
Hydrogen evolution catalysis by molybdenum sulfides (MoSx): are thiomolybdate clusters like [Mo3S13]2− suitable active site models?
Marie-Luise
Grutza†
a,
Ashwene
Rajagopal†
b,
Carsten
Streb
 *b and
Philipp
Kurz
*a
*b and
Philipp
Kurz
*a
aInstitut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: philipp.kurz@ac.uni-freiburg.de
bInstitut für Anorganische Chemie I, Universität Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: carsten.streb@uni-ulm.de
First published on 13th June 2018
Abstract
Molybdenum sulfides are highly active hydrogen evolution reaction (HER) catalysts based on earth-abundant elements. In their most active forms, amorphous solid-state MoSx show HER activity comparable to noble metals. Due to the amorphous structure of the catalysts, insights into the reaction mechanism, the nature of the catalytic site(s) and catalyst deactivation are difficult to obtain. This perspective summarizes recent developments in our understanding of molybdenum sulfide HER catalysis and explores whether molecular molybdenum sulfido clusters – so-called thiomolybdates – are suitable models to study catalytic processes of Mo–S compounds. Further, the perspective raises fundamental questions relating to the reactivity, degradation and repair of thiomolybdate HER catalysts and discusses whether lessons can be learned from related moieties, in particular the iron molybdenum cofactor (FeMoco) in the enzyme nitrogenase.
Introduction
The reduction of protons to molecular hydrogen is a key reaction for the production of “renewable fuels” or “sustainable chemicals” via artificial photosynthesis, power-to-gas technologies, microbial electrochemistry and other routes.1 The product, H2, can either be used directly as a fuel or fed into established industrial processes such as the synthesis of ammonia, methanol or hydrogen chloride, petrochemical refining or various hydrogenation reactions.2 Amongst the proton-coupled multi-electron transfer reactions relevant in the context of energy technologies (e.g. water oxidation or carbon dioxide reduction), the hydrogen evolution reaction (HER) is relatively simple as only two electrons and two protons are involved (eqn (1)):| 2H+ + 2e− → H2 | (1) |
Nevertheless, suitable catalysts are required which operate with high HER reaction rates at redox potentials close to the thermodynamic potential for reaction (1), E0(H+/H2) = 0 V (note: all redox potentials in this contribution are given vs. the reversible hydrogen electrode, RHE). Many d-block metals are active HER catalysts (often in their elemental forms). Of special technological importance are platinum, used already ∼150 years ago by von Hofmann as HER electrocatalyst,3 and nickel. The two metals currently are the standard cathode materials in acidic (proton exchange membrane, PEM) or alkaline water electrolysers, respectively.4 Despite their excellent catalytic properties, these metals also have limitations: nickel (like some other metals) can only be used in alkaline media as it corrodes at low pH, while platinum is economically not viable due to its low abundance on earth, resulting in high costs of ∼5300 € per mol.5 Thus, an upscaling of H2 production, especially from acidic media, e.g. by PEM electrolysis or microbial electrolysis cells (MECs) will most likely require the use of HER catalysts other than Pt.
An attractive class of materials for this task are molybdenum sulfides (MoSx), which have been studied as earth-abundant alternatives to Pt starting from initial work by Tributsch et al. in the 1970s.6 Over the last 15 years, substantial research efforts have resulted in MoSx-based HER catalysts which show Pt-like stabilities in acidic media and require only ∼200 mV more reductive potentials than Pt to reach comparable H2 evolution rates.7–10 This slightly inferior performance might be justified by the fact that MoSx is about 2500 times cheaper than platinum (Mo ∼ 2 € per mol;5 S ≪ 1 € per mol) and thus much more suitable for large-scale applications. Furthermore, some of us recently found that amorphous molybdenum sulfides showed better long-term stability for HER catalysis than Pt in wastewater electrolytes,11 indicating that MoSx might be an ideal choice if robust cathode materials for “real life conditions” are required.
Although many researchers agree that molybdenum sulfides fulfil a number of key requirements for a possible technological application as HER catalyst, there are widely differing opinions about the mechanism by which MoSx actually catalyses the formation of H2. As will be described in the following sections of this perspective, one reason for this is the fact that many of the most active molybdenum sulfides are amorphous, non-stoichiometric solids – a fact also indicated by the commonly used abbreviation “MoSx”. This makes mechanistic investigations for these HER catalysts particularly difficult.
To fill this gap, we propose molecular molybdenum sulfido clusters (thiomolybdates), such as the well-known complex [Mo3S13]2−, as structurally well-defined model compounds for the HER active sites of molybdenum sulfides. This has been suggested before and indeed the decoration of MoSx surfaces with [Mo3S13]2− has resulted in enhanced catalytic activities.9,12 Surprisingly, H2 formation by [Mo3S13]2− under homogeneous conditions has only recently been reported,13,14 but we will argue that studying this reaction might be an especially rewarding approach to unravel the mechanism of proton reduction by molybdenum sulfides. Finally, we will suggest that synergistic mechanistic insights could be obtained by comparing reactivities of man-made molybdenum sulfides with a related biological catalyst, the iron molybdenum cofactor (FeMoco) of the enzyme nitrogenase. Nitrogenases catalyse both the reduction of N2 to NH3 and the reduction of H+ to H2. In addition, there are notable structural and chemical similarities between MoSx, [Mo3S13]2− and the FeMoco, so that the extensive knowledge about the enzyme's active site should be of additional help to understand HER catalysis by MoSx.
Structures of molybdenum sulfides and thiomolybdates
Crystalline MoS2
Crystalline molybdenum disulfide (MoS2), which occurs in the lithosphere in form of the mineral molybdenite, has a layered, sandwich-like structure.17 It consists of planes of hexagonally packed molybdenum cations (commonly described as Mo4+, 4d2) surrounded by two layers of sulfide (S2−) anions. In this way, [MoS6] trigonal prisms with Mo–S-distances of 2.41 Å are formed, suggesting largely covalent, polar Mo–S bonds.18 The two most common crystalline MoS2 polytypes both contain these trigonal prismatic [MoS6]-units and only differ in the stacking arrangement of adjacent layers (Fig. 1). This results in hexagonal 2H-MoS2 (space group P63/mmc, two layers per unit cell) and rhombohedral 3R-MoS2 (space group R3m, three layers per unit cell), both featuring Mo–Mo- and S–S-distances of 3.17 Å along the plane and 3.16 Å between two S2− at opposite sides of each sheet.18,19 As the layers in MoS2 are only held together by weak van der Waals interactions, the interlayer distance is rather large (3.49 Å) and it is possible to separate the sheets using methods similar to graphene exfoliation (which is also the basis for the large scale commercial use of MoS2 as lubricant).20,21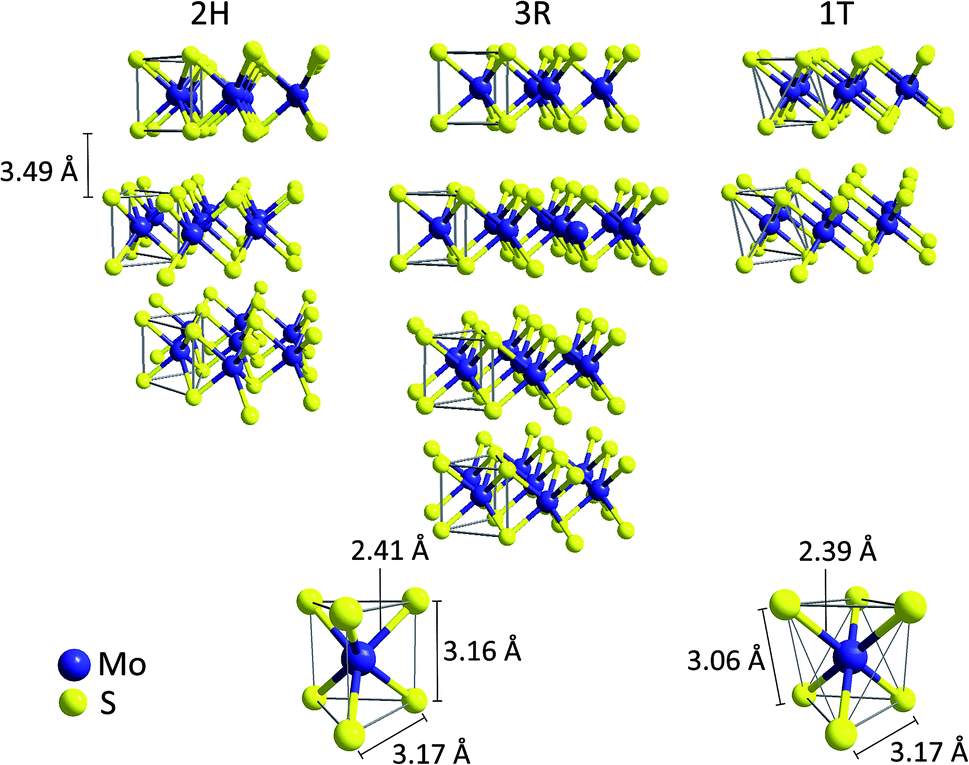 | ||
