
Photocatalytic hydrogen evolution from neutral aqueous solution by a water-soluble cobalt(II) porphyrin†
Belete B.
Beyene
 ab and
Chen-Hsiung
Hung
*a
ab and
Chen-Hsiung
Hung
*a
aInstitute of Chemistry, Academia Sinica, Nankang, Taipei-11529, Taiwan, Republic of China. E-mail: chhung@gate.sinica.edu.tw
bDepartment of Chemistry, Bahir Dar University, P.O. Box 79, Bahir Dar, Ethiopia
First published on 25th June 2018
Abstract
Efficient storage of solar energy via light-driven hydrogen evolution is an attractive and promising strategy to address challenges related to increasing global energy demand. Herein, we report a highly active homogeneous photocatalytic hydrogen evolution system comprising a water-soluble cobalt(II) porphyrin, CoTPPS, as a molecular catalyst, [Ru(bpy)3]2+ as a photosensitizer, and ascorbic acid as a sacrificial electron donor. Under optimized conditions, a hydrogen evolution rate of 1.3 μmol min−1, a TOF of 120.8 min−1, and a TON of 6410 are obtained in 1 M phosphate buffer at pH 6.8 by irradiation with an LED light source at 420 nm and 0.6 W cm−2. The studies on the effects of pH and catalyst concentration showed that optimal H2 evolution efficiency was achieved at a pH of 6.8 and a catalyst concentration of 1.5 μM. Moreover, the kinetics of the fluorescence emission quenching experiment indicates that the rate of reductive quenching of [Ru(bpy)3]2+* by ascorbate (7.5 × 106 s−1) is higher than that of oxidative quenching by CoTPPS (1.7 × 104 s−1). As also supported by redox potential energy level analysis, a mechanistic pathway was proposed in which reduction of photo-excited [Ru(bpy)3]2+* by ascorbic acid produces [Ru(bpy)3]1+, which is a strong reductant and will subsequently reduce CoTPPS to generate cobalt-hydride for hydrogen evolution after protonation.
Introduction
Solar energy, water, and carbon dioxide are converted into organic compounds and oxygen in photosynthesis, which sustains life on Earth. To produce fuel from solar energy, researchers have paid much attention to the development of artificial photosynthetic systems.1–3 It is believed that solar water splitting is a clean, sustainable, and renewable way to store solar energy for future fuel and energy supply.4–8 Moreover, it has attracted great interest because of the ever-increasing global energy demand and serious environmental problems due to emission of CO2 upon combustion of fossil fuels.1,2,9A number of highly efficient photocatalytic H2 evolution systems have been developed since the pioneering work of Honda and Fujishima,10 who demonstrated the first photo-electrochemical (PEC) cell for water splitting.9,11 The first multicomponent solar-driven H2 generation system using Ru(bpy)32+, where bpy stands for 2,2′-bipyridine, as a photosensitizer, colloidal Pt as the catalyst and triethanolamine (TEOA) as the sacrificial electron donor was reported by Lehn and co-workers.12 Although their report was encouraging because of an excellent H2 evolution activity of Pt, the high cost and low abundance presented problems for designing catalysts based on precious metals. Thus, efforts have been devoted to exploring cheap and earth-abundant transition metal complexes with superior activity and stability for use as the catalyst for solar water splitting. In line with this, a variety of macrocyclic cobalt, nickel, and iron complexes have been extensively studied in homogeneous photochemical H2 production from water in conjugation with a photosensitizer and sacrificial electron donor.13–25 Among them, cobalt complexes are the predominant candidates due to the unique redox and electronic properties of cobalt that favor H2 evolution.
Cobalt porphyrins have also received great attention as molecular catalysts for reduction of protons or splitting H2O to generate H2.26–31 However, most studies were conducted under electrochemical conditions using acids as proton sources. Only very limited reports are available for H2 evolution from neutral aqueous solution employing porphyrin-based catalysts.28,32 To the best of our knowledge, up to now only two reports have been documented in the literature for homogeneous photocatalytic H2 evolution in aqueous solution using cobalt porphyrins.32,33 Indeed, porphyrins or metalloporphyrins were more frequently used as photosensitizers in photochemical H2 evolution in combination with diverse catalysts such as colloidal Pt, Pt-supported materials, and enzymes.34–36
Inspired by our previous results using cobalt porphyrins as electrocatalysts for H2 evolution,37 we herein report a highly active photocatalytic hydrogen evolution system in neutral aqueous solution using a water-soluble cobaltous meso-tetrakis(p-sulfonylphenyl)porphyrin complex (CoTPPS) as the catalyst, Ru(bpy)32+ as the light absorbing photosensitizer, and ascorbic acid (AscH) as the sacrificial electron donor (Scheme 1). Substantial H2 evolution was observed by irradiating the solution containing CoTPPS, Ru(bpy)32+, and AscH with an LED light source at 420 nm and 0.6 W cm−2. A turnover number (TON) of 6.4 × 103 and a turnover frequency (TOF) of 120.8 min−1 with a reaction rate of 1.3 μmol min−1 are achieved. These values are better than those of most of the systems that contain a cobalt catalyst, Ru(bpy)32+ as the photosensitizer, and AscH as the electron donor. Moreover, our experimental results show that the catalyst is quite stable toward H2 evolution and the photochemical performance is limited by the degradation of the sensitizer upon photo-irradiation.
