copper nitrate were dissolved in a minimal amount of deionized water (18 MΩ) and impregnated into the carbon support by finely mixing the slurry using a glass mortar and pestle followed by drying at T = 60 °C. The dried powder was then reduced in a hydrogen atmosphere (7 at%, 100 sccm) at T = 550 °C for 60 minutes. Once cooled, the reactor (1′′ I.D. quartz tube) was left for 8–12 hours under a small partial pressure of oxygen (∼0.1 at%) for the passivation of the pyrophoric NiCu/KB catalyst. Pd/XC72R (30 wt% of Pd) was used as the cathode catalyst for MEA tests and its synthesis is described elsewhere.20
:5 with a metal loading on Ketjenblack EC600J of 50 wt%. Necessary amounts of nickel nitrate and copper nitrate were dissolved in a minimal amount of deionized water (18 MΩ) and impregnated into the carbon support by finely mixing the slurry using a glass mortar and pestle followed by drying at T = 60 °C. The dried powder was then reduced in a hydrogen atmosphere (7 at%, 100 sccm) at T = 550 °C for 60 minutes. Once cooled, the reactor (1′′ I.D. quartz tube) was left for 8–12 hours under a small partial pressure of oxygen (∼0.1 at%) for the passivation of the pyrophoric NiCu/KB catalyst. Pd/XC72R (30 wt% of Pd) was used as the cathode catalyst for MEA tests and its synthesis is described elsewhere.20
Catalyst characterization
X-ray diffraction patterns were obtained using a Rigaku Smartlab diffractometer equipped with a D-Tex Ultra silicon strip detector in a Bragg–Brentano focusing geometry. Cu K-α was used as the X-ray source with a K-β receiving slit filter. The catalyst was mounted on a zero-background quartz holder and scanned from 10°–140° as 2θ at a scan rate of 7° min−1. MDI JADE 2010 software was used to perform whole-pattern-refinement using Ni phase data from the ICSD database as a reference [98-001-4691].Transmission Electron Microscopy (TEM) was performed using a Joel 2010 TEMF microscope with an accelerating voltage of 200 keV. Samples were dispersed in isopropyl alcohol then sonicated for 30 minutes before being impregnated onto a carbon mesh TEM grid. Analysis of particle size and particle size distributions was performed using ImageJ software.
XPS spectra were obtained using a Kratos Axis Ultra DLD X-ray photoelectron spectrometer using a monochromatic Al Kα source operating at 125 W. All powders were conductive and mounted using a conductive double-sided tape. No use of charge neutralization was necessary. Three areas for each sample were analyzed. High-resolution spectra were acquired at pass energies of 20 eV. Data analysis and quantification were performed using CasaXPS software. Sensitivity factors provided by the manufacturer were utilized. High-resolution Ni 2p and Mo 3d spectra were acquired. Ni 2p spectra were fitted using a combination of line shapes for Ni metal, NiO, and Ni(OH)2 developed by Biesinger et al.22
Electrochemical evaluation with a rotating disk electrode
HOR measurements were performed in a 3-electrode cell consisting of a glassy carbon rotating disk electrode (5 mm in diameter) as the working electrode, graphite counter electrode, and an eDAQ Hydroflex™ reversible hydrogen reference electrode. Catalyst inks were prepared using 5 mg of the catalyst, 150 μL of 0.5 wt% Nafion in deionized water and 850 μL of isopropyl alcohol. After sonication for 30 minutes, an appropriate loading of the catalyst (100 μgmetal, 200 μgmetal, 300 μgmetal, 400 μgmetal, and 500 μgmetal total metal loading) was drop-cast onto the glassy carbon electrode and allowed to air dry. Cyclic voltammetry tests were performed in 0.1 M KOH solution saturated in N2 or H2. Before electrochemical testing, the NiMo/KB catalyst was conditioned by scanning in the potential interval from −0.20 to 0.40 V at a sweep rate 100 mV s−1 for approximately 100 cycles in a N2-saturated electrolyte. All HOR measurements were performed with a sweep rate of 5 mV s−1. All potentials are reported vs. RHE.From the CV data obtained in the N2 saturated electrolyte, the electrochemical surface area was calculated from the total anodic charge associated with Ni(OH)2 formation (0.514 mC cm−2) and was normalized to the loading of metal on the working electrode. The exchange current density was then calculated from the linear polarization region of the CV performed in the H2 saturated electrolyte, −0.01 to 0.01 V vs. RHE.
MEA fabrication and fuel cell tests
Catalyst inks for MEA fabrication were prepared by ball-milling the NiCu/KB catalyst with the ionomer (Tokuyama AS4) and isopropyl alcohol at 350 rpm for 30 minutes. The nominal amount of solid AS4 in catalytic layers was: anode (NiCu/KB) 45 wt% and cathode (30 wt% Pt/XC72R) 30 wt%. An aerosol hand-spray setup was used to deposit 4 mg cm−2 of the anode NiCu/KB and 0.2 mgPd cm−2 of the cathode Pd/XC72R catalyst onto the active surface of the 5 cm2 membrane (Tokuyama, A201). Following the preparation of the CCM, the CCM was placed in de-aerated 1 M KOH for 12 h. This step is necessary to fully exchange Cl− in the Tokuyama ionomer with OH− to transfer the ionomer and membrane from Cl− to OH− conductivity. It was then rinsed with DI water and was assembled inside of the cell hardware (Fuel Cell Technology Inc, 5 cm2) at room temperature using 4.4 Nm torque.The cell was tested under the following conditions: H2/O2, Tcell = 60–80 °C, relative humidity (RH) of 100% or 50%, flow rates for anode and cathode were 250 ccm and 200 ccm, respectively, and with a backpressure of 20 psig at both the cathode and anode.