| Fig. 1 Top: Illustration of the ion arrangement in the three common MoS2 polytypes 2H, 3R, 1T. Bottom: trigonal-prismatic molybdenum coordination found in the 2H- and 3R-MoS2 modifications (left) compared to the [MoS6] octahedra found in 1T-MoS2.15,16 | ||
The similar energies of the S 3p and the Mo 4d orbitals enable a facile internal electron transfer and MoS2 is thus a semiconductor with a small bandgap of ∼1.3 eV.22 As a result, samples of 2H- and 3R-MoS2 are generally black and quite conductive. Furthermore, a third, metallic polytype (1T, space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1) is known, which can be described as a distorted layer structure of octahedrally coordinated molybdenum atoms with one layer per unit cell (CdI2 type).21 Both 1T- and 3R-MoS2 are metastable phases and can be converted into the thermodynamically favoured polytype 2H-MoS2 at temperatures of approx. 100 °C.16,21,23
m1) is known, which can be described as a distorted layer structure of octahedrally coordinated molybdenum atoms with one layer per unit cell (CdI2 type).21 Both 1T- and 3R-MoS2 are metastable phases and can be converted into the thermodynamically favoured polytype 2H-MoS2 at temperatures of approx. 100 °C.16,21,23
The thiomolybdate clusters [Mo3S13]2− and [Mo2S12]2−
In addition to solid-state (crystalline and amorphous) molybdenum sulfides, their molecular counterparts, i.e. molecular molybdenum sulfides or thiomolybdates24 have also recently been explored for the hydrogen evolution reaction.9,13,25 Thiomolybdates are synthetically accessed starting from their oxido-analogues ((poly-)oxomolybdates). Under aqueous reaction conditions, the treatment of polyoxomolybdates with sulfide sources like H2S or solutions containing polysulfide (Sn2−, n ≈ 2–25) leads to the exchange of oxido ligands for sulfide or disulfide (S22−) and the formation of thiomolybdates.Thiomolybdate formation is often accompanied by changes of the molybdenum oxidation state as the sulfur-containing reagents can act both as ligands and as reducing agents for the molybdenum cations. This is exemplified in Fig. 2: thiomolybdate synthesis using classical oxomolybdate precursors featuring MoVI centres typically leads to one- or two-electron reductions in addition to the ligand exchange.24–26 Alternatively, thiomolybdate clusters can also be accessed starting from the mononuclear MoVI-precursor [MoS4]2− (tetrathiomolybdate), a reaction where some of the initial sulfide ligands are oxidized (e.g. to elemental sulfur) and then eliminated from the complex. [MoS4]2− has also been suggested as a first reaction intermediate for the formation of thiomolybdates from (poly)oxomolybdates.27 Typical products which can be obtained from either of the two reaction routes are the thiomolybdate clusters [MoV2S12]2− (={Mo2}) and [MoIV3S13]2− (={Mo3}). The latter will be in the focus of this article as it has been most thoroughly studied in the context of HER catalysis by “MoSx”.28–31 Historically, there has also been much interest in clusters like {Mo2} and {Mo3} in the late 20th century when they were investigated as potential model compounds for the active site of the FeMoco of the enzyme nitrogenase, as will be discussed in more detail later.24
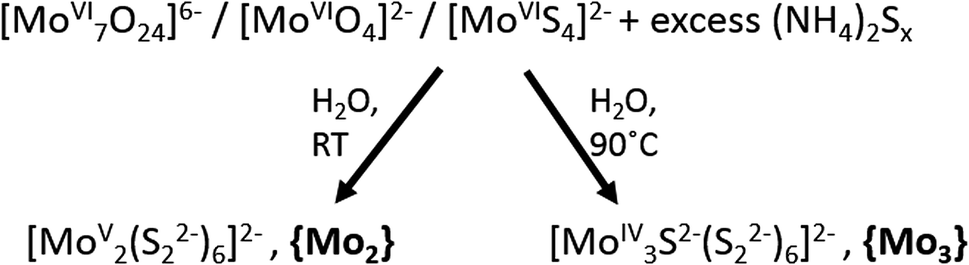 | ||
| Fig. 2 Synthetic routes for the preparation of the thiomolybdates [Mo2S12]2−, {Mo2}, and [Mo3S13]2−, {Mo3}, from (poly-)oxomolybdate precursors. | ||
While the related molecular formulae of {Mo2} and {Mo3} at a first sight suggest structural similarities, closer analysis reveals significant differences between the geometries of these molecules (which will become relevant for example when discussing redox-properties in the context of HER catalysis, see below): as shown in Fig. 3, {Mo2} features two Mo5+ ions linked by two bridging disulfido ligands (η2-μ-S22−) with Mo–S bond lengths of 2.4–2.5 Å and Mo–S–Mo bond angles of ∼71°.32 In addition, each Mo centre is bound to two terminal, side-on coordinated disulfides (η2-S22−) with Mo–S bond distances of 2.4–2.5 Å. For both the bridging and the terminal disulfides, the S–S bond distances are in the range of 2.03–2.05 Å. So each Mo centre is coordinated by four η2-S22− ligands in a pseudo-tetrahedral fashion (and therefore by eight S in total) and the Mo–Mo distance has been determined to be 2.8 Å.
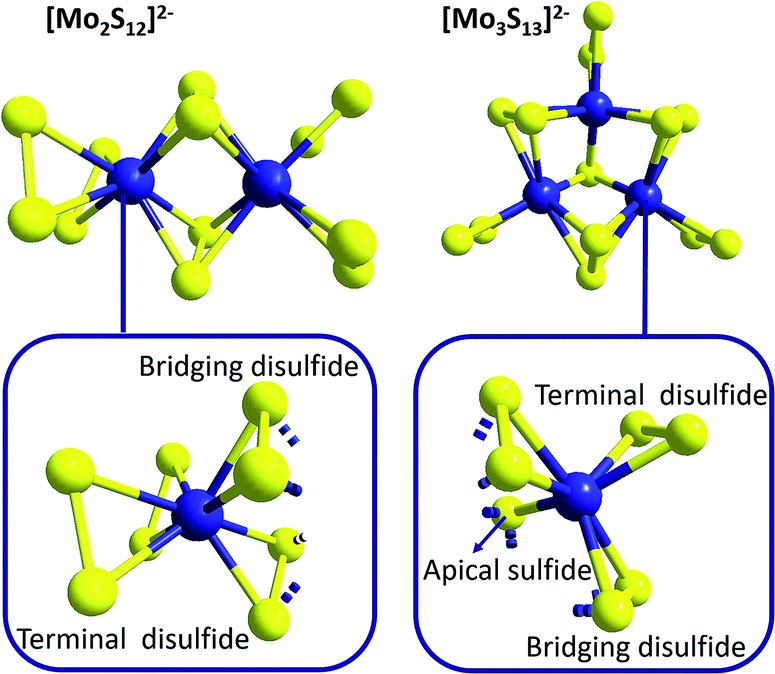 | ||
| Fig. 3 Molecular structures of [Mo2S12]2−, {Mo2}, and [Mo3S13]2−, {Mo3}, highlighting the different arrangements of the sulfur anions around the molybdenum cations for both species. | ||
The trinuclear anion {Mo3} features a central, apical bridging sulfido ligand (μ3-S2−), around which three Mo centres (oxidation state +IV) are arranged in form of a regular triangle with Mo–S bond lengths of ∼2.35 Å and Mo–S–Mo bond angles of ∼71° (Fig. 3).32 In contrast to {Mo2}, the neighbouring Mo4+ cations of {Mo3} are bridged only by one disulfido ligand (η2-μ-S22−). Nevertheless, the geometries of the resulting [Mo–(μ-S2)–Mo]-moieties are very similar to those of {Mo2}: the Mo–S bond lengths are 2.4–2.5 Å, the Mo–S–Mo bond angles ∼67° and the S–S bond distances 2.00–2.03 Å. The Mo4+ cations of {Mo3} are also coordinated to terminal disulfide ligands (η2-S22−), but here there is only one terminal S22− per Mo4+ with Mo–S bond lengths of 2.4–2.5 Å and S–S bond lengths of 2.04–2.05 Å. Unlike {Mo2}, the Mo centres of {Mo3} are only in direct contact with seven sulfur atoms (the apical μ3-S2− and six S atoms located in three η2-S22−). Due to the geometry of the Mo–S-triangle, the environment around each Mo cannot be described by common coordination polyhedra. The Mo–Mo distances for {Mo3} are ∼2.7 Å and thus – despite the lower oxidation state of Mo – slightly shorter than in {Mo2}, which might be explained by the special interaction between the three Mo4+ ions caused by the central μ3-sulfido ligand.
Amorphous MoSx
Unlike MoS2, compounds of the approximate composition “MoS3” can only be prepared in amorphous form (for example as intermediates from the synthesis of MoS2 from solutions containing [MoS4]2−).35,36 As the Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S-ratios in these compounds vary greatly, we will use the more general term “MoSx“ (with 2 < x < 3) for the rest of this article to identify all compounds of this type (alternatively, they have also been described as “amorphous MoS3”).36 The amorphous and non-stoichiometric properties of such materials make the acquisition of structural data more difficult. Concluding from a number of experimental studies involving X-ray absorption (XAS),37,38 photoelectron and vibrational spectroscopy,33,39 X-ray diffraction/radial distribution function analysis40 and chemical excision,41 there are currently two models for the arrangement of the atoms in MoSx.