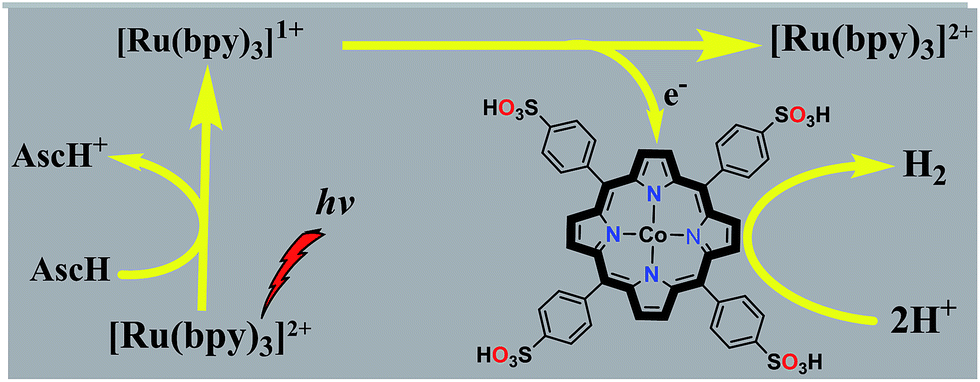 | ||
| Scheme 1 Photocatalytic system for H2 generation in this study. | ||
Results and discussion
Synthesis of the catalyst
The molecular catalyst was prepared by modifying literature procedures.38–40 The sulfonation of free-base tetraphenylporphyrin (TPPH2) was carried out in H2SO4 (98%) at 90 °C followed by cobalt metallation at 60 °C to afford CoTPPS. The identity and purity of the cobalt complex were confirmed by spectroscopic methods that we reported previously.32 The complex is highly soluble in water and moderately soluble in DMSO and DMF, but it is insoluble in all other organic solvents. The absorption spectra of CoTPPS in DMSO and water displayed different profiles. Moreover, the absorption pattern was unchanged when CoTPPS was left to stand for more than one day in aqueous solution or in the presence of an acid (with the amount more than 100 equivalents of our molecule), indicating its stability in aqueous solution and acidic media.The optical properties of the photocatalytic system
The optical properties of the mixed solution of Ru(bpy)32+, CoTPPS, and AscH in 1 M phosphate buffer at pH 6.8 were investigated by UV-vis and emission spectroscopies upon irradiation with monochromatic light with a wavelength of 420 nm. The UV-vis spectrum shows a broad absorption band ranging from 380 to 560 nm with its absorbance decreased and the color changed from yellow to light-red upon extended irradiation (Fig. 1a and b). The emission intensity of the solution also decreased dramatically upon long-term irradiation as presented in Fig. 1c. Since AscH has no absorption in the range of 380 to 560 nm, the absorption spectroscopic changes indicate that either the photosensitizer or the catalyst is photo-bleached upon irradiation. The absorbance of CoTPPS is also within the absorption range of [Ru(bpy)3]2+ and individual photostability tests were carried out in order to identify the photo-degradable component. UV-vis absorption spectroscopy before and after light irradiation indicated that CoTPPS undergoes only minor changes when irradiation is performed for 2 h, but [Ru(bpy)3]2+ displays photo-degradation after irradiation for 2 h, as also reported in the literature (Fig. 2a and b).16,41–43 Moreover, CoTPPS has no emission at all and the intense emission peak of the three-component reaction mixture, which decreased upon light irradiation (Fig. 1c), is presumably due to Ru(bpy)32+. Therefore, the spectral changes upon light irradiation are mainly due to the photosensitizer, although the catalyst undergoes minor changes. In the photochemical H2 evolution study section, we confirmed that the reduced species of CoTPPS formed in the three-component system upon light irradiation is also stable. This remarkable photostability and high solubility of CoTPPS in water motivated us to investigate photocatalytic H2 evolution in neutral aqueous solution using Ru(bpy)32+ as the sensitizer and ascorbic acid as the sacrificial electron donor.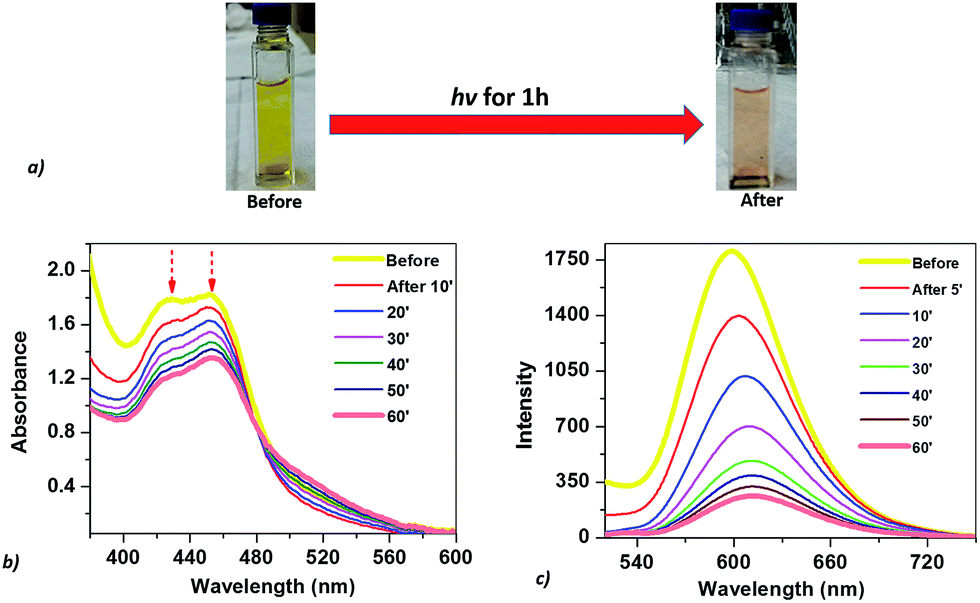 | ||
| Fig. 1 (a) Photograph of the solution before and after irradiation for 1 h; (b) UV-vis spectral changes; and (c) emission intensity decay of the solution in 1 h. Conditions: 1.2 mM Ru(bpy)32+, 0.3 M AscH, and 30 μM CoTPPS in pH 6.8 phosphate buffer under irradiation at 420 nm. | ||
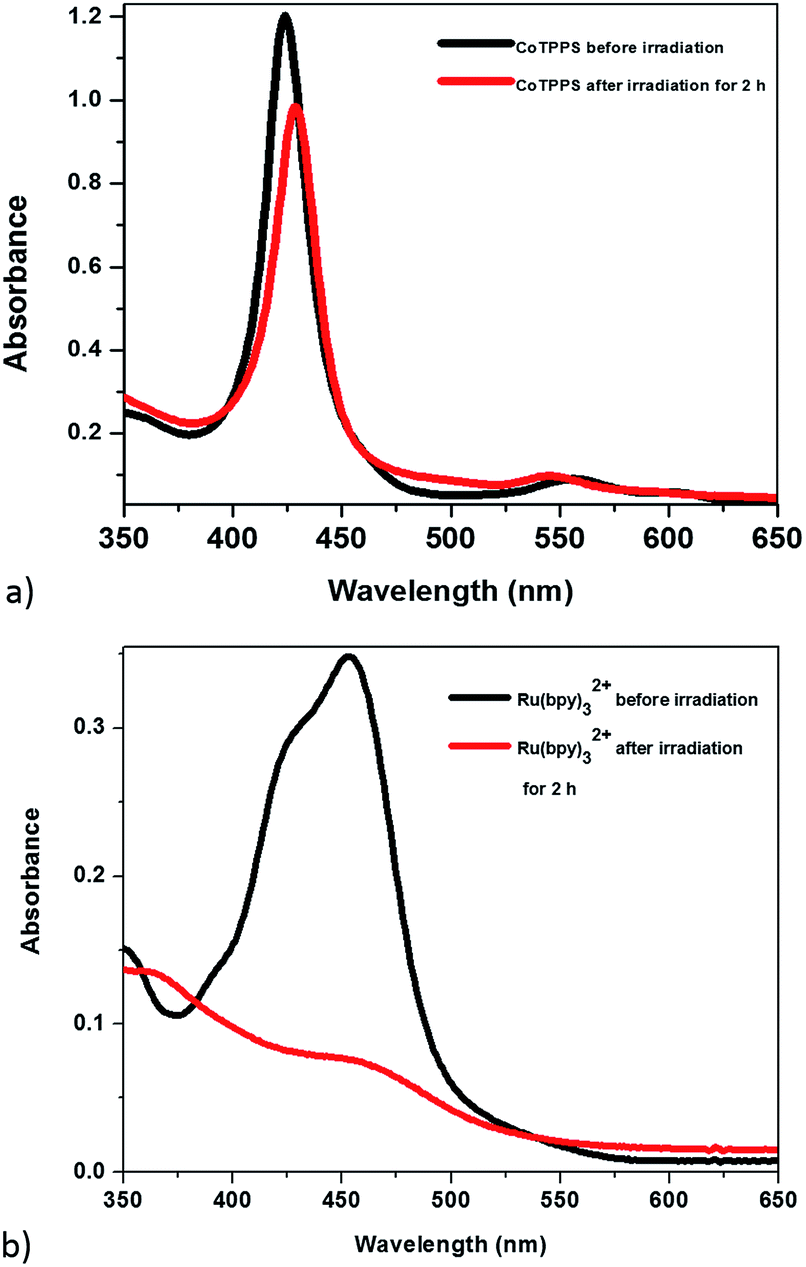 | ||
| Fig. 2 UV-visible absorption in H2O before (black) and after (red) irradiation with the 420 nm light source for 2 h: (a) CoTPPS; (b) Ru(bpy). | ||
Photochemical H2 evolution studies
The light-driven hydrogen evolution experiments were carried out by continuous irradiation with LED light (420 nm) of a N2 purged aqueous solution containing a mixture of CoTPPS, Ru(bpy)32+, and AscH in a quartz glass photo-reactor with a volume of 4 mL. The headspace H2 gas was periodically checked using a gas chromatograph equipped with a TCD detector. As shown in Fig. 3, a significant amount of H2 evolved from a solution containing 0.3 M AscH, 1.2 mM Ru(bpy)32+ and 30 μM CoTPPS at pH = 6.8.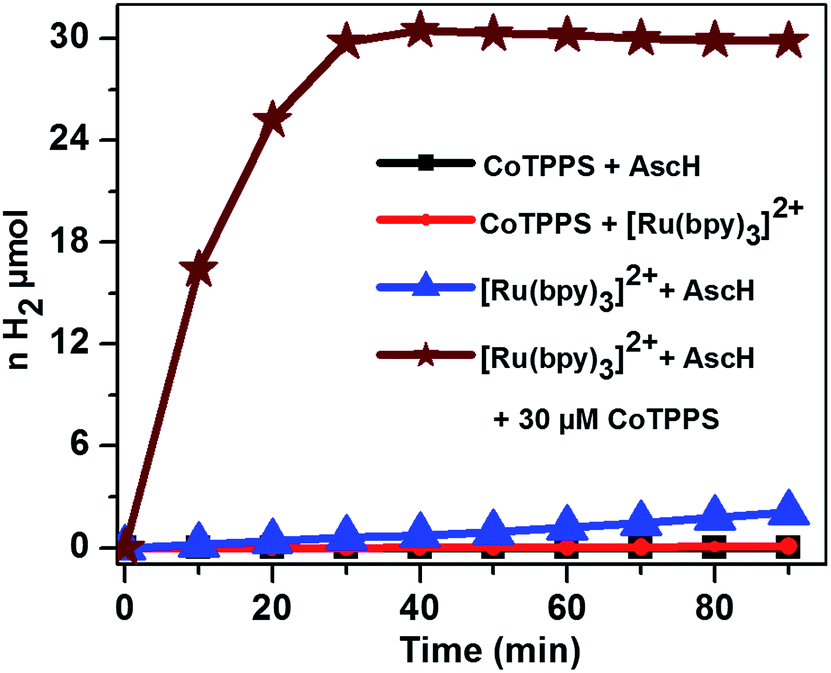 | ||