X-ray computed tomography
The micro-X-ray CT was performed at Beamline 8.3.2 at Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (LBNL). A double-multilayer monochromator was used to select 28 keV X-rays, and the detection was with a LuAG scintillator and 5× lenses with a sCMOS PCO Edge camera, resulting in a 1.33 μm pixel dimension and a 3.3 mm horizontal field of view (FOV). For each scan, a 200 ms exposure time was used with 1025 acquired projections over a 180° rotation range. Five scans along the channel were used to observe full 1.5 lengths of the channel. 15% overlap was used between these 5 FOVs. A single serpentine 1 mm × 1 mm channel sample holder was used for the in operando experiment. The active area of the MEA was 1 cm2. The operating conditions were the OCV and a cell temperature of 45 °C and humidity bottle temperatures of 60 °C and 40 °C, resulting in 58% and 120% RH, respectively.Nano-X-ray CT images were acquired at Beamline 32-ID at APS at Argonne National Laboratory (ANL). The energy was set at 8 keV with 1 s exposure time per projection. A Fresnel zone plate (FZP) with a 30 nm outermost zone width was used and the phase ring was placed in a back focal position to enhance soft materials contrast. 1800 back projections were acquired during the scan. An approximately 80 μm sample was cut with a micromanipulator to fit into the FOV and mounted onto a flat pin (which was then mounted onto the stage). The room RH was 50% RH; to reach 100% RH a droplet of water was placed onto the sample with a pin.
Image analysis
TomoPy (open-source software developed at ANL) was used for phase retrieval and tomographic reconstructions with the Gridrec algorithm for micro-CT and the ASTRA toolbox for nano-CT.22–25 Our previous studies provide more details on image reconstruction.26,27,29 Image segmentation and analysis were performed with Fiji/ImageJ and Avizo Fire 8.1. For nano-CT data, the image was cropped to 52 × 48 μm. The manual threshold was selected, which was the same for both 50% RH, and 100% RH to separate the void/ionomer and solid. The NiCu catalyst was easier to segment because of its brightness compared to the softer carbon material. Avizo Fire 8.1 was used to align the 50% and 100% image stacks and ImageJ with BoneJ plug-in was used to fit pores and solid-size distributions. Volume rendering of nano-CT data was achieved with Avizo Fire 8.1.Results and discussion
The morphology of the as synthesized NiCu/KB catalyst was investigated by TEM, where it was observed that the NiCu particles range in the size from 8 nm to >50 nm with an average particle size (τ) of 22 nm and a standard deviation of 9 nm. Fig. 1a shows NiCu nanoparticles well dispersed on the carbon black support. However, regions of higher particle density reveal significant agglomeration and a tendency towards forming larger particles. Such a broad particle distribution can be explained by a high loading of the NiCu alloy on the carbon (50 wt%) and a relatively high temperature reduction (T = 550 °C).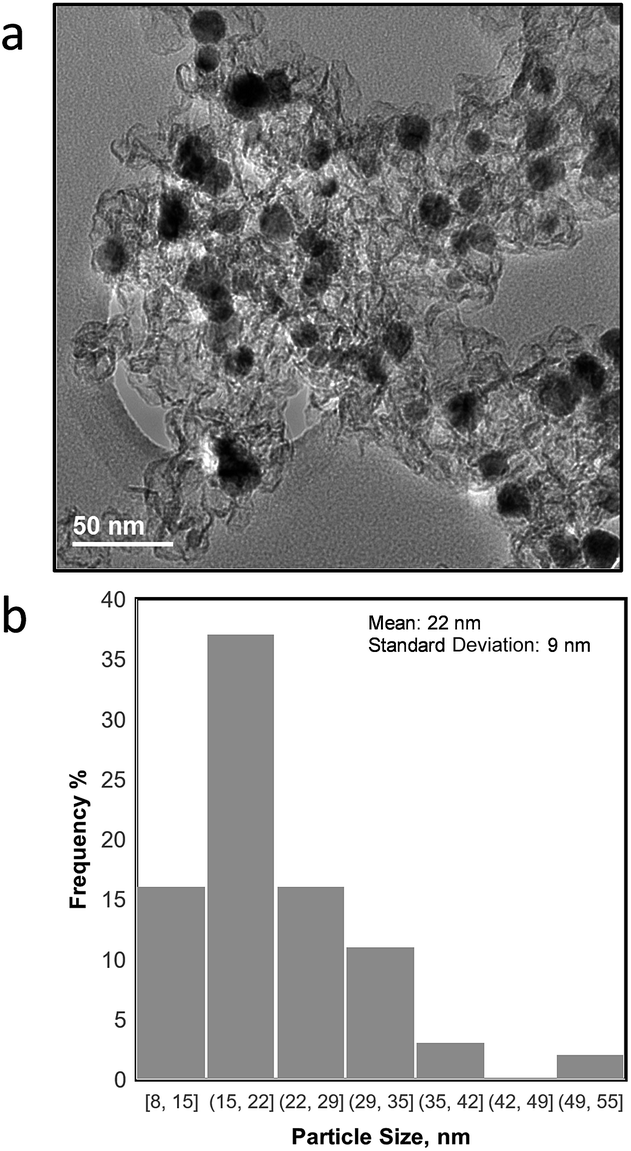 | ||
| Fig. 1 a) TEM image of NiCu/KB and (b) particle size distribution for all NiCu particles observed in TEM micrographs. | ||
Analysis of the X-ray diffraction pattern for NiCu/KB shows a single FCC Ni phase with a lattice constant of 3.52 Å and an average crystallite size of 27 nm, calculated from the whole pattern refinement (Fig. 2). This is in good correspondence with the TEM observations. Here, the lattice constant for a reference sample of 50 wt% Ni/KB was calculated as 3.52 Å. Applying the Vegard's rule, the amount of Cu in solid solution with Ni can be calculated from the linear combination of the pure FCC lattice constants of Ni [98-001-4691] and Cu [98-001-3667], being 3.516 Å and 3.615 Å, respectively. The resulting lattice constant for a ratio of Ni to Cu of 95:5 is 3.520 Å. Due to random errors, the lattice expansion due to alloying Ni with Cu could not be measured. However, the absence of the amorphous or crystalline Cu phases suggests that all copper is incorporated into the FCC Ni phase presenting a true alloy at every particle size. The presence of amorphous Ni oxide and hydroxide phases is also evident in the Ni/KB XRD pattern, ∼37° in 2-theta.