:S-ratios in these compounds vary greatly, we will use the more general term “MoSx“ (with 2 < x < 3) for the rest of this article to identify all compounds of this type (alternatively, they have also been described as “amorphous MoS3”).36 The amorphous and non-stoichiometric properties of such materials make the acquisition of structural data more difficult. Concluding from a number of experimental studies involving X-ray absorption (XAS),37,38 photoelectron and vibrational spectroscopy,33,39 X-ray diffraction/radial distribution function analysis40 and chemical excision,41 there are currently two models for the arrangement of the atoms in MoSx.
One model describes the compounds as networks of triangular [Mo3(μ3-S2−)]-clusters, the common (and therefore apparently very stable) central unit in molecular clusters such as [Mo3S13]2− ({Mo3}, see above), [Mo3S4(CN)9]5−, [Mo3S7Cl6]2− and others (Fig. 4). All of these are thiomolybdates with Mo in a formal oxidation state of +IV.42,43 Chemical excision experiments showed that triangular Mo3-clusters can be extracted in the form of [Mo3S4(CN)9]5− from MoSx by treatment with CN− in alkaline solution, leading to the hypothesis that these units are also important components of MoSx.33,34,41 Furthermore, mass spectrometry studies on isotopically labelled 92MoSx or 100MoSx indicated that the Mo3-clusters must already be present in the amorphous solid,34 while photoelectron spectroscopy confirmed a molybdenum oxidation state of +IV and vibrational spectroscopy revealed small amounts of μ3-S2− similar to [Mo3S13]2−.33,44 Overall, this model describes MoSx as a MoIV-compound with a general formula of MoIV(S2−)(S22−). One should note that this formula cannot be obtained simply by condensation of [Mo3S13]-clusters, meaning that other Mo–S building blocks must be present in the material as well.44
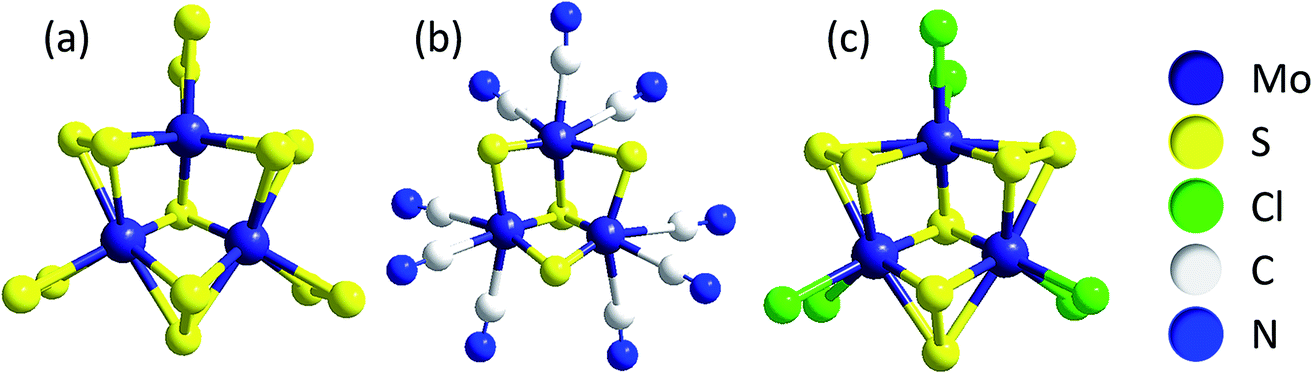 | ||
| Fig. 4 Anionic clusters containing triangular [Mo3(μ3-S2−)]-units, a possible building block of MoSx: (a) [Mo3S13]2− ({Mo3}); (b) [Mo3S4(CN)9]5− and (c) [Mo3S7Cl6]2−.33,34 | ||
An alternative model describes the structure of MoSx as an assembly of chains built up from distorted, trigonal-prismatic [MoS6]-units, which are linked on opposite triangular faces between alternate pairs of molybdenum atoms to form [Mo2S9]-units (Fig. 5).33 A pronounced covalent Mo–Mo-bond exists between the two metal centres of this unit, explaining why MoSx is diamagnetic.44,45 From XAS results the Mo–Mo distances in MoSx were determined to be ∼2.7 Å, which also fits to the geometry expected for such [Mo2S9]-moieties (however, similar Mo–Mo distances are also observed in {Mo2} or {Mo3}, see above).42,46 Many of these units are then connected to form chains, which feature longer Mo–Mo-distances of 3.1 Å at every second Mo–Mo-contact, indicating the absence of covalent Mo–Mo bonds.36 Vibrational spectroscopy showed clear evidence for the presence of disulfide bonds, so that the formula MoV(S2−)2(S22−)1/2 was proposed as best representation of this model of MoSx.37,39 This suggestion is also in accordance with studies combining X-ray diffraction and theoretical methods as well as neutron diffraction data.36,47 Moreover, the presence of MoV and polysulfides was indicated by electron spin resonance.48
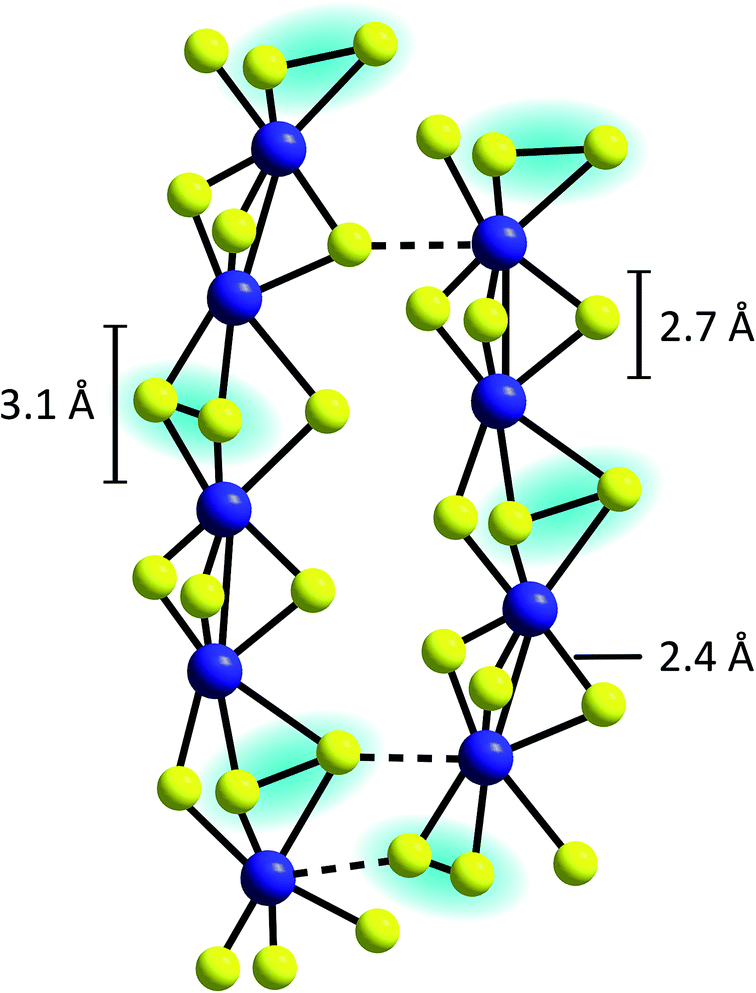 | ||
| Fig. 5 Proposed chain structure of MoSx.33,37 | ||
Adding further to these diverging structural suggestions, it has been reported that the arrangement of the atoms in MoSx is strongly dependent on the preparation method. As both MoSx and poorly crystalline MoS2 contain a large fraction of disulfide anions (S22−) at edge positions, it is nearly impossible to establish clear distinctions between these different amorphous molybdenum sulfides.49 Due to the polymorphism of molybdenum disulfide described above, it is also likely that the transformation of MoSx to MoS2 involves a number of intermediate structures as well as the change of a structure constructed from Mo–S clusters or Mo–S chains into a structure based on Mo–S layers.
The complexity of the system was further highlighted in a recent study by Tran et al. on amorphous MoSx. Based on resonance Raman, XPS and EPR measurements as well as electron microscopy, the authors suggested that the material under investigation had a polymeric structure where [Mo3S13]2− clusters are linked by disulfide bonds to form one-dimensional chains as well as two-dimensional networks.50 However, as the Raman spectra also showed signals typical for Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-units in addition to the Mo–S- and S–S-vibrations, the authors concluded that such O-/S-exchange might be a common feature of amorphous molybdenum sulfides, especially when exposed to water. As we will see in the following section, it could even be a reaction step of general importance for HER catalysis by “MoSx”.