| Fig. 3 The plot of the amount of light-driven H2 evolution with time in 1 M KPi (pH 6.8) solution containing 1.2 mM Ru(bpy)32+, 0.3 M AscH and 30 μM CoTPPS (brown curve) and the control experiments at 420 nm. | ||
Control experiments under the same experimental conditions but in the dark as well as upon irradiation with light in the absence of Ru(bpy)32+, AscH, or CoTPPS showed only trace amounts of H2 being evolved, suggesting that light and the three components are required for efficient evolution of H2. In order to explore the effects of parameters such as light wavelength, concentration of the sacrificial electron donor, photosensitizer and catalyst, and pH of the solution on H2 evolution activity, various studies were performed.
The initial optimization study was conducted by irradiation with a light source at different wavelengths, i.e. 365 nm, 420 nm, and 525 nm. The study of the dependence of H2 evolution on light wavelength reveals that 420 nm is the most suitable for absorption by the photo-catalytic system to generate H2. This is because of the absorption wavelength of the photosensitizer, which lies in the range between 370 and 520 nm. Hence, irradiation with either 365 or 525 nm produces an insignificant amount of H2 (Fig. S5†). Using a light source of 420 nm, we then aimed at exploring the dependence of H2 evolution on the amounts of AscH and Ru(bpy)32+. The photo-catalytic H2 evolution using AscH as the sacrificial electron donor, ruthenium-based photosensitizers, and molecular catalysts was reported to be highly dependent on the concentration of ascorbic acid, the photosensitizer and the catalyst.15 At AscH concentrations <0.1 M, increasing the concentration increases the amount of H2 produced and hence the TOF or TON, because an increase in AscH increases the quenching efficiency, which also increases the efficiency of the photochemical process. However for [AscH] ≥0.1 M, increasing the concentration has no effect on the amount of H2 produced, meaning that similar rates, TOFs and TONs are obtained for all concentrations of AscH ≥0.1 M. This is consistent with the fact that the efficiency of photochemical H2 evolution (absorption efficiency multiplied by quenching efficiency) is comparable. This is because the same amount of Ru(bpy)32+ is used (meaning the same absorption efficiency) and the quenching efficiency, which is determined by AscH concentration, is identical at all concentrations ≥0.1 M. In fact, the increased amount of H2 evolved with higher TON and TOF values upon addition of a large excess of AscH is due to a more efficient reductive quenching of excited Ru(bpy)32+* to generate Ru(bpy)31+ species. In a similar fashion, when using a large amount of Ru(bpy)32+ (≥0.8 mM), a larger amount of H2 is produced since the absorption efficiency is high or more photosensitizers reached the excited state (Fig. S6†). But, at concentrations higher than 0.8 mM, the reaction rate and thus the TOF (determined in the linear region of the catalysis plot) are similar. This implies that, at these higher concentrations, the photochemical processes have the same absorption efficiency. In addition, since the same amount of AscH is used, the quenching efficiencies are also similar and hence the overall efficiencies of the photochemical processes are identical. At lower concentrations of Ru(bpy)32+, increasing the concentration increases the reaction rate, TOF and TON as more photosensitizers reach the excited state and are then quenched by AscH and reduced to Ru(bpy)31+, which effectively reduces our catalyst for efficient hydrogen evolution (Fig. S7†).
The hydrogen evolution activity with 0.3 M AscH and 1.2 mM Ru(bpy)32+ at 420 nm was then optimized against pH values from 5.8 to 8.0 as shown in Fig. 4.