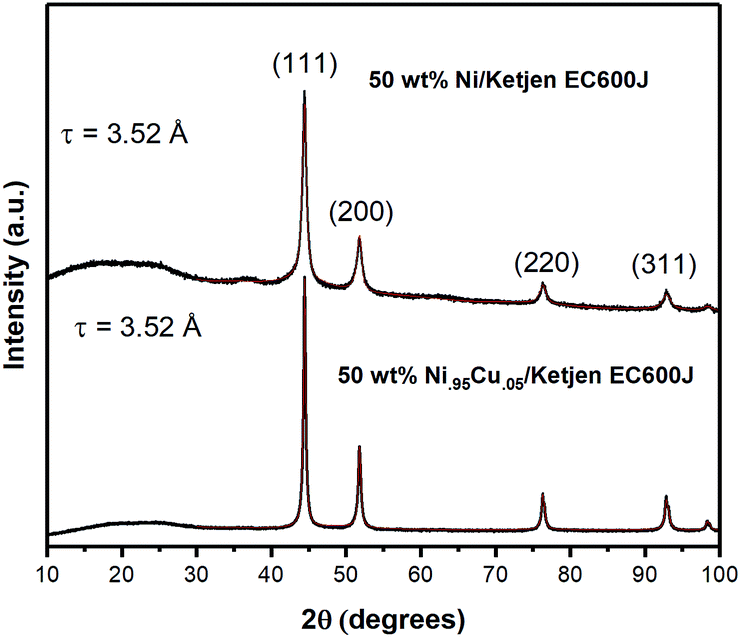 | ||
| Fig. 2 X-ray diffraction pattern for NiCu/KB (bottom) and Ni/KB (top) prepared by thermal reduction at T = 550 °C. The peak intensities are normalized to the (111) reflection. Pattern fits are overlayed on experimental data (red). | ||
Fig. 3 shows high-resolution Ni 2p and Cu 2p photoelectron spectra obtained for Ni–KB and NiCu/KB samples. Ni 2p spectra were fitted using reference photo-peaks for metallic Ni, NiO and Ni(OH)2 established previously.28 The elemental composition and relative distribution of nickel species are shown in Fig. 3c. The bimetallic sample has a smaller amount of oxygen detected, which is also reflected in the absence of hydroxides in the high-resolution Ni 2p spectrum for the NiCu sample. The main type of nickel in both the Ni/KB and NiCu/KB samples is nickel oxide. The surface concentration of reduced metallic or alloyed Ni, however, is much larger for the alloyed sample. From the high-resolution Cu 2p spectrum, it is seen that Cu is mainly present as metallic Cu with a small amount of oxides.
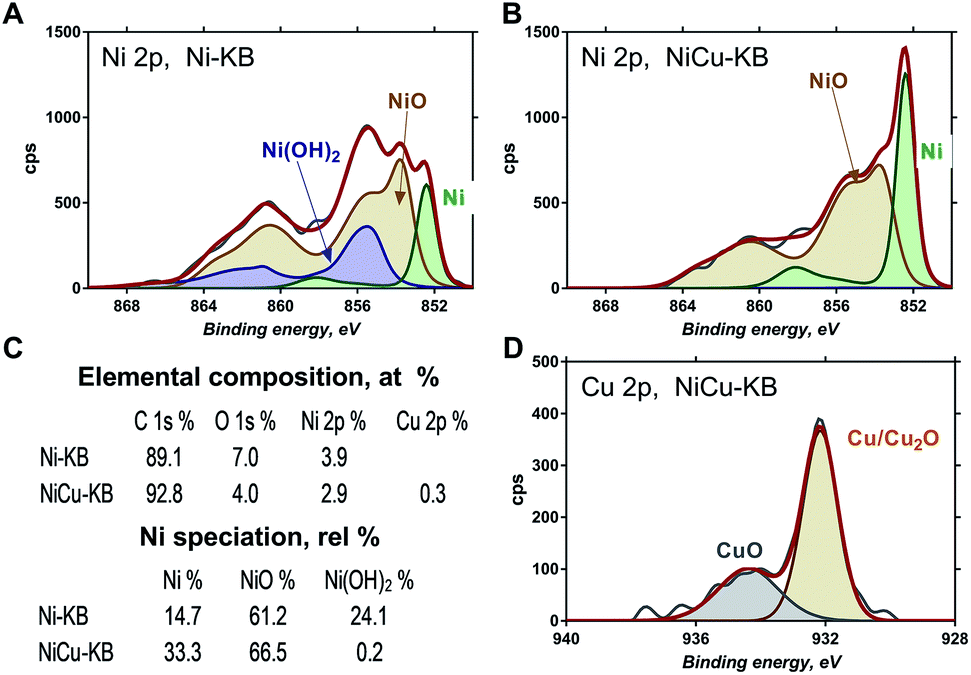 | ||
| Fig. 3 High-resolution Ni 2p spectrum from (A) Ni/KB and (B) NiCu/KB. (D) High-resolution Cu 2p spectrum from NiCu/KB. (C) Table showing elemental composition and Ni speciation. | ||
Cyclic voltammetry was used to characterize the intrinsic activity towards the HOR for NiCu/KB. Fig. 4 summarizes the results of the electrochemical evaluation of the catalyst by cyclic voltammetry in a rotating disk electrode (RDE). All voltammograms are being shown after the subtraction of the capacitance current associated with Ketjenblack, as measured in 0.1 M KOH with a scan rate of 5 mV s−1. Fig. 4a shows a comparison of cyclic voltammograms (CV) in N2vs. H2 saturated electrolyte, confirming the activity of NiCu/KB towards the HOR. At anodic potentials greater than 0.2 V vs. RHE, a decreasing limiting current is observed, which is due to the formation of Ni(OH)2, which passivates the catalyst surface and hinders the HOR. This finding is consistent with previously reported observations.18
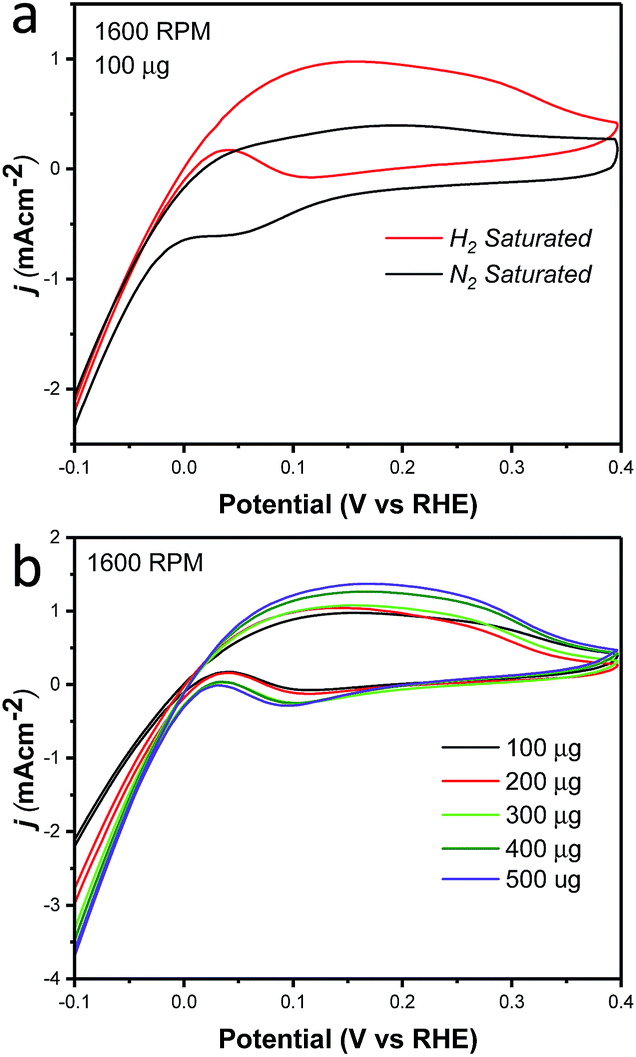 | ||
| Fig. 4 Cyclic voltammograms showing (A) comparison of NiCu/KB in N2 saturated 0.1 M KOH (black) and in H2 saturated 0.1 M KOH and (B) NiCu/KB in H2 saturated 0.1 M KOH with various metal loadings on the electrode with a sweep rate of 5 mV s−1 and a rotation speed of 1600 rpm. | ||
Table 1 compares the specific activity of the catalyst reported here to the NiCu and NiMo electrocatalysts reported previously. The exchange current density normalized to the specific surface area is in strong agreement with previous studies of the NiMo system with a ratio of Ni to Mo of 90:10. Furthermore, the specific activity for NiCu in this work is 1.8–3.6 times higher than that of previously reported NiCu electrocatalysts by the Savinova's group.18 The mass-weighted activity and specific surface area are lower than the values reported previously for the NiCu system. One critical difference between this work and Savinova's work is that we have synthesized a true NiCu nanoparticle alloy, as opposed to a surface-segregated nanocomposite. One can hypothesize that in a true alloy the number of Ni atoms on the surface immediately exposed to a Cu atom is larger. This should facilitate a bi-functional mechanism of the HOR on true allowed NiCu catalysts. This difference may also result from the larger average particle size of ∼22 nm, compared to ∼11 nm in the referenced work.18 By reducing particle sizes it is possible to even further improve the activities and surface area of this electrocatalyst.