O-units in addition to the Mo–S- and S–S-vibrations, the authors concluded that such O-/S-exchange might be a common feature of amorphous molybdenum sulfides, especially when exposed to water. As we will see in the following section, it could even be a reaction step of general importance for HER catalysis by “MoSx”.
HER catalysis by molybdenum sulfides and thiomolybdates
Methods to measure HER activity
How is the HER activity of “MoSx” materials or molecules normally probed? The most common way are electrochemical measurements, for which the compounds to be studied have to be immobilized on conductive supports like metal electrodes, conductive transparent oxides (FTO/ITO) or carbon supports. Numerous methods such as simple drop-casting of MoSx suspensions, impregnation using thiomolybdate solutions, electrodeposition starting from [MoS4]2− and many more have been used to prepare “MoSx-electrodes” and it comes as no surprise that the catalytic results obtained depend strongly on the parameters chosen for electrode fabrication.11,45,50–53 The electrochemical measurements are typically performed in strongly acidic aqueous media, e.g. 0.5 M H2SO4 or 1 M HClO4 (pH = 0–0.3) and the activity of the catalysts is generally described by listing the onset potential, the overpotential η necessary to reach certain current densities (e.g. j = 10 mA cm−2) and/or the Tafel slope. Advantages of this method are the virtually free choice of reduction potential and electrolyte, the high precision of the electrochemical measurements, the similarity between this HER activity test and the intended application (which is mainly electrolysis) and the fact that stability tests can easily be carried out for days or even longer. Drawbacks are the fact that the catalyst immobilisation step involves a whole new set of experimental parameters and that detailed investigations of immobilised MoSx particles or molecules on electrode surfaces are not at all trivial. In consequence the knowledge about the structure and composition of MoSx catalysts on electrodes and their changes during electrolysis is often very limited.An alternative approach to assess HER-activity are photochemical experiments, which have been particularly popular in the field of artificial photosynthesis for decades. The typical photosensitizers used here are ruthenium complexes based on [Ru(bpy)3]2+.54 The dye is combined with a sacrificial electron donor such as ascorbic acid or triethylamine and the MoSx catalyst in “one-pot” reaction mixtures with acetonitrile, methanol, dimethyl formamide, water or mixtures of these as most common solvents.55 When illuminated, light-driven electron transfer from the donor via the photosensitizer to the catalyst occurs.54 If this results in the reduction of protons, hydrogen can be detected as reaction product and HER rate can be calculated. Advantages of this system are the fact that the MoSx catalysts can be used “as prepared” and dissolved molecular species can be monitored “in operando” (using UV/vis spectroscopy etc.). Mechanistic studies, e.g. concerning the electron transfer steps between the different species, are easier, too. On the other hand, such “one-pot systems” involve many molecules (and also their decomposition products as the reaction proceeds), they operate under diffusion control (and are thus normally slow) and the instability of photosensitizers in solution usually limits such studies to few hours of continuous catalysis. In addition, photosensitizer-catalyst aggregation is a well-known side reaction, which can limit the catalytic concentration range.56
Heterogeneous HER catalysis by MoSx solids
Molybdenum sulfide (MoS2) has long been known for its reactivity towards hydrogen and has been employed industrially as catalyst to reductively remove sulfur from petrochemical feedstocks (oil, gas) using H2 by catalytic hydrodesulfurization (HDS). Notably, while the mechanism of this process is still not fully understood, it has been suggested that catalytic turnover occurs at the edges or rim sites of the catalyst while the basal crystalline planes of MoS2 are much less active.57These features are reflected when considering the HER activity of crystalline MoS2: none of the crystalline modifications, 2H-, 3R- or 1T-MoS2, show substantial activity in HER catalysis. As a possible explanation, DFT studies showed that analogous to the situation for HDS the extended basal planes found in the structures of MoS2 (Fig. 1) are catalytically inactive, leaving only the few edges of the planes or sulfur vacancies as possible reaction sites for HER.58,59 These edges can either contain molybdenum or sulfur as outermost atoms and can additionally have different sulfur coverages. Raybaud et al. calculated that the most favourable environment for HER catalysis were sulfur-terminated edges, as they showed the smallest reconstruction energies for the binding of hydrogen and thus these calculations support a “sulfur-based” HER mechanism.60 In addition, Li et al. demonstrated by experimental studies that sulfur vacancies on crystalline MoS2 are HER active sites and their catalytic activity is dependent on vacancy density and the crystallinity around the vacancies.59 However, it has to be mentioned that these studies were based on ideal crystalline MoS2 and are thus not necessarily correct for the structurally quite different, disulfide-containing amorphous MoSx-materials.49,50
In comparison to crystalline MoS2, amorphous MoSx materials proved to be excellent materials for HER catalysis.10,61 As already mentioned in the introduction, some optimized MoSx-cathodes require only ∼200 mV more reductive potentials than Pt to reach comparable electrocatalytic H2 evolution rates, show very good stabilities in acidic media and are also more “tolerant” than Pt against the catalyst poisoning substances present in wastewater electrolytes – all at a fraction of the cost of platinum!7–11
Electrochemical HER is often described to involve the following elementary reaction steps:62
| H3O+ + e− ⇌ H* + H2O | (2) |
| H* + H3O+ + e− ⇌ H2 + H2O | (3) |
| H* + H* ⇌ H2 | (4) |
Based mainly on the analyses of electrochemical and spectroscopic data, there are currently at least two theories on how HER might take place on amorphous MoSx materials. Both involve catalytic sites which are not present in crystalline MoS2 and thus both offer an explanation why MoSx shows HER activity while MoS2 does not. On the other hand, the two mechanisms differ fundamentally in their assignment of the catalytically active species, as will be described in the following.
The first mechanistic proposal is “molybdenum-based” and was put forward by Tran et al. after a thorough investigation of electrodeposited amorphous molybdenum sulfide.50 As described above, the experimental data of this study pointed to a description of the material as polymers of {Mo3}-clusters linked by disulfido ligands (Fig. 6). Additionally, MoVO units could be detected and these and/or unsaturated MoIV sites originating from the loss of terminal disulfides were identified as the sites were HER catalysis is most likely to occur. As shown in Fig. 7, MoIV-□-sites are also accessible from the proton-coupled reduction of MoVO units. A further proton-coupled reduction could convert MoIV-□ into the key intermediate MoV–H−, from which H2 can be generated either after H–H-coupling on the surface or by protonation of this hydride species. The latter leads to unsaturated MoV-□ and from here an electron transfer could regenerate MoIV-□ either directly or via the MoVO-species formed after hydration of MoV-□. In summary, in this mechanism molybdenum acts as redox active component and also as substrate binding site, involving the MoV/MoIV-pair as the critical redox couple. Molecular hydrogen might be formed by a comproportionation reaction between H+ and H− and thus in a way related to the formation/cleavage of H2 by [FeFe]- or [NiFe]-hydrogenases.63 Alternatively, the combination of two H atoms to H2 might occur on or close to the catalyst surface, a mechanism resembling models for HER catalysis by platinum.