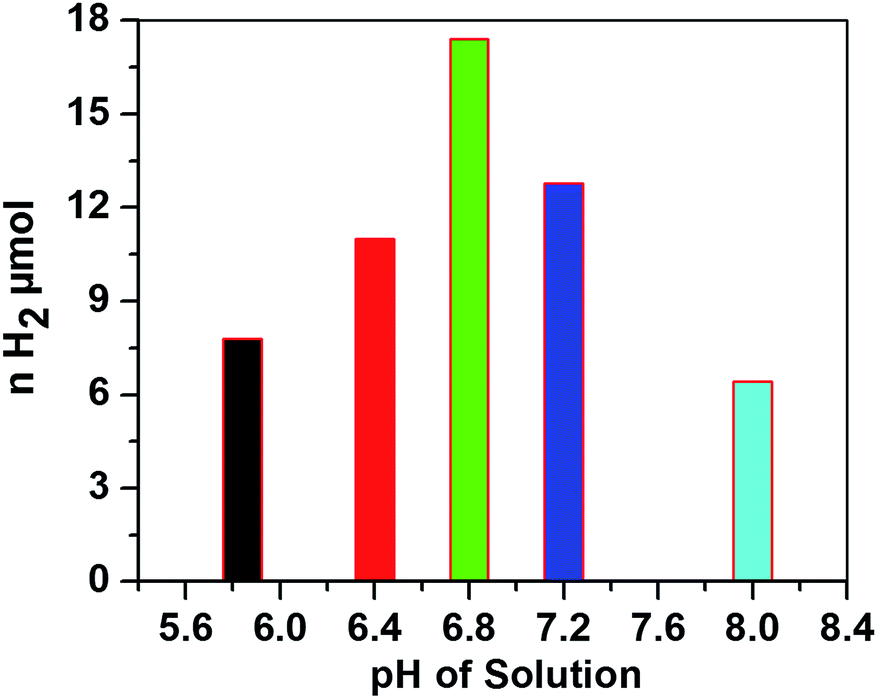 | ||
| Fig. 4 Amount of H2 evolved versus pH of the solution after being irradiated for 1.5 h. Conditions: [CoTPPS] = 2.9 μM, [AscH] = 0.3 M and [Ru(bpy)32+] = 1.2 mM. | ||
In the literature, the highest TONs and TOFs were obtained at various pH values depending on the photocatalytic conditions and the electronic properties of the molecular catalyst.15,32,44–46 Interestingly, in most of the reports on systems similar to that in this study, the H2 evolution ability was hindered at pH < 5 and diminished at pH > 8. Under highly acidic conditions, the ability of ascorbic acid to quench excited Ru(bpy)32+* is very low and the generated active Co(I) is negligible. This is attributed to the presence of an acid–base equilibrium between ascorbic acid (AscH) and ascorbate (Asc1−) with pKa = 4.1. On the other hand, the decrease in H2 evolution activity at higher pH is ascribable to the disfavored protonation of the reduced Co(I) catalyst; the acid–base equilibrium constant between Co(I) and Co(III)–H was reported to be pKa = 7.7.47 In our system, the highest H2 evolution activity and maximum turnover number were obtained at pH 6.8. Moreover, the amount of H2 evolved at a pH close to neutral conditions is higher than that at both relatively low and relatively high pH values (Fig. 4), giving an inverted V shape profile.48
It was also reported that the H2 evolution activity also depends on the amount of catalyst.15,32,46 In order to understand the dependence of the H2 evolution activity on the concentration of CoTPPS, we carried out photolysis with different catalyst concentrations (0.98 to 30 μM) at pH 6.8 using the abovementioned optimized conditions. As can be seen from Fig. 5, when the amount of catalyst in solution is increased from 1.5 μM to 30 μM, a sharp increase in the amount of H2 produced is observed.
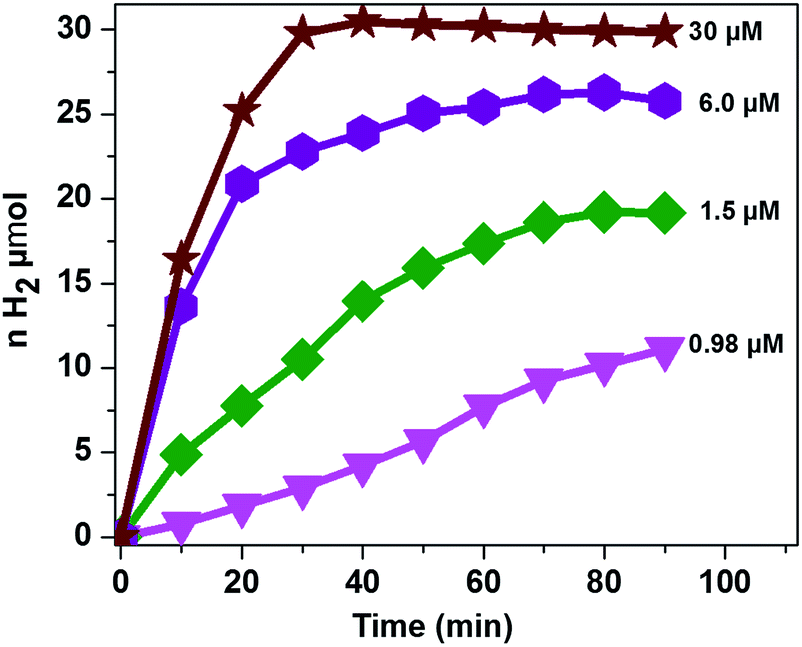 | ||
| Fig. 5 Dependence of the H2 evolution activity at pH 6.8 on catalyst concentration with [AscH] = 0.3 M and [Ru(bpy)32+] = 1.2 mM. | ||
However, addition of a larger excess of CoTPPS will retard H2 evolution. For instance, when 100 μM or more of CoTPPS was used, the amount of H2 produced diminished to a value less than that obtained using 0.98 μM CoTPPS. This [CoTPPS]-dependent activity may be related to the competition of light absorption between Ru(bpy)32+ and CoTPPS due to the strong Soret band absorption of CoTPPS at 424 nm. A similar phenomenon has been reported for photocatalytic H2 evolution employing a cationic cobalt porphyrin, Ru(bpy)32+ and AscH.32
Analysis of the activity at different concentrations of the catalyst shows that the largest H2 evolution was observed when using 30 μM CoTPPS as shown in Fig. 5. However, the optimal reaction rate of 1.3 μmol min−1 with the highest TON and TOF of 6410 and 120.8 min−1, respectively, was obtained from an ensemble with 1.5 μM CoTPPS (Fig. 6 and 7). From the plots of TON versus reaction time obtained from the stability test at various concentrations of CoTPPS, a continuous increase of the TON with time was observed but a plateau was reached after irradiation for around 1 h as can be seen from Fig. 6. The TON and TOF dropped at lower and higher concentrations of the catalyst. Overall, the best conditions to achieve the highest photocatalytic activity (TON and TOF) in this ensemble are a light source at 420 nm with pH 6.8 in 1 M phosphate buffer with 1.2 mM Ru2+, 0.3 M AscH, and 1.5 μM CoTPPS. Noticeably, this ensemble surpassed the maximum TON of 725 and TOF of 10.9 min−1 in 1 M pH 7 phosphate buffer solution using cationic Co(II)NMeTPyP4+ as the catalyst with the same photosensitizer and sacrificial electron donor.32