Previous work reporting the high catalytic activity of Ni95Cu5 towards the HOR in alkaline media demonstrated an electronic and ensemble effect due to alloying of Ni with Cu and the segregation of Cu islands during thermal reduction at T = 250 °C.18 At this temperature, the formation of a solid solution of Ni and Cu is not favorable, leading to phase segregation. In the present work, the synthesis was carried out at T = 550 °C and X-ray diffraction data do not suggest Ni and Cu segregation. The high activity of Ni96Cu5 in this work is thought to be caused purely by a chemical effect.
Recent understanding of the HOR mechanism in alkaline media on Pt14,15 revealed a bifunctional mechanism consisting of (i) the adsorption of Had intermediates on a primary active site, and (ii) the 1 e− reduction of water forming Pt–Had and OHq−aq, thus facilitating the Volmer step in the HOR mechanism. It was suggested that a strategy for lowering the overpotential for the HOR in alkaline media could involve alloying Pt or the primary active metal site, with a base metal, which forms a passivated hydroxide layer. In doing so, the Volmer step of the reaction may be facilitated, as it involves the approach of intermediate OH− through the outer Helmholtz plane towards a negatively charged electrode, which is unfavorable for negatively charged species. In the present work, Ni is considered as the primary active site for the generation of Had while Cu acts as a base metal which may form a hydroxide passivation layer. As surface analysis had shown, the Ni surface in the Ni/KB catalyst sample has a passivated hydroxide layer as an “overcoat”, which makes it not favorable for initial hydrogen adsorption. In contrast, in the alloyed NiCu/KB catalyst sample, the analysis shows the Ni surface free of hydroxides, and thus the exposed Ni surface could serve as primary sites for the first step of the reaction, while Cu acts as the site for the second step of the reduction of water forming Ni–Had and NiCu–OHq−aq (see Fig. 5). We hypothesize here that the NiCu fully alloyed nanoparticle catalyst supports a bi-functional Tafel–Volmer mechanism for hydrogen oxidation in the alkaline electrolyte,13 which is outlined for this specific material selection in eqn (1–3):
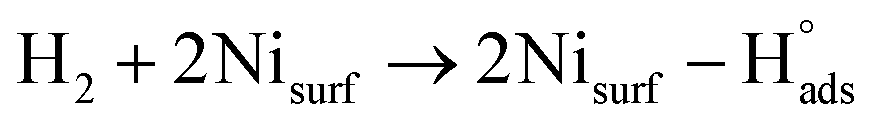 | (1) |
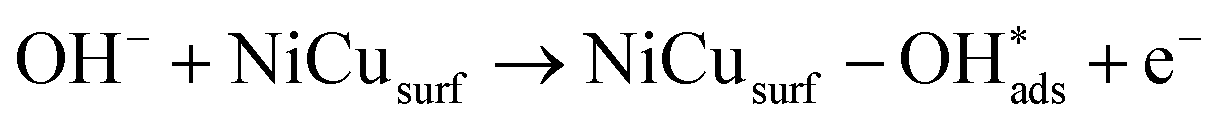 | (2) |
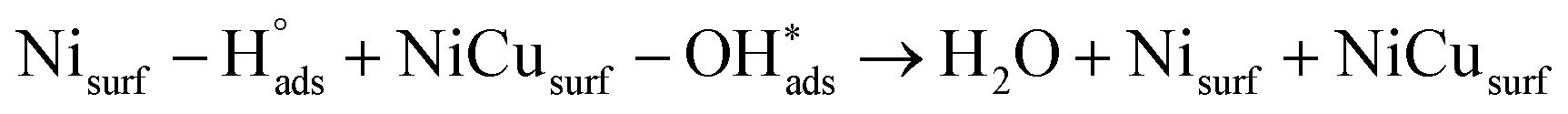 | (3) |
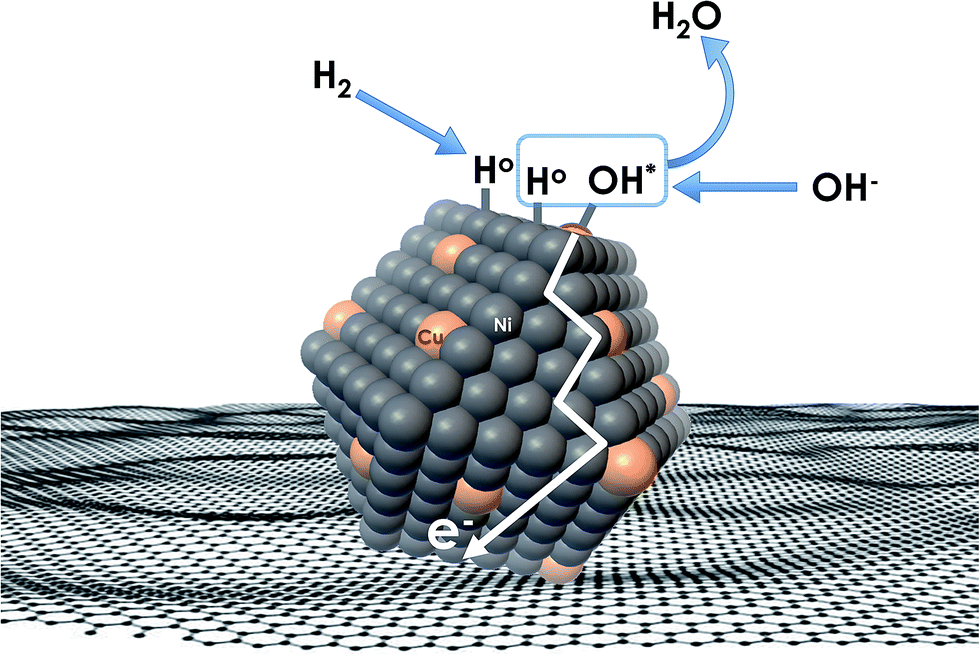 | ||
| Fig. 5 Conceptual schematic of the mechanism of the HOR on alloyed NiCu nanoparticles, supported on a carbonaceous surface. | ||
This suggested mechanism largely agrees with those proposed for the PGM metal catalysts (such as Pd), which are alloyed with a transition metal (e.g. Ni). The observations allow us to draw an analogy between the NiCu alloyed nano-particle catalysts and PdNi in their HOR reactivities and mechanisms.31