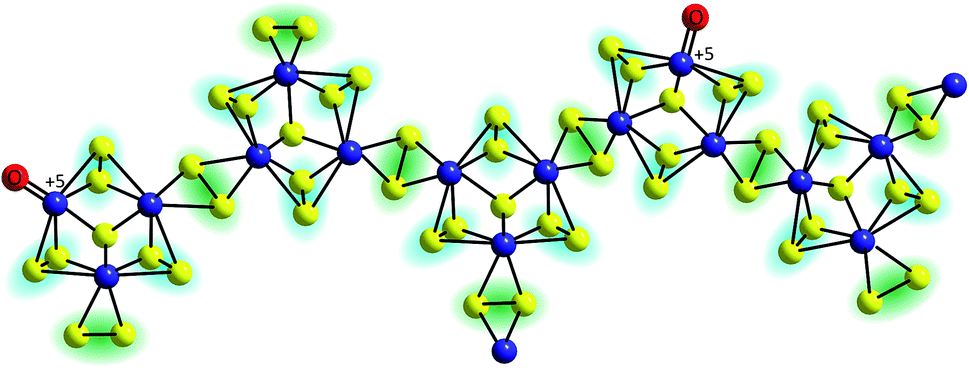 | ||
| Fig. 6 Polymeric structure of amorphous “MoSx” proposed by Tran et al. on the basis of EPR, Raman and electron microscopy data. Important features are [Mo3(μ3-S2−)]-units as well as terminal oxido ligands bound to MoV centres.50 | ||
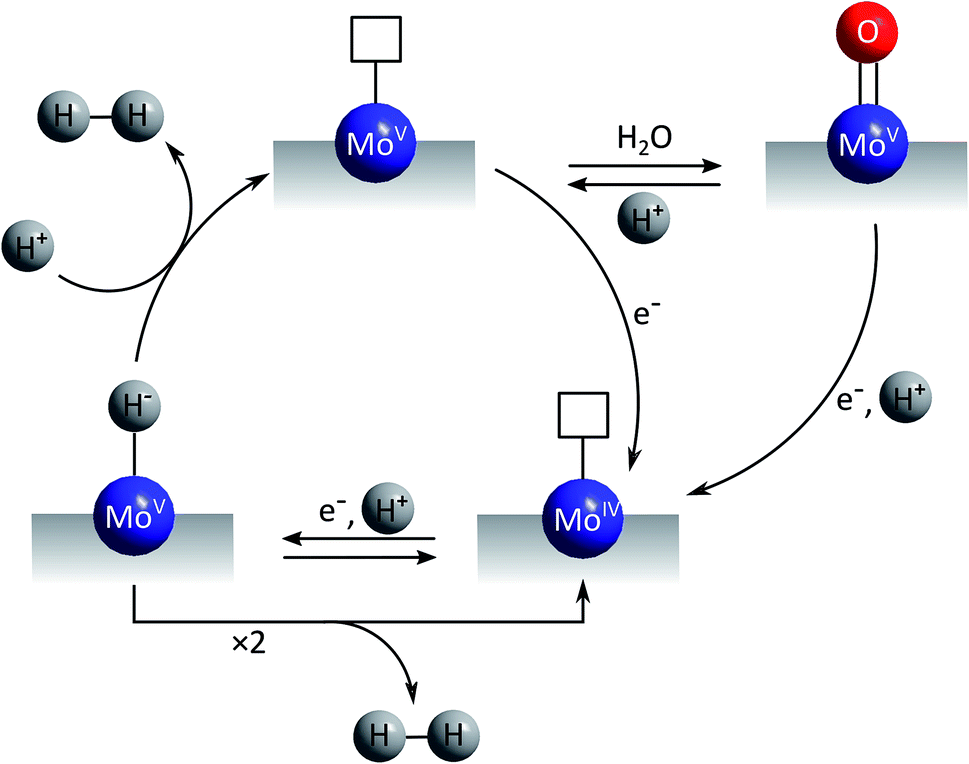 | ||
| Fig. 7 “Molybdenum-based” mechanism for HER catalysis by MoSx proposed by Tran et al.50 | ||
A very different, “sulfur-based” mechanism for HER catalysis by MoSx was put forward by Lassalle-Kaiser et al. following a detailed in situ study of MoSx electrodeposited on indium-doped tin oxide (ITO).53 Using XAS at the Mo K-edge in combination with XPS, the material obtained in this way was identified as amorphous MoSx covered with a thin layer of molybdenum oxide (MoOx, Fig. 8).64 By applying a weakly reducing potential of +300 mV vs. RHE, the MoSx core presumably releases disulfides and/or sulfides and the MoOx surface layer is transformed into amorphous MoSx with oxidation state assignments of MoIV and S-II best fitting the spectroscopic results. Under catalytic conditions (applied potential of −300 mV), a further elimination of sulfur occurs, generating a MoSx material with x < 3, but Mo still mainly in an oxidation state of +IV. Additionally, small amounts of MoIII and terminal disulfides could be detected and the authors considered these units as most probable active sites for HER catalysis. In the proposed catalytic cycle, the protonation and reduction of such MoIII–(S22−)-sites causes a weakening or even the cleavage of the S–S-bond, as the reduction is sulfur-centred. A second proton-coupled reduction generates [MoIII(SH)2] as key intermediate, which releases H2 to reform the initial MoIII–(S22−)-state.53 Additional evidence supporting this mechanism could be obtained by in operando Raman measurements of the H/D isotopic exchange: as Mo–H vibrations were not detected, Mo–(S22−)-units were assigned as the more likely catalytically active species. Furthermore, Ting et al. recently detected Mo–S–H species on MoSx under HER conditions using vibrational spectroscopy, while confirming the absence of Mo–H or Mo–D vibrations.52
 | ||
| Fig. 8 “Sulfur-based” mechanism for HER catalysis by MoSx proposed by Lassalle-Kaiser et al.53 | ||
To summarise, in contrast to the “Mo-centred” model, here (di)sulfides are acting both as proton binding and as redox active entities with the consequence that S-I/S-II-transitions are crucial for the catalytic cycle. Even as the MoIV/MoIII-pair could be involved in the reaction, the formation of the H–H-bond does also not involve molybdenum but occurs “sulfur-based” from two neighbouring Mo–S–H units. Unlike the “Mo-centred” model above, this pathway does not resemble any established HER mechanism and would thus be in some way “unique”. On the other hand, we would also like to mention that both presented mechanisms also show some common aspects: both involve a rearrangement of the catalyst under reducing conditions which is needed to generate the actual active catalytic species and both attribute a crucial role to terminal disulfides – either as “leaving group” to generate vacant molybdenum coordination sites or as redox-active component directly involved in reactions resulting in H2-formation.
Reactivity differences between oxo- and thiomolybdates
When gauging the HER activity of thiomolybdates, it is worthwhile to compare and contrast their fundamental reactivity with their well-known oxomolybdate counterparts. While small polyoxomolybdates are almost exclusively based on high-valent MoVI ions, thiomolybdates often contain lower-valent molybdenum in its MoIV and MoV oxidation states. The differences arise from the improved stabilization of the oxidation state +VI by the very electronegative, “hard” oxido ligands, while the larger, more polarizable sulfido and disulfido ligands lead to a structural and electronic stabilization of lower-valent molybdenum. Please also note that disulfido ligands (S22−) are particularly attractive bulky ligands, which formally occupy two coordination sites while introducing the same electrical charge as the mono-sulfido (S2−) units – this is a key stabilization route for the small MoIV/V thiomolybdate prototypes {Mo2} and {Mo3} discussed before (see Fig. 4).The different electronic structures of the metal centres in thio- and oxomolybdates also result in strikingly different reactivities. While polyoxomolybdates are generally highly active oxidation catalysts with unique stability even under highly oxidizing conditions, their stability under reductive conditions is limited as “over-reduction” of the Mo centres would lead to molecules with high negative charge and thus limited thermodynamic stability. Treatment of oxomolybdates with strong reducing agents therefore often leads to reductive cluster degradation.65
In contrast, thiomolybdates are significantly more stable under reductive conditions which is a prerequisite for their use in HER catalysis. In contrast, under oxidative conditions, thiomolybdates are unstable both due to the oxidative degradation of the sulfide/disulfide ligands as well as the MoIV centres.28–31 Finally, when comparing the redox-activity of thio- vs. oxomolybdates, it is noteworthy that the high redox-activity of oxomolybdates is associated with metal-based redox transitions (mainly MoVI ⇌ MoV), while in thiomolybdates, the Mo 4d electrons are stabilized in covalent Mo–Mo bonds and redox-activity is often associated with the sulfide/disulfide redox couple (S-II ⇌ S-I). The facile internal electron transfer in thiomolybdates is a manifestation of the ability of sulfide/polysulfide ligand systems to exhibit energy levels both above and below the 4d levels of molybdenum.22
HER reactivity of immobilized thiomolybdates
The desire to rationalize reaction steps of the enzyme nitrogenase (see below) triggered an interest in thiomolybdates as potential active site models already ∼40 years ago,24 as these rather simple compounds can be studied in detail both experimentally and theoretically. Following this approach again, we believe that in-depth insights into the hydrogen evolution mechanisms for molecular thiomolybdates might also prove very useful to rationalize HER catalysis by solid-state molybdenum sulfides.10,61,66Along these lines, a few mechanistic studies have already been performed, where {Mo3} was deposited on electrode surfaces to explore its electrochemical HER reactivity. In 2014, a ground-breaking study by Kibsgaard et al. explored the electrochemical HER activity of submonolayers of {Mo3} deposited on carbon electrodes.9 The system showed excellent electrocatalytic HER activity under acidic conditions and Tafel slopes in the range of 40 mV dec−1, the latter being indicative of a Volmer–Heyrovsky mechanism (see eqn (2)–(4), above). These findings were in line with earlier reports on amorphous MoSx, where similar Tafel slopes also indicated that a Heyrovsky-type H2 formation step could be involved.67
A subsequent study by Huang et al. explored the electrochemical HER activity of the dinuclear thiomolybdate {Mo2} deposited on fluoride-doped tin oxide (FTO) as conductive catalyst support.12 The authors again reported Tafel slopes for the HER in the range of 40 mV dec−1, thus suggesting that HER catalysis by {Mo2} also follows a Volmer–Heyrovsky reaction mechanism. Density functional theory (DFT) calculations by the authors further supported this suggestion and indicated that Volmer–Heyrovsky steps are energetically favoured over the alternative Volmer–Tafel reaction route. The authors propose that H atom binding during the Volmer step occurs at the bridging disulfide ligands (formally leading to a pair of –SH and a –S˙ radicals). However, it should be noted that this study did not explore the effects of disulfide ligand loss (which could enable the formation of vacant MoIV sites capable of forming hydride species), so it is currently not clear whether a hydride mechanism might also be possible for {Mo2}.