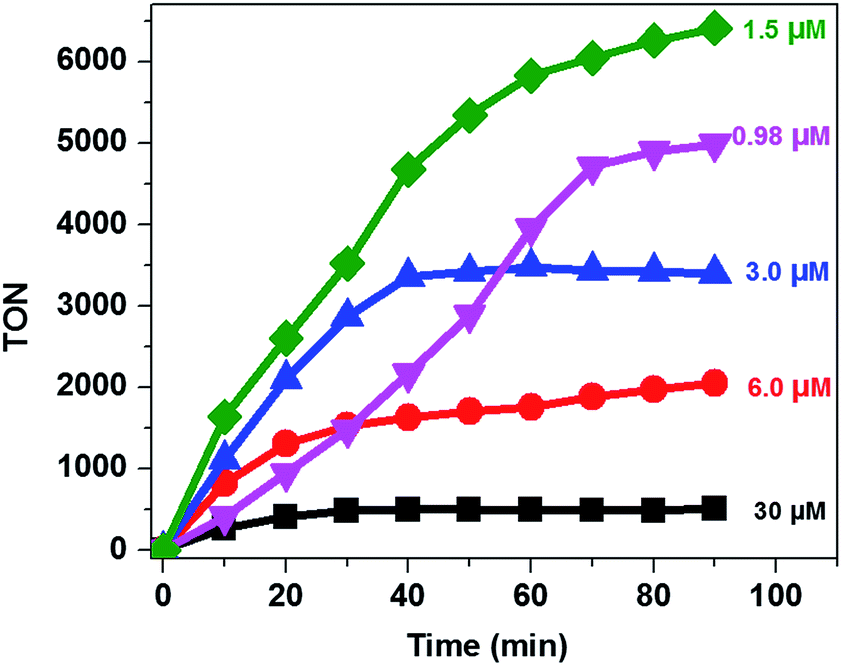 | ||
| Fig. 6 TON vs. reaction time at different [CoTPPS] in pH 6.8 buffer solution, with [AscH] = 0.3 M and [Ru(bpy)32+] = 1.2 mM at 420 nm. | ||
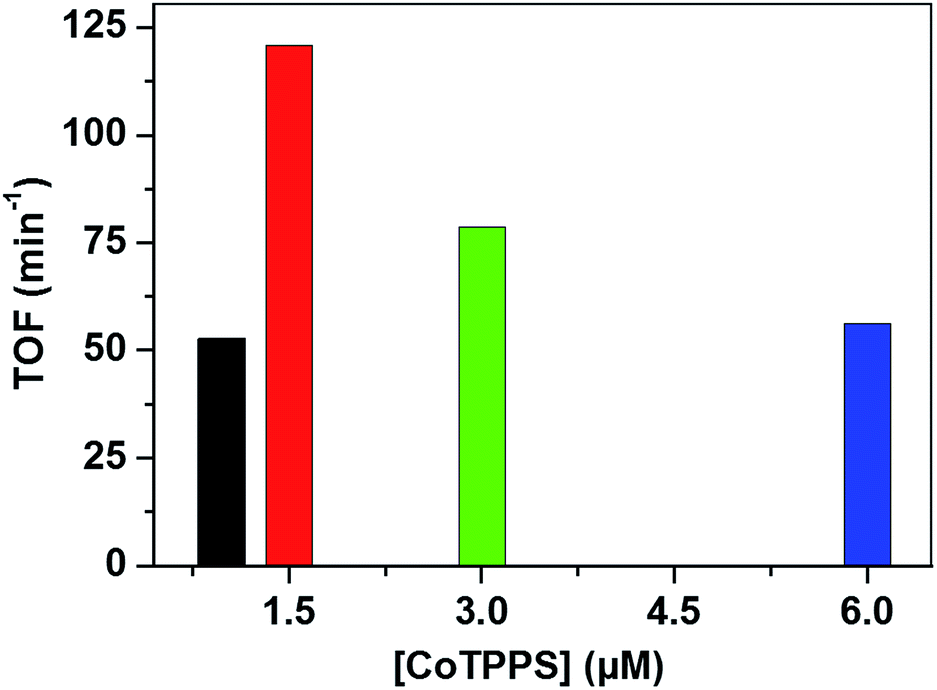 | ||
| Fig. 7 TOF obtained from different [CoTPPS] in pH 6.8 buffer solution, with [AscH] = 0.3 M and [Ru(bpy)32+] = 1.2 mM at 420 nm. | ||
It is believed that the better photo-stability of CoTPPS should be the reason for observing an over eightfold enhancement in the TON in our system. The photocatalytic performance in this study has been compared with that of other reported cobalt-photocatalyst/[Ru(bpy)3]2+/AscH systems in Table 1.
| Catalyst | Rate (μmol min−1) | TOF (min−1) | TON | Ref. | |
|---|---|---|---|---|---|
| a Indicates the highest values obtained in this work. | |||||
| CoDMG | 4.3 | 8.6 | 214 | 45 | |
| [Co(TPMA)]2+ | — | 3.3 | 94 | 44 | |
| [Co(ppq)-aqua]2+ | — | 9.7 | 333 | 49 | |
| [Co(NMeTPyP)]6+ | 0.54 | 10.9 | 725 | 32 | |
| [Co(DPA-BPY)]2+ | — | 66.7 | 4400 | 15 | |
| [Co(bpy2PYMe)]2+ | ∼1.7 | 11 | 1630 | 16 | |
| [Co(bpy2PYMe)]2+ | ∼2.1 | 53.3 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
14 | |
| CoTPPS | 0.98 μM | 0.13 | 52.5 | 5663 | This work |
| 1.5 μM | 0.4 | 120.8 | 6410 | ||
| 3.0 μM | 0.71 | 78.8 | 3403 | ||
| 6.0 μM | 1.05 | 56.2 | 2190 | ||
| 30.0 μM | 1.3 | 16.01 | 508 | ||
Although the different designs of these systems, e.g. source of photons and type of the buffer solution, make a direct comparison difficult, we can see that CoTPPS demonstrates promising catalytic activity and is only inferior to Co(bpy2PYMe)2+, which contains a polypyridyl as the supporting ligand. To confirm that the decrease of catalytic activity is due to the photo-degradation of Ru(bpy)32+, we added an additional portion of each reagent into the reaction solution after the H2 evolution had ceased and observed that only after replenishing it with fresh Ru(bpy)32+ did the H2 evolution resume (Fig. 8), whereas no significant amount of H2 was produced upon resupply of AscH or CoTPPS. This study again confirms that CoTPPS is a photo-stable and highly active catalyst, whose activity is limited by the decomposition of the photosensitizer. We are currently aiming at employing this catalyst with photochemically robust sensitizers to obtain a stable, efficient, and highly active H2 evolution system.
 | ||
| Fig. 8 Re-activation of H2 generation from the addition of a fresh aliquot of 0.05 mM Ru(bpy)32+ into the solution after the H2 evolution had ceased. Photolysis conditions: pH = 6.8, [AscH] = 0.06 M, light wavelength = 420 nm. | ||
To assess the source of protons for the H2 evolution, the photochemical H2 evolution was performed under the optimized conditions using D2O as the solvent. Our study using D2O as the solvent shows a much smaller amount of H2 evolved as compared to that in H2O, suggesting that the primary proton source is water and ascorbic acid behaves as an electron donor to reduce the excited photosensitizer (Fig. 9).