Additionally, the effect of the catalyst layer thickness was further examined by comparing the specific activity and mass-weighted activity at each catalyst loading (Fig. S2†). A trend of decreasing specific activity as well as mass-weighted activity was evident and is consistent with the less than proportional increase in the current density with the increased catalyst loading (Fig. 4b). As the exchange current density calculated from the micro-polarization region is not influenced by mass transfer characteristics of the electrode, it seems evident that an increase in the catalyst layer thickness results in an increase in the electrochemical impedance. The reaction impedance associated with the Volmer step of the HOR mechanism and the formation of an intermediate and surface OH− ions, can then explain why the Ni95Cu5 performance in both the RDE and MEA methods does not reach that of PGM catalysts. This factor also highlights a critical design point in optimizing the catalyst layer in MEA and fuel cell tests.
These results, in combination with our previous results obtained for NiMo/KB for the HOR in alkaline media,17 suggest the affinity of the alloying base metal to hydroxyl adsorption to be in an optimal range (not be too strong, nor too weak) in order to facilitate both abundance of surface hydroxyl groups available in proximity to atomically adsorbed hydrogen for water formation, and subsequent water release. This concept should be further verified and studied in detail by the DFT and other modelling tools.
MEA fuel cell tests
MEA studies were performed using the catalyst coated membrane (CCM) method of MEA fabrication with Tokuyama membranes and ionomers. The CCM method was selected due to the poor thermal stability of the Tokuyama membrane and ionomer at temperatures above 80 °C, typically used for the hot-pressed MEA gas-diffusion electrode (GDE) method of fabrication. The poor stability of AEM ionomers at elevated temperatures is due to the susceptibility of the quaternary ammonia sites in the crosslinked polymer to nucleophilic attack by OH−.7Steady-state polarization curves (see Fig. 6) were obtained at various temperatures under fully humidified conditions producing a peak power density of ∼350 mW cm−2 at 80 °C. Decreasing RH did not result in higher current densities (see Fig. 8), as was observed for the NiMo electrocatalyst in the previous study.17 This is indicative that in this anode catalyst layer, water management is more efficient. For three temperatures, the OCV achieved was high – above 1.0 V. As the temperature was raised, due to the faster reaction kinetics the current density increased, particularly in the activation region. Ohmic and mass-transport current densities also improved at higher temperatures due to the improved OH− conductivity and improved water removal by evaporation and phase-change induced flow.17 The peak power density obtained is, at present, the highest reported for any PGM-free anodic material integrated as the HOR catalyst in an AEM fuel cell tested as the MEA.