However, this question was to some extent addressed by Tran et al., who concluded that amorphous MoSx is composed of polymeric {Mo3} chains (Fig. 6).50 As described above, the authors suggest the formation of MoV–H− hydride species as key intermediates of HER catalysis (Fig. 7). These could be formed in the Volmer step after the loss of a terminal disulfide ligand under reaction conditions. DFT calculations also indicate that hydrogen formation via MoV–H species is energetically favoured by 18 kcal mol−1 over an alternative path where the H atom binds at a bridging disulfide ligand. On the other hand, the already mentioned investigations by Lassalle-Kaiser et al. and Ting et al. offered spectroscopic evidence that H atom binding and H2 formation most likely occurs at disulfide groups (Fig. 8).52,53
These selected results show that there is significant evidence that the HER active sites of MoSx resemble thiomolybdate-like units like {Mo2} or {Mo3}. Additionally, they highlight the need for in-depth studies on such clusters under HER conditions in order to identify key reaction intermediates and thus to arrive at a conclusion which of the catalytic cycles shown in Fig. 7 and 8 better represents reality.
Homogeneous HER activity of thiomolybdates
Based on this motivation, 2018 saw the first studies on HER catalysis by {Mo3} in solution.13,14 By moving from electrocatalytic to homogeneous reaction conditions, detailed analyses of {Mo3} reactivity without having to consider interactions between the clusters and the electrode supports became possible, because now the many experimental and theoretical tools used to investigate homogeneous catalysis could be deployed. In one of these studies, Dave et al. explored the light-driven HER activity of {Mo3} in combination with the photosensitizer [Ru(bpy)3]2+ and the electron/proton donor ascorbic acid.13 The authors reported outstanding turnover numbers (TON ∼ 41000 after 24 h irradiation) and high turnover frequencies (TOF > 150 min−1), demonstrating the potential of HER catalysis by molecular thiomolybdates. In addition, initial insights into the catalytic mechanism were provided by a combination of spectroscopy and density functional theory. The authors report that a stepwise exchange of the terminal disulfido ligands with aqua ligands is observed under catalytic conditions. Notably, experimental data and DFT calculations indicate that hydrogen evolution is energetically favoured for partially aqua-exchanged species like [Mo3S13−x(H2O)x](2−x)− (x = 2 or 4), so that these species are suggested as the most active HER catalysts in solution. This is also supported by the observation of an initial TOF increase which is followed by a decreasing TOF, explained by the fact that the fully aqua-substituted species ([Mo3S7(H2O)6]4+) shows non-favourable hydrogen formation energetics (Fig. 9). A similar loss of the catalytic activity is also observed in the presence of competing coordinating ligands, e.g. chloride, and the fact that catalysis by the chlorido-complex [Mo3S7Cl6]2− (Fig. 4) is very slow. Finally, the hydrogen evolution energetics for all studied {Mo3} derivatives suggested a Volmer–Heyrovsky mechanism and hydrogen formation via H-atom binding at the bridging disulfide or formation of a MoV–H− were calculated to be energetically very similar (ΔΔG ∼ 1 kcal mol−1). Thus, in principle, both mechanisms could be operational (possibly even simultaneously) and the exact route could be dependent on the exact reaction conditions.
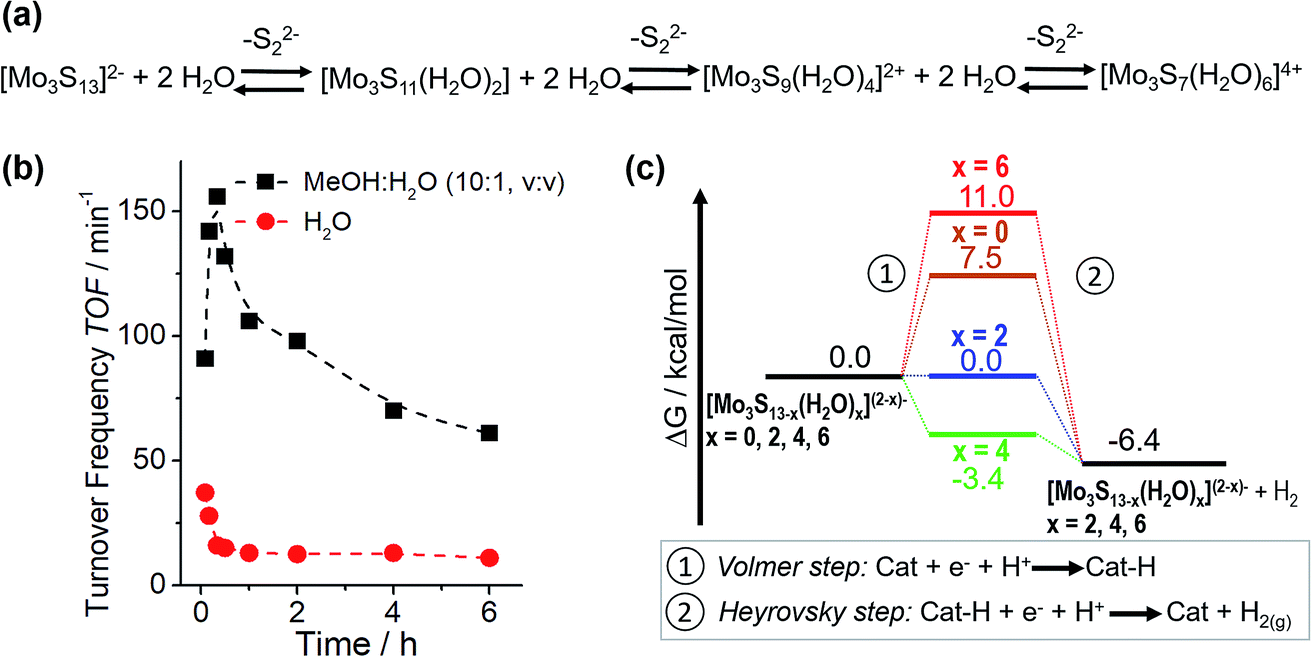 | ||
| Fig. 9 (a) Proposed ligand exchange mechanism for aqueous solutions of {Mo3} leading to aquo-thiomolybdate species. The structure of the “completely exchanged” complex [Mo3S7(H2O)6]4+ is assumed to closely resemble that of the chlorido-analogue [Mo3S7Cl6]2− shown in Fig. 4; (b) changes in turnover frequencies over reaction time for light-driven HER catalysis by {Mo3} in H2O and a MeOH/H2O mixture, indicating a link between reactivity and the exchange of disulfido for aqua ligands. (c) Calculated hydrogen formation energetics for {Mo3} derivatives featuring different numbers of water ligands. All figures modified from ref. 13. | ||
Catalyst stabilization and possible repair
The first results obtained for the homogeneous HER catalysis by {Mo3} described in the last section allow us to suggest stabilization concepts which could lead to enhanced performance of Mo–S HER catalysts. Under homogeneous conditions, the simplest approach is to prevent or slow down a complete exchange of the terminal disulfide ligands, e.g. by removal of competing ligands (like buffer anions or chelating ligands) or by a careful choice of the solvent. A more elegant route could be the design of ligands which mimic the electronic properties of terminal disulfides while offering stronger coordination to the Mo centers.9,10,17,37 For light-driven catalysis, these ligands could even be used to directly link catalyst and photosensitizer in form of a covalent dyad.38 Computational modelling could be a promising approach to facilitate identification of suitable ligands.39 A third option could be to reverse disulfide substitution using Le Chatelier's principle: providing additional disulfide ligands in solution should shift the equilibrium towards mixed disulfido–aqua complexes which seem to be the most active catalysts. Initial experiments have indicated that this approach might be feasible as an addition of polysulfide to the previously described photocatalytic system enhanced H2 evolution rates in long term experiments (total reaction time of 6 h).13Under heterogeneous conditions, the deposition of {Mo3} and related species on suitable substrates (e.g. (photo-)electrodes) could lead to a slower ligand exchange due to stabilizing surface interactions. This would be in line with pioneering studies where electrocatalysts based on deposited {Mo3} catalysts showed promising long-term performance even when experiments were carried out in water, indicating that surface deposition results in a more stable catalyst performance.6,9,15–19 In addition, if partial substitution of the disulfido ligands is desired to optimize HER activity (e.g. to enable the formation of Mo hydrides, Fig. 7), the oriented deposition of {Mo3} on substrates could be beneficial, so that one Mo(η2-S2) unit faces away from the surface to facilitate disulfide ligand exchange. In general, questions related to {Mo3}-substrate interfacing remain largely unexplored but could prove to be a useful way for tuning the HER activity and stability of immobilized thiomolybdates.