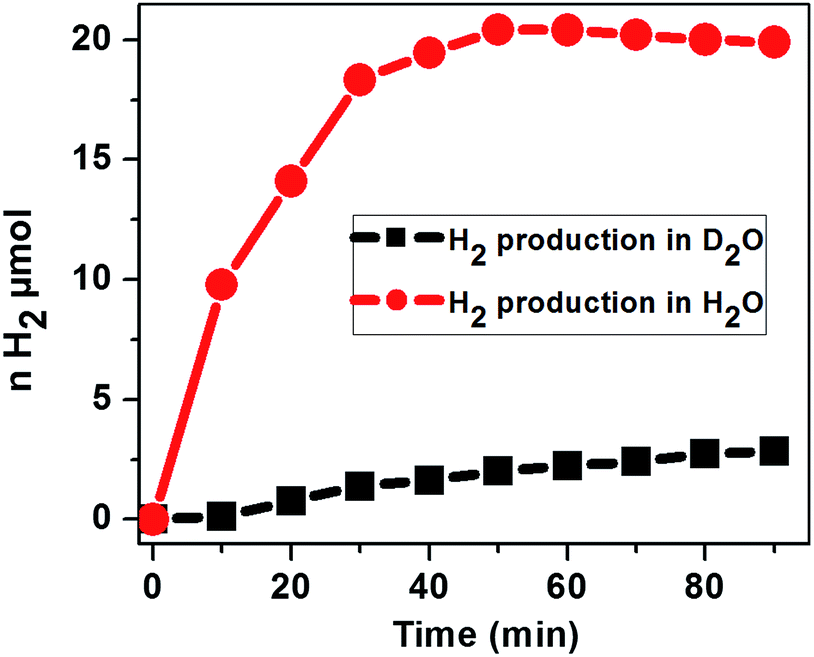 | ||
| Fig. 9 Photocatalytic H2 evolution with the time course of light irradiation in N2 saturated H2O and D2O containing 2.9 μM CoTPPS, 0.3 M AscH, and 1.2 mM Ru(bpy)32+ under irradiation at 420 nm. | ||
Mechanism of photochemical H2 evolution
In principle, homogeneous photocatalytic H2 evolution reactions involving a molecular catalyst, photosensitizer, and sacrificial electron donor are supposed to occur via either an oxidative or reductive quenching pathway. In oxidative quenching, the photosensitizer (Ru(bpy)32+) is first excited upon irradiation and it transfers an electron to the catalyst to generate an oxidized sensitizer (Ru(bpy)33+). This oxidized photosensitizer is then reduced by AscH to regenerate (Ru(bpy)32+). Under reductive quenching, however, the excited photosensitizer (Ru(bpy)32+*) will be reduced to [Ru(bpy)3]1+ by AscH. This reduced species then reduces the catalyst, enhancing the water reduction activity, and regenerates the original photosensitizer. In this study, we investigated both the reductive quenching of the excited state of the photosensitizer (Ru(bpy)32+*) by the sacrificial electron donor (AscH) as shown in Fig. 10 and oxidative quenching of the excited state of the PS (Ru(bpy)32+*) by the catalyst (CoTPPS) as shown in Fig. S9.† However, the dominant mechanism for the CoTPPS–PS–AscH-based system has been investigated to deduce a likely mechanism by fluorescence emission quenching and electrochemical redox potential energy level analysis experiments. In the fluorescence emission experiment, the corresponding Stern–Volmer plots gave a quenching constant of kq = 2.5 × 107 M−1 s−1 for Ru(bpy)32+* for quenching by AscH (Fig. 10) and kq = 2.6 × 1010 M−1 s−1 for quenching by CoTPPS (Fig. S9 and S10†). However, taking into consideration that the real H2 evolution experiment was conducted with an [AscH] of 0.3 M, the Ru(bpy)32+* reductive quenching rate is 7.5 × 106 s−1. The rate of oxidative quenching of Ru(bpy)32+* by CoTPPS (using 1.5 μM CoTPPS) is 1.7 × 104 s−1. Hence, the rate of quenching by AscH is several orders of magnitude higher than the rate of quenching by CoTPPS, indicating that the photocatalytic system operates via a reductive quenching pathway.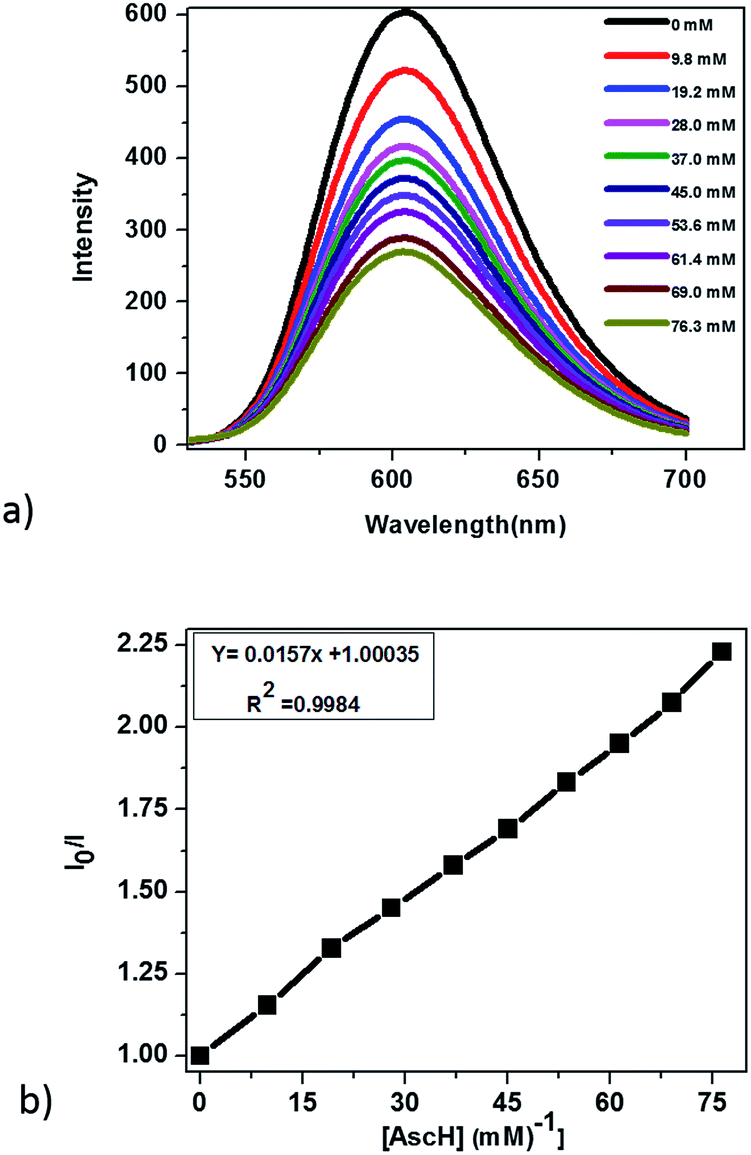 | ||
| Fig. 10 (a) Fluorescence emission quenching of Ru(bpy)32+* by addition of AscH, and (b) Stern–Volmer plot for (a). Conditions: pH = 6.8, [Ru(bpy)32+] = 1.2 mM, [AscH] = 0 to 76.3 mM, light wavelength = 420 nm. | ||
Electrochemical potential energy level analysis of the ensemble comprising Ru(bpy)32+, CoTPPS, and AscH also supports the mechanism deduced from the kinetics of the fluorescence emission quenching experiment. Although the redox potential energy levels of AscH, Ru(bpy)32+ and its excited state [Ru(bpy)32+]* in water have been well documented,14,50–52 we aimed to measure them under our experimental conditions. Accordingly, the redox potentials of the Ru-photosensitizer, AscH and CoTPPS are obtained from electrochemical (CV) measurements in aqueous phosphate buffer solution at pH 7 and in 0.1 M Bu4NPF6/DMSO solution (Fig. S11–S14†). The oxidation potential energy level of AscH in H2O (+0.53 V vs. SHE) is less positive than the Ru(II/III) redox couple energy level (+1.32 V vs. SHE), confirming the capability of AscH for donating electrons to [Ru(bpy)3]2+* (Fig. 11). From literature reports, the redox potential difference between Ru(II/III) and its excited state [Ru(II/III)*] is calculated to be 0.44 V vs. SHE.45–47 Since we