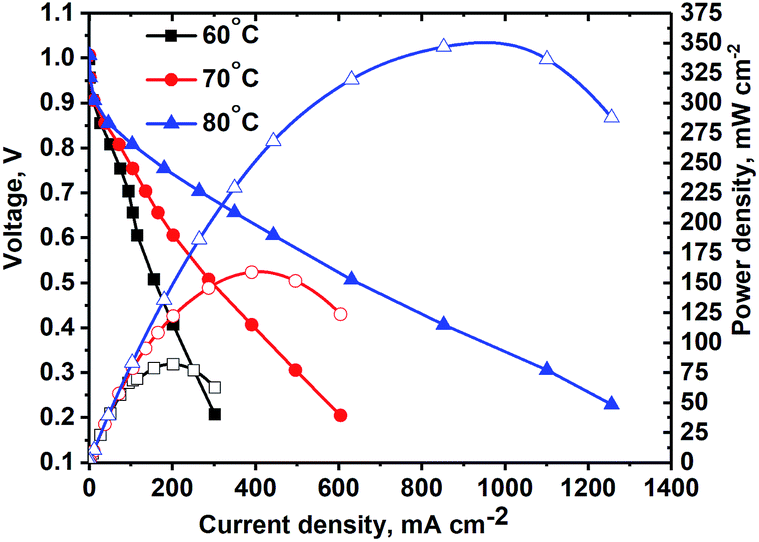 | ||
| Fig. 6 Polarization curve (no IR-corrections) of the MEA with the NiCu/KB anode. Conditions: Tcell = 60, 70 and 80 °C, RH = 100%, H2 and O2 backpressure = 20 psig, anode: NiCu/KB, 4 mg cm−2, cathode: Pd/XC72R, 0.2 mg cm(Pd)−2. | ||
Micro-X-ray CT
Fig. 7a and b show a three-dimensional representation of grey-scale tomography data for the cell with 58% and 120% RH feeds and OCV conditions. The anode NiCu/KB catalyst layer is the thickest (140 μm) and brightest part within the cell. For a PGM-free CCM 140 μm is the moderate thickness. The catalyst layer is observed to be densely packed, crack-less and fairly uniform in thickness. The cathode catalyst layer showed some mud cracks. The comparison of data at 58 and 120% RH reveals no apparent micro-scale swelling and no liquid condensation. In our previous work, we showed that NiMo/KB acts hydroscopically – absorbing large amounts of water.17 In that study, polarization behavior was strongly dependent on the RH, where the peak power density was reached at 70% RH. Here, water flooding is not an issue at 100% RH, as observed by both polarization data and X-ray CT images. Areas of high local hydrophilicity found in our previous study are possibly due to the secondary phase of Mo, or due to the chemical state of Ni. Opposite wettability behavior was found in a recent study of model electrodes and in theory, suggesting that β-Ni(OH)2 is hydrophobic, whereas β-NiOOH (001) is hydrophilic.18,19 These changes in wettability were ascribed to changes in chemical compositions of the surface. | ||
| Fig. 7 Micro-CT representation of the MEA within the in operando hardware for (a) 58% and (b) 120% RH at a temperature of 45 °C. The scale-bar is 500 μm. | ||
Nano-X-ray CT
No apparent difference between 58 and 120% RH was observed from the micro-X-ray CT data for larger macropores (>1 μm). To probe water distribution in smaller macropores (<1 μm), we utilize nano-X-ray CT imaging. Fig. 8 shows three-dimensional NiCu distribution within carbon black. The average carbon agglomerate radius was 0.55 μm with unimodal distribution. At the same time, the NiCu catalyst had a wider distribution of sizes. From micro-X-ray CT in Fig. 7, NiCu distribution on a micro-scale is uniform. Within 52 μm of the nano-CT domain probed, the NiCu average cluster size was found to be 1.23 μm. Furthermore, NiCu occupies 8% of the electrode volume, as shown in Fig. 8f, where the remaining of the volume is either the carbon support or the void space.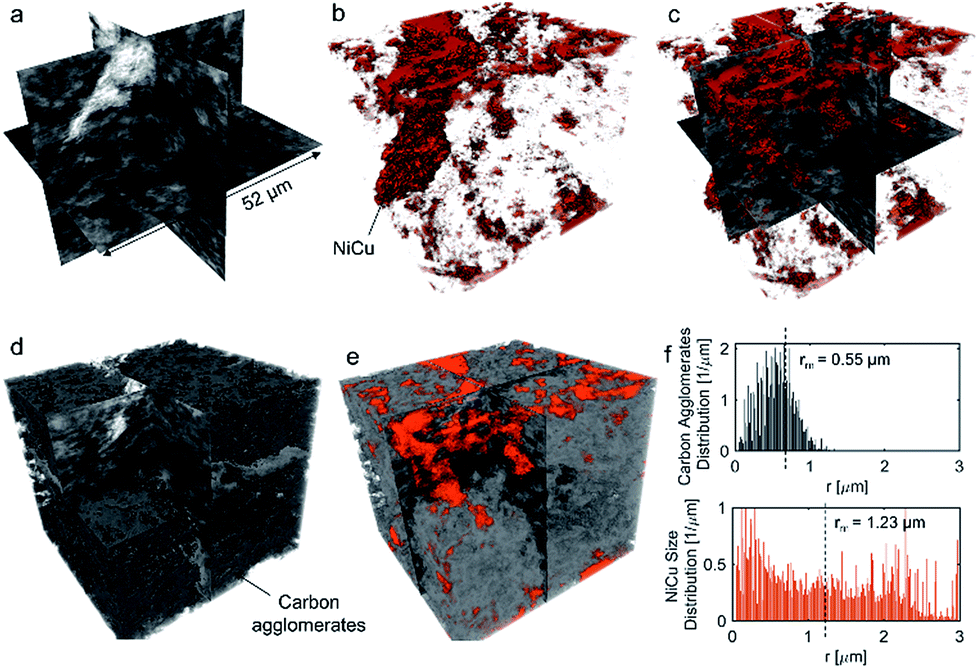 | ||
| Fig. 8 Three-dimensional representation of the NiCu/KB electrode, with (a) grey-scale, (b) volume-rendered NiCu (shown in red), (c) overlaying (a) and (b), (d) carbon agglomerates segmented, (e) overlaying (d) and (b). The carbon agglomerate distribution and NiCu size distribution are shown in (f). | ||
Fig. 9 shows a comparison of void/ionomer distribution between 50% and 100% RH conditions. The difference between these identical location nano-X-ray CTs is subtle. However, the darker domains expand when water is introduced into the sample. Darker domains correspond to both void and ionomer spaces. Fig. 9c and d show the locations where ionomer expanded and its schematic, respectively. The volume fraction of the void/ionomer space increased from 12 to 17% as the RH increased from 50 to 100%.