Parallels of MoSx-clusters with the FeMoco of nitrogenase
Finally, we would like to draw the reader's attention to a related entity in bioinorganic chemistry: the FeMoco, active site of the Mo-nitrogenase enzyme. The FeMoco is a complex, asymmetric unit with an overall elemental composition of [MoFe7S9C], whose architecture can be described as two distorted, corner-sharing [(Mo,Fe)4S3C] cubes connected by three μ2-sulfido ligands and an unusual, only recently discovered μ6-carbon centre (Fig. 10).70 While the biological function of this moiety is the reduction of molecular nitrogen from the air to ammonia – a reaction of fundamental importance for the entire biosphere – one equivalent of H2 is also formed as (biologically unwanted) by-product for each catalytic turnover (eqn (5)):| N2 + 8e− + 8H+ + energy → 2NH3 + H2 | (5) |
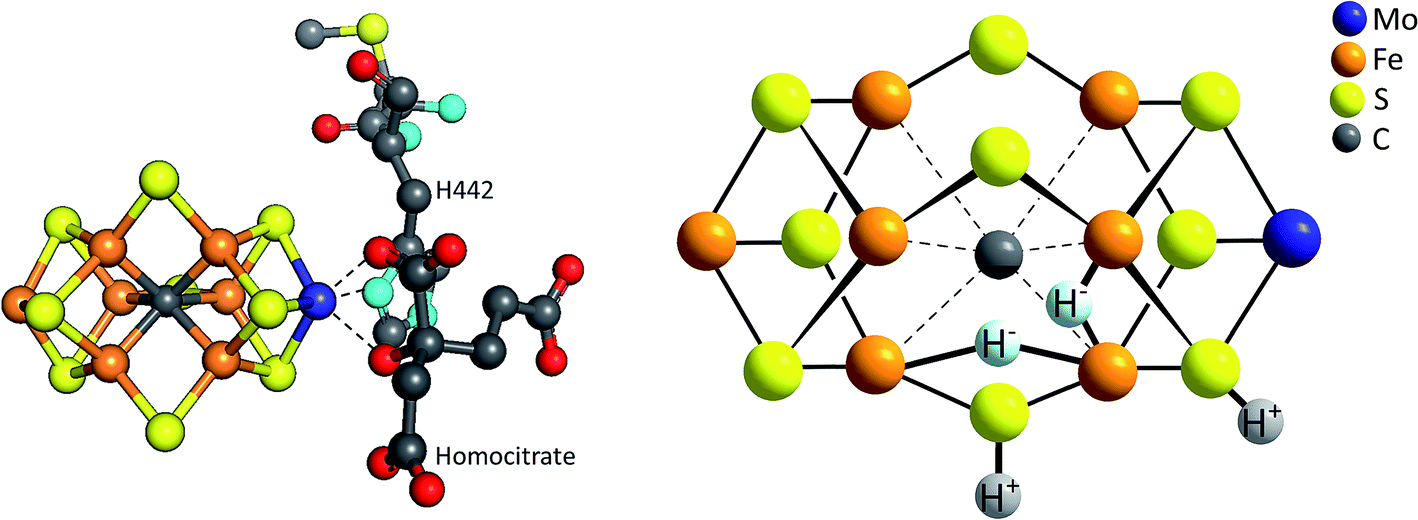 | ||
| Fig. 10 Left: structure of the FeMoco, the [MoFe7S9C] cluster which forms the active site for N2 and H+ reduction within the enzyme Mo-nitrogenase;68 right: model for the E4 intermediate of the FeMoco's catalytic cycle showing the formation of two hydrides (H−) bound to Fe and two protons (H+) bound to μ-sulfido ligands.69 | ||
Both the FeMoco and synthetic polynuclear complexes like [Mo3S13]2− are thus molybdenum sulfido clusters able to catalyse the reduction of protons to molecular hydrogen. In addition, there are further parallels between the two: (1) the coordination number of molybdenum in both cases is at least six (unlike e.g. in other Mo-enzymes such as sulfite oxidase where a CN of 5 is also often observed); (2) an oxidation state of +IV has usually been assigned to the Mo in the FeMoco, which corresponds to the Mo oxidation state in [Mo3S13]2− and (3) the stepwise reduction of the FeMoco during its overall eight-electron reaction (eqn (5)) is accompanied by the stepwise protonation of the cluster and these steps most likely also involve the sulfido ligands coordinated to Mo and Fe, as shown by the model for the so-called E4 state of the FeMoco's shown in Fig. 10.
The E4 state can be seen as a “halfway intermediate” of the enzyme's catalytic cycle because four of the eight electrons and protons have been added since the start (E0). Most models also identify the E4 form of the FeMoco as the species from which H2 formation occurs (and N2 reduction begins, see full catalytic cycle in Fig. 11). The chemical situation at E4 is thus especially important in the context of HER catalysis. However, despite decades of intense research, the complexity of the reaction sequence as well as the active site have so far prohibited a complete understanding of the enzymatic mechanism on an atomic level and so catalytic cycles for Mo-nitrogenase presented in the literature have to be considered as preliminary. Nevertheless, suggestions for the H2-forming step like the one shown in Fig. 11 contain a number of parallels related to the reaction steps presented above for HER by MoSx-materials and/or molecular Mo/S-clusters: (1) H2 formation involves protons bound to S2−; (2) important intermediates most likely feature hydrido ligands (however, here apparently always bound to Fe and never to Mo); (3) while there is still a debate whether the oxidation state of Mo in the FeMoco is better described as +IV or +III, it is most likely constant during the entire cycle, so that molybdenum-centred redox-chemistry is not essential for the overall reaction.71 On the other hand, the fact that Fe is almost certainly involved in both the FeMoco's redox chemistry as well as the formation of the H–H bond points at another interesting possibility for future research: the use and investigation of mixed molecular Fe/Mo/S-clusters as “nitrogenase-inspired” HER catalysts.
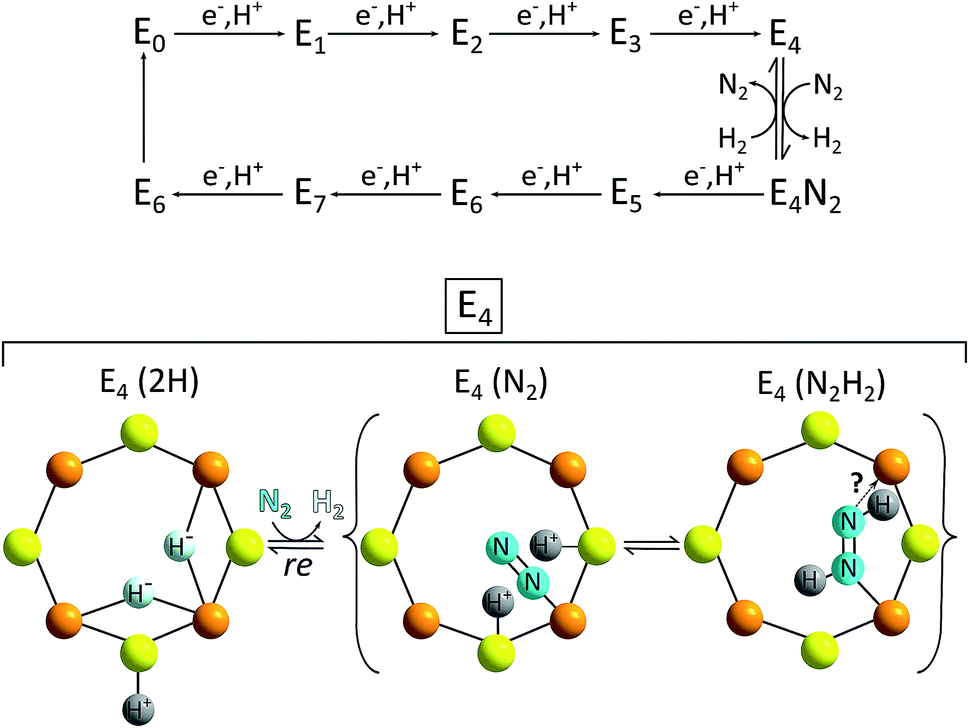 | ||
| Fig. 11 Top: simplified scheme of the Lowe–Thorneley model for the catalytic cycle of N2/H+-reduction by the FeMoco of the enzyme nitrogenase; bottom: proposal for the reaction of the E4 intermediate with N2 – a step possibly involving both the reductive elimination of H2 and the transfer of H+ and e− to N2. The latter marks the start of the second half of the catalytic cycle leading to ammonia formation.69 | ||
Concluding remarks and outlook
Over the last decade, the use of amorphous molybdenum sulfides (MoSx, 2 < x < 3) has opened up a very promising approach to develop earth-abundant catalysts for the hydrogen evolution reaction. Significant progress has been made in understanding structural differences between crystalline (MoS2) and amorphous (MoSx) molybdenum sulfides. Despite this structural information, to-date many questions concerning the catalytic mechanism of HER by MoSx remain, largely due to the experimental and theoretical complexities involved in studying these amorphous materials of varying composition, particularly in operando. As some recent studies propose molecular thiomolybdates as key building blocks of MoSx, investigations of prototypes such as [Mo3S13]2− and [Mo2S12]2−, especially in homogeneous solution, could very well lead to a much better understanding of HER catalysis by MoSx. Recent pioneering studies of a photocatalytic system involving [Mo3S13]2− as homogeneous HER catalyst have already demonstrated that this approach allows a detection of reaction intermediates and their theoretical treatment in an atomic detail hardly possible for the related heterogeneous MoSx materials. As a final thought, this perspective proposes that the extensive knowledge which is already available for the structurally related Fe/Mo/S-active site of the Mo-nitrogenase enzyme could also prove very useful for researchers wishing to unravel and improve HER catalysis by thiomolybdates or MoSx.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. and C. S. gratefully acknowledge financial support by Ulm University, the German Research Foundation (DFG Research Training Group GRK 1626 “Chemical Photocatalysis” and TRR234 “CataLight”) and the Ministerium für Wissenschaft, Forschung und Kunst of Baden-Württemberg (seed funds for the collaborative research initiative SFB-TRR234 “CataLight”). Work in Freiburg was supported by the DFG (priority program SPP 1613, grant KU2885/2-2) and the Austrian Klima- und Energiefonds (project no. 853639, MolSulCat).References
- (a) H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228 CrossRef PubMed; (b) W. Leitner, E. A. Quadrelli and R. Schlögl, Green Chem., 2017, 19, 2307 RSC; (c) J. Messinger, W. Lubitz and J.-R. Shen, Phys. Chem. Chem. Phys., 2014, 16, 11810 RSC; (d) J. Turner, G. Sverdrup, M. K. Mann, P.-C. Maness, B. Kroposki, M. Ghirardi, R. J. Evans and D. Blake, Int. J. Energy Res., 2008, 32, 379 CrossRef; (e) B. E. Logan and K. Rabaey, Science, 2012, 337, 686 CrossRef PubMed; (f) A. Kadier, Y. Simayi, P. Abdeshahian, N. F. Azman, K. Chandrasekhar and M. S. Kalil, Alexandria Eng. J., 2016, 55, 427 CrossRef.