obtain +1.32 V vs. SHE for Ru(II/III), we can estimate the redox potential of [Ru(II/III)*] by subtracting 0.44 V vs. SHE. Hence, the oxidation potential of Ru(II/III)*, +0.88 V vs. SHE, is more positive than the AscH oxidation potential. The electrochemical measurements were repeated in a 0.1 M Bu4NPF6/DMSO electrolyte system to confirm the consistency of the potential energy levels predicted in H2O. In DMSO, AscH showed two oxidation peaks at +0.5 and +1.39 V vs. SHE, which are also less positive than the redox potential of Ru(II/III), +1.65 V vs. SHE (Fig. S12†). Therefore, this analysis shows that the excited state of the photosensitizer [Ru(bpy)32+*] is reductively quenched by the strong reducing agent (AscH), while it is thermodynamically unfavorable for oxidative quenching by CoTPPS and this prompts us to propose steps 1 and 2 in Scheme 2. Following an efficient reductive quenching of [Ru(bpy)32+*] by AscH, Ru(bpy)31+ is formed as a reactive intermediate (as also reported in many literature studies).45–47 The electron transfer from Ru(bpy)31+ to Co(II)TPPS produces Co(I)TPPS with re-generation of the original photosensitizer, [Ru(bpy)32+]. In order to support this hypothesis, we carried out electrochemical (CV) measurements of Ru(bpy)32+ and CoTPPS both in aqueous phosphate buffer at pH 7 and a 0.1 M Bu4NPF6/DMSO electrolyte system. The electrochemical experiment in DMSO indicated that the first reduction potential energy level of the photosensitizer [Ru(II/I), −0.91 V vs. SHE] lay above or was more negative than the first reduction potential energy level of our catalyst [Co(II/I)TPPS, −0.47 V vs. SHE] as shown in Fig. S10.† This shows the feasibility of electron transfer from Ru(I) to Co(II) to generate Co(I) and Ru(II) under photochemical conditions. Moreover, it is easily noticeable from the CV experiment that further reduction of Co(I) to Co(0) under photochemical conditions is unfavourable because from potential energy level analysis, the Co(I/0) reduction potential of CoTPPS is more negative (−1.54 V vs. SHE) than all three redox potentials of the Ru-based complex (−0.91, −1.09, and −1.35 vs. SHE). The study was also repeated in H2O and it shows a consistent result. For the Ru-photosensitizer, the cathodic current starts to rise at a more negative potential (−1.32 V vs. SHE) than that of CoTPPS (−0.85 V vs. SHE) indicating the possibility of electron transfer from Ru(I) to Co(II) under photochemical conditions (Fig. S14†). Based on this result, we predicted step 3 in Scheme 2. Accordingly, protonation of Co(I) seems to be a presumable pathway to produce Co(III)–H, followed by a 2nd protonation to generate H2 and Co(III) as we proposed in step 4. In the last step, Co(III) is reduced back to Co(II) by excess AscH, which has a less positive potential energy level. Hence, potential energy level analysis is an important tool to elucidate the possible mechanism in the photochemical process.
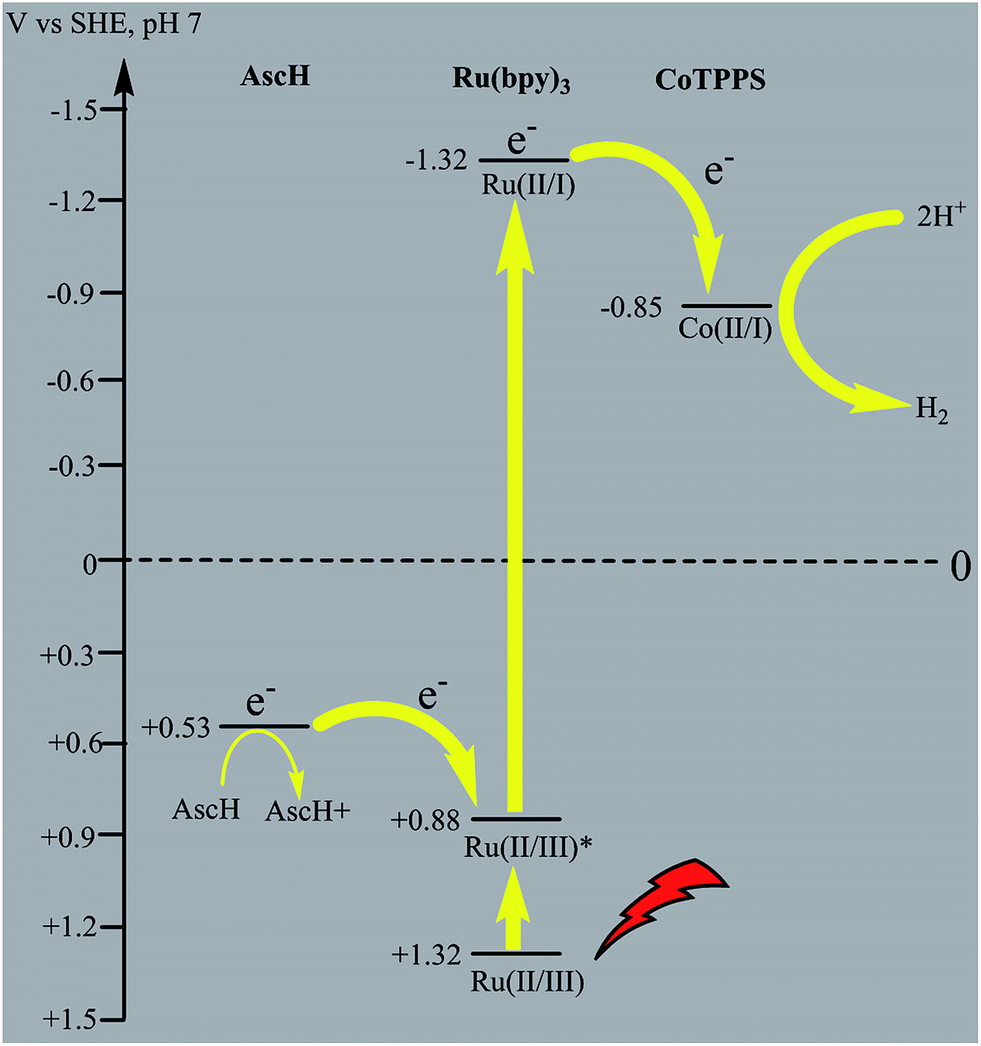 | ||
| Fig. 11 Thermodynamic redox potential analysis of the molecules involved in the photocatalytic H2 evolution process. | ||
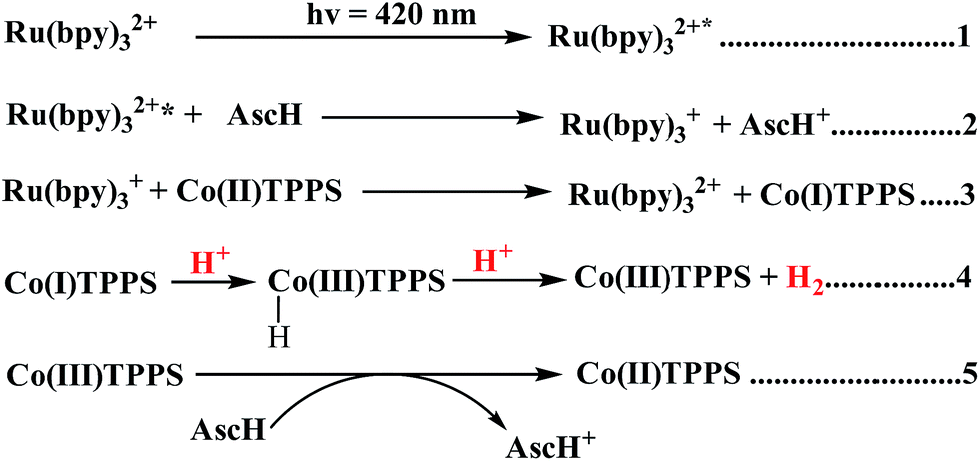 | ||
| Scheme 2 Proposed mechanism of photocatalytic H2 evolution using CoTPPS, [Ru(bpy)3]2+, and AscH under light irradiation (λ = 420 nm). | ||
In general, both the kinetic (fluorescence emission quenching experiments) and thermodynamic (potential energy level analysis) studies suggest that reductive quenching is the predominant pathway in our photocatalytic H2 evolution system as shown in Scheme 2.