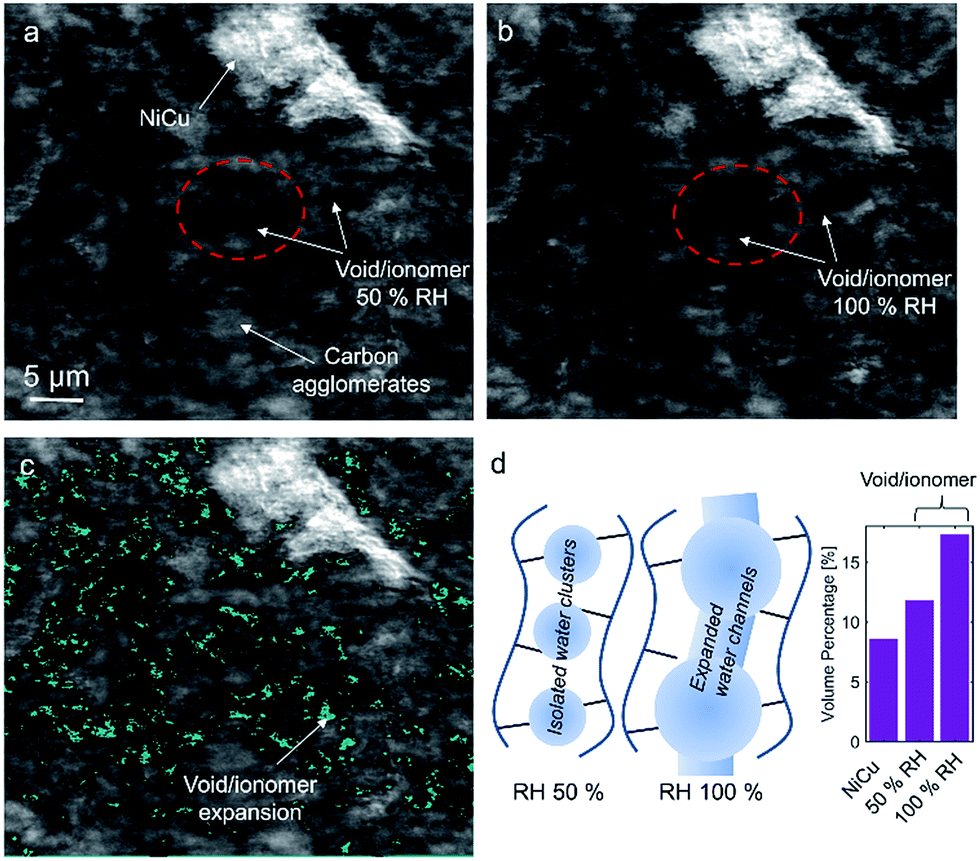 | ||
| Fig. 9 Nano-X-ray CT cross-section tomographs of (a) 50% RH and (b) 100% RH, where carbon agglomerates, the void/ionomer space, and NiCu are marked. (c) The difference in the void/ionomer space after subtracting (a) from (b). (d) A schematic of ionomer swelling and volume percentage for NiCu, and void/ionomer at two RHs. | ||
The volume-rendered void/ionomer space is shown in Fig. 10 for both RHs. From the size distribution, the mean radius of voids at 50% RH is 0.36 μm, whereas for 100% RH it is larger being 0.43 μm. From subtracting the two void/ionomer distributions, it is apparent that the ionomer swells uniformly from smaller to larger voids. The critical pore radius where this transition happened is 0.6 μm.
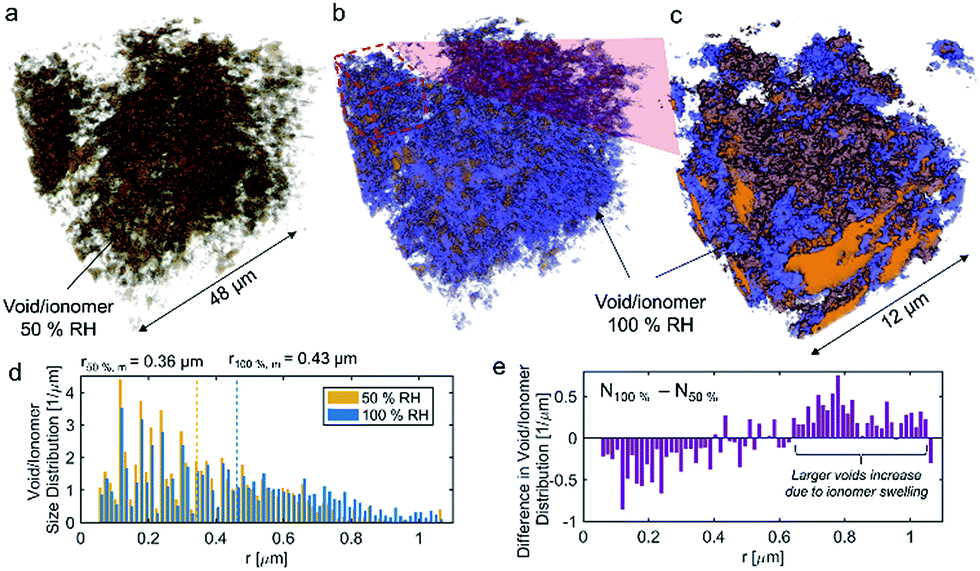 | ||
| Fig. 10 Volume-rendered void/ionomer space for (a) 50% RH, (b) combined 50% RH and the difference between 100% and 50% RH, (c) zoom-in into 12 μm of the FOV. The void/ionomer size distributions as well as mean radii are shown in (d). The difference between 100% and 50% RH size distributions of void/ionomer is shown by (e). | ||
From the water-management perspective, if we assume a uniform surface tension corresponding to the more hydrophobic nature of the material, having water in larger macropores is advantageous. Large macropores present lower-resistance pathways for water removal into the gas-flow channel avoiding local flooding. Comparing this study to our previous one17 it is evident that surface properties of the electrocatalysts themselves dictate the local wettability, as the carbon support used in these two studies was identical. Hence, the effective MEA design requires more homogeneous catalyst dispersion, low thickness and the hierarchical morphology and also understanding of the surface chemistry and its effect on local wettability, which has not been discussed in the literature. X-ray CT observations reported here on the NiCu/KB catalyst layer are in contrast with our previous findings when the NiMo/KB catalyst was used.17 One can speculate that the increased oxophilic character of Mo moieties provides a favourable local micro-environment for water nucleation and thus results in prominent liquid water formation in the anode catalyst layer, observed as “flooding” in NiMo catalysts. Such behaviour can be expected across the sub-category of bi-metallic30,31 or composite32 HOR catalysts, employing oxophilic moieties as co-catalysts.