- K. H. Büchel, H.-H. Moretto, P. Woditsch and M. Bertau, Industrielle Anorganische Chemie, Wiley-VCH, Weinheim, Germany, 4th edn, 2013 Search PubMed.
- A. W. von Hofmann, Introduction to Modern Chemistry: Experimental and Theoretic, Walton and Maberly, London, 1866 Search PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901 CrossRef.
- London Metal Exchange, LME Metal Prices, http://www.lme.com, accessed April 1st 2018.
- (a) H. Tributsch, J. Electroanal. Chem., 1977, 81, 97 CrossRef; (b) W. Jaegermann and H. Tributsch, Prog. Surf. Sci., 1988, 29, 1 CrossRef.
- B. Seo and S. H. Joo, Nano Convergence, 2017, 4, 19 CrossRef PubMed.
- Y. Yan, B. Xia, Z. Xu and X. Wang, ACS Catal., 2014, 4, 1693 CrossRef.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248 CrossRef PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878 RSC.
- M. Kokko, F. Bayerköhler, J. Erben, R. Zengerle, P. Kurz and S. Kerzenmacher, Appl. Energy, 2017, 190, 1221 CrossRef.
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Angew. Chem., Int. Ed. Engl., 2015, 54, 15181 CrossRef PubMed.
- M. Dave, A. Rajagopal, M. Damm-Ruttensperger, B. Schwarz, F. Nägele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020 RSC.
- Y. Lei, M. Yang, J. Hou, F. Wang, E. Cui, C. Kong and S. Min, Chem. Commun., 2018, 54, 603 RSC.
- I. Song, C. Park and H. C. Choi, RSC Adv., 2015, 5, 7495 RSC.
- E. Benavente, Coord. Chem. Rev., 2002, 224, 87 CrossRef.
- R. G. Dickinson and L. Pauling, J. Am. Chem. Soc., 1923, 45, 1466 CrossRef.
- B. Schönfeld, J. J. Huang and S. C. Moss, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 404 CrossRef.
- (a) R. J. J. Newberry, Am. Mineral., 1979, 64, 758 Search PubMed; (b) A. B. Anderson, Z. Y. Al-Saigh and W. Keith Hall, J. Phys. Chem., 1988, 92, 803 CrossRef; (c) S. Bertolazzi, J. Brivio and A. Kis, ACS Nano, 2011, 5, 9703 CrossRef PubMed.
- (a) S. G. Benka, Phys. Today, 2005, 58, 9 Search PubMed; (b) Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef PubMed.
- F. Wypych and R. Schöllhorn, Chem. Commun., 1992, 1386 RSC.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- (a) F. Wypych, T. Weber and R. Prins, Chem. Mater., 1998, 10, 723 CrossRef; (b) Q. Tang and D.-e. Jiang, Chem. Mater., 2015, 27, 3743 CrossRef.
- A. Müller, E. Diemann, R. Jostes and H. Bögge, Angew. Chem., Int. Ed. Engl., 1981, 20, 934 CrossRef.
- D. Recatalá, R. Llusar, A. L. Gushchin, E. A. Kozlova, Y. A. Laricheva, P. A. Abramov, M. N. Sokolov, R. Gómez and T. Lana-Villarreal, ChemSusChem, 2015, 8, 148 CrossRef PubMed.
- A. V. Virovets, M. Laege, B. Krebs, O. A. Geras'ko, V. E. Fedorov, O. V. Shishkin and Y. T. Struchkov, J. Struct. Chem., 1996, 37, 666 CrossRef.
- S. H. Laurie, D. E. Pratt and J. H. L. Yong, Inorg. Chim. Acta, 1984, 93, L57–L59 CrossRef.
- A. Müller, R. G. Bhattacharyya and B. Pfefferkorn, Chem. Ber., 1979, 112, 778 CrossRef.
- R. Jostes, A. Müller and E. Diemann, J. Mol. Struct., 1986, 137, 311 CrossRef.
- A. Müller, W.-O. Nolte and B. Krebs, Angew. Chem., Int. Ed. Engl., 1978, 17, 279 CrossRef.
- V. P. Fedin, J. Czyzniewska, R. Prins and T. Weber, Appl. Catal., A, 2001, 213, 123 CrossRef.
- A. Müller, W. Jaegermann and J. H. Enemark, Coord. Chem. Rev., 1982, 46, 245 CrossRef.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Phys. Chem., 1995, 99, 9194 CrossRef.
- A. Müller, V. P. Fedin, K. Hegetschweiler and W. Amrein, J. Chem. Soc., Chem. Commun., 1992, 1795 RSC.
- H. W. Wang, P. Skeldon and G. E. Thompson, Surf. Coat. Technol., 1997, 91, 200 CrossRef.
- S. J. Hibble, R. I. Walton, D. M. Pickup and A. C. Hannon, J. Non-Cryst. Solids, 1998, 232–234, 434 CrossRef.
- D. R. Huntley, T. G. Parham, R. P. Merrill and M. J. Sienko, Inorg. Chem., 1983, 22, 4144 CrossRef.
- (a) S. P. Cramer, K. S. Liang, A. J. Jacobson, C. H. Chang and R. R. Chianelli, Inorg. Chem., 1984, 23, 1215 CrossRef; (b) S. J. Hibble, D. A. Rice, D. M. Pickup and M. P. Beer, Inorg. Chem., 1995, 34, 5109 CrossRef.
- C. H. Chang and S. S. Chan, J. Catal., 1981, 72, 139 CrossRef.
- E. Diemann, Z. Anorg. Allg. Chem., 1977, 432, 127 CrossRef.
- S. J. Hibble, M. R. Feaviour and M. J. Almond, J. Chem. Soc., Dalton Trans., 2001, 935 RSC.
- A. Müller, S. Sarkar, R. G. Bhattacharyya, S. Pohl and M. Dartmann, Angew. Chem., Int. Ed. Engl., 1978, 17, 535 CrossRef.
- V. P. Fedin, M. N. Sokolov, Y. V. Mironov, B. A. Kolesov, S. V. Tkachev and V. Y. Fedorov, Inorg. Chim. Acta, 1990, 167, 39 CrossRef.
- S. J. Hibble and G. B. Wood, J. Am. Chem. Soc., 2004, 126, 959 CrossRef PubMed.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916 CrossRef.
- (a) A. Müller, W. O. Nolte and B. Krebs, Inorg. Chem., 1980, 19, 2835 CrossRef; (b) A. Müller, R. Jostes, W. Jaegermann and R. G. Bhattacharyya, Inorg. Chim. Acta, 1980, 41, 259 CrossRef.
- F. Z. Chien, S. C. Moss, K. S. Liang and R. R. Chianelli, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 1 CrossRef.
- L. Busetto, A. Vaccari and G. Martini, J. Phys. Chem., 1981, 85, 1927 CrossRef.
- P. Afanasiev, C. R. Chim., 2008, 11, 159 CrossRef.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640 CrossRef PubMed.
- Y. Deng, L. R. L. Ting, P. H. L. Neo, Y.-J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 7790 CrossRef.
- L. R. L. Ting, Y. Deng, L. Ma, Y.-J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 861 CrossRef.
- B. Lassalle-Kaiser, D. Merki, H. Vrubel, S. Gul, V. K. Yachandra, X. Hu and J. Yano, J. Am. Chem. Soc., 2015, 137, 314 CrossRef PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef PubMed.
- (a) J. Djamil, S. A. Segler, A. Dabrowski, W. Bensch, A. Lotnyk, U. Schürmann, L. Kienle, S. Hansen and T. Beweries, Dalton Trans., 2013, 42, 1287 RSC; (b) U. Gupta and C. N. R. Rao, Nano Energy, 2017, 41, 49 CrossRef; (c) B. Han and Y. H. Hu, Energy Sci. Eng., 2016, 4, 285 CrossRef.
- B. Kirchhoff, S. Rau