Conclusion
In summary, we reported a water-soluble cobalt(II) porphyrin as a highly active, photo-stable and inexpensive molecular catalyst for H2 production in neutral aqueous solution. Under properly optimized conditions, LED light irradiation of a three-component system comprising a catalyst, photosensitizer, and sacrificial electron donor produces H2 with a maximum TON of 6410 and a TOF of 120.8 min−1. The catalyst is photo-stable and the catalytic activity is mainly limited by photo-degradation of the sensitizer. The kinetic (fluorescence emission quenching) and thermodynamic (redox potential energy level) studies revealed the mechanism of H2 evolution to be reductive quenching of the excited photosensitizer by AscH, followed by electron transfer to Co(II)TPPS to effectively evolve H2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Science and Technology of Taiwan (106-2113-M-001-031 and 103-2113-M-001-004) and Academia Sinica for financial support.Notes and references
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef PubMed.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef.
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef.
- H. Chen, Z. Sun, S. Ye, D. Lu and P. Du, J. Mater. Chem. A, 2015, 3, 15729–15737 RSC.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 RSC.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef PubMed.
- M. Nippe, R. S. Khnayzer, J. A. Panetier, D. Z. Zee, B. S. Olaiya, M. Head-Gordon, C. J. Chang, F. N. Castellano and J. R. Long, Chem. Sci., 2013, 4, 3934–3945 RSC.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef PubMed.
- C. L. Hartley, R. J. DiRisio, M. E. Screen, K. J. Mayer and W. R. McNamara, Inorg. Chem., 2016, 55, 8865–8870 CrossRef PubMed.
- S. Troppmann, E. Brandes, H. Motschmann, F. Li, M. Wang, L. Sun and B. König, Eur. J. Inorg. Chem., 2016, 2016, 554–560 CrossRef.
- K. Han, M. Wang, S. Zhang, S. Wu, Y. Yang and L. Sun, Chem. Commun., 2015, 51, 7008–7011 RSC.
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603–631 RSC.
- Y.-J. Yuan, J.-R. Tu, H.-W. Lu, Z.-T. Yu, X.-X. Fan and Z.-G. Zou, Dalton Trans., 2016, 45, 1359–1363 RSC.
- J. Zhao, Y. Ding, J. Wei, X. Du, Y. Yu and R. Han, Int. J. Hydrogen Energy, 2014, 39, 18908–18918 CrossRef.
- X. Du, J. Zhao, J. Mi, Y. Ding, P. Zhou, B. Ma, J. Zhao and J. Song, Nano Energy, 2015, 16, 247–255 CrossRef.
- Y. Xu, Y. Ye, T. Liu, X. Wang, B. Zhang, M. Wang, H. Han and C. Li, J. Am. Chem. Soc., 2016, 138, 10726–10729 CrossRef PubMed.
- R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2373–2377 CrossRef.
- R. M. Kellett and T. G. Spiro, Inorg. Chem., 1985, 24, 2378–2382 CrossRef.
- J. G. Kleingardner, B. Kandemir and K. L. Bren, J. Am. Chem. Soc., 2014, 136, 4–7 CrossRef PubMed.
- M. M. Roubelakis, D. K. Bediako, D. K. Dogutan and D. G. Nocera, Energy Environ. Sci., 2012, 5, 7737–7740 RSC.
- C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775–8777 CrossRef PubMed.
- D. Huang, J. Lu, S. Li, Y. Luo, C. Zhao, B. Hu, M. Wang and Y. Shen, Langmuir, 2014, 30, 6990–6998 CrossRef PubMed.
- M. Natali, A. Luisa, E. Iengo and F. Scandola, Chem. Commun., 2014, 50, 1842–1844 RSC.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 15145–15148 RSC.
- M. Zhu, Y. Dong, Y. Du, Z. Mou, J. Liu, P. Yang and X. Wang, Chem.–Eur. J., 2012, 18, 4367–4374 CrossRef PubMed.
- M. Zhu, Y. Lu, Y. Du, J. Li, X. Wang and P. Yang, Int. J. Hydrogen Energy, 2011, 36, 4298–4304 CrossRef.
- W. Kim, T. Tachikawa, T. Majima, C. Li, H.-J. Kim and W. Choi, Energy Environ. Sci., 2010, 3, 1789–1795 RSC.
- B. B. Beyene, S. B. Mane and C.-H. Hung, Chem. Commun., 2015, 51, 15067–15070 RSC.
- K. Yamamoto, S. Nakazawa, A. Matsufuji and T. Taguchi, J. Chem. Soc., Dalton Trans., 2001, 251–258, 10.1039/b006619m.
- X.-T. Zhou, G.-Q. Ren and H.-B. Ji, J. Porphyrins Phthalocyanines, 2013, 17, 1104–1112 CrossRef.
- G. Abellán, E. Coronado, C. J. Gómez-García, C. Martí-Gastaldo and A. Ribera, Polyhedron, 2013, 52, 216–221 CrossRef.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef.
- L. J. Henderson, M. Ollino, V. K. Gupta, G. R. Newkome and W. R. Cherry, J. Photochem., 1985, 31, 199–210 CrossRef.
- T. K. Foreman, J. B. S. Bonilha and D. G. Whitten, J. Phys. Chem., 1982, 86, 3436–3439 CrossRef.
- M. Natali, E. Badetti, E. Deponti, M. Gamberoni, F. A. Scaramuzzo, A. Sartorel and C. Zonta, Dalton Trans., 2016, 45, 14764–14773 RSC.
- E. Deponti and M. Natali, Dalton Trans., 2016, 45, 9136–9147 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef PubMed.
- M. Natali, ACS Catal., 2017, 7, 1330–1339 CrossRef.
- F. Wang, W.-G. Wang, X.-J. Wang, H.-Y. Wang, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193–3197 CrossRef PubMed.
- L. Tong, R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2014, 136, 4881–4884 CrossRef PubMed.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef.
- W. R. McNamara, Z. Han, C.-J. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15594–15599 CrossRef PubMed.
- C. V. Krishnan and N. Sutin, J. Am. Chem. Soc., 1981, 103, 2141–2142 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV, CV, CPE, and GC calibration data. See DOI: 10.1039/c8se00253c |
| This journal is © The Royal Society of Chemistry 2018 |