Conclusions
NiCu fully alloyed nanoparticles, supported on medium surface area mesoporous carbon black (Ketjenbalck) are described here as a new, scalable and practical HOR catalyst suitable for integration in a practical AMFC anode. Structure-to-property relationship studies reveal a bi-functional mechanism of the HOR on the NiCu catalyst, with the atomically dispersed Cu moiety playing a substantial role in balancing the surface hydroxyl availability and forming and releasing water as a reaction product. Consequently, this new catalyst demonstrates a superior area specific activity in the RDE and a record-setting power density for the PGM-free AMFC anode approaching 350 mW cm−2 at 80 °C.The nano- and micro-X-ray CT results suggest that the nickel state in NiCu is hydrophobic in nature, where the NiCu surface may be isostructural with β-Ni(OH)2. The improved water distribution supports this conclusion observed in operando for NiCu/KB compared to NiMo/KB, resulting in improved water management in the MEA for NiCu/KB.
Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
The project is supported by DOE EERE program, Incubator Award DE-EE0006962 “Development of PGM-free Catalysts for Hydrogen Oxidation Reaction in Alkaline Media”. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef.
- S. Maurya, S.-H. Shin, Y. Kim and S.-H. Moon, RSC Adv., 2015, 5, 37206–37230 RSC.
- R. B. Kaspar, M. P. Letterio, J. A. Wittkopf, K. Gong, S. Gu and Y. S. Yan, J. Electrochem. Soc., 2015, 162, F483–F488 CrossRef.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177–181 RSC.
- R. C. T. Slade, J. P. Kizewski, S. D. Poynton, R. Zeng and J. R. Varcoe, in Fuel Cells: Selected Entries from the Encyclopedia of Sustainability Science and Technology, ed. K.-D. Kreuer, Springer, New York, NY, 2013, pp. 9–29, DOI: 10.1007/978-1-4614-5785-5_2 Search PubMed.
- A. Serov, I. V. Zenyuk, C. G. Arges and M. Chatenet, J. Power Sources, 2018, 375, 149–157 CrossRef.
- E. F. Holby, G. Wu, P. Zelenay and C. D. Taylor, J. Phys. Chem. C, 2014, 118, 14388–14393 CrossRef.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 RSC.
- K. Strickland, E. Miner, Q. Jia, U. Tylus, N. Ramaswamy, W. Liang, M.-T. Sougrati, F. Jaouen and S. Mukerjee, Nat. Commun., 2015, 6, 7343 CrossRef PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasche, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z.-F. Ma and Q. Jia, Angew. Chem., Int. Ed., 2017, 56, 15594–15598 CrossRef PubMed.
- N. Ramaswamy, S. Ghoshal, M. K. Bates, Q. Jia, J. Li and S. Mukerjee, Nano Energy, 2017, 41, 765–771 CrossRef.
- Z. Zhuang, S. A. Giles, J. Zheng, G. R. Jenness, S. Caratzoulas, D. G. Vlachos and Y. Yan, Nat. Commun., 2016, 7, 10141 CrossRef PubMed.
- S. Kabir, K. Lemire, K. Artyushkova, A. Roy, M. Odgaard, D. Schlueter, A. Oshchepkov, A. Bonnefont, E. Savinova, D. C. Sabarirajan, P. Mandal, E. J. Crumlin, I. Zenyuk, P. Atanassov and A. Serov, J. Mater. Chem. A, 2017, 5, 24433–24443 RSC.
- A. G. Oshchepkov, P. A. Simonov, O. V. Cherstiouk, R. R. Nazmutdinov, D. V. Glukhov, V. I. Zaikovskii, T. Y. Kardash, R. I. Kvon, A. Bonnefont, A. N. Simonov, V. N. Parmon and E. R. Savinova, Top. Catal., 2015, 58, 1181–1192 CrossRef.
- Y.-H. Chang, N. Y. Hau, C. Liu, Y.-T. Huang, C.-C. Li, K. Shih and S.-P. Feng, Nanoscale, 2014, 6, 15309–15315 RSC.
- M. J. Eslamibidgoli, A. Gross and M. Eikerling, Phys. Chem. Chem. Phys., 2017, 19, 22659–22669 RSC.
- A. Serov, N. I. Andersen, S. A. Kabir, A. Roy, T. Asset, M. Chatenet, F. Maillard and P. Atanassov, J. Electrochem. Soc., 2015, 162, F1305–F1309 CrossRef.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef.
- D. Gürsoy, F. De Carlo, X. Xiao and C. Jacobsen, J. Synchrotron Radiat., 2014, 21, 1188–1193 CrossRef PubMed.
- D. M. Pelt, D. Gürsoy, W. J. Palenstijn, J. Sijbers, F. De Carlo and K. J. Batenburg, J. Synchrotron Radiat., 2016, 23, 842–849 CrossRef PubMed.
- F. De Carlo, D. Gürsoy, F. Marone, M. Rivers, D. Y. Parkinson, F. Khan, N. Schwarz, D. J. Vine, S. Vogt and S.-C. Gleber, J. Synchrotron Radiat., 2014, 21, 1224–1230 CrossRef PubMed.
- V. De Andrade, A. Deriy, M. J. Wojcik, D. Gürsoy, D. Shu, K. Fezzaa and F. De Carlo, SPIE Newsroom, 2016 DOI:10.1117/2.1201604.006461.
- I. V. Zenyuk, D. Y. Parkinson, L. G. Connolly and A. Z. Weber, J. Power Sources, 2016, 328, 364–376 CrossRef.
- T. Asset, A. Roy, T. Sakamoto, M. Padilla, I. Matanovic, K. Artyushkova, A. Serov, F. Maillard, M. Chatenet, K. Asazawa, H. Tanaka and P. Atanassov, Electrochim. Acta, 2016, 215, 420–426 CrossRef.
- A. D. Shum, D. Y. Parkinson, X. Xiao, A. Z. Weber, O. S. Burheim and I. V. Zenyuk, Electrochim. Acta, 2017, 256, 279–290 CrossRef.
- M. H. Tang, C. Hahn, A. J. Klobuchar, J. W. Desmond Ng, J. Wellendorff, T. Bligaard and T. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 19250–19257 RSC.
- M. Alesker, M. Page, M. Shviro, Y. Paska, G. Gershinsky, D. R. Dekel and D. Zitoun, J. Power Sources, 2016, 304, 332–339 CrossRef.
- H. A. Miller, F. Vizza, M. Marelli, A. Zadick, L. Dubau, M. Chatenet, S. Geiger, S. Cherevko, H. Doan, R. K. Pavlicek, S. Mukerjee and D. R. Dekel, Nano Energy, 2017, 33, 293–305 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00261d |
| This journal is © The Royal Society of Chemistry 2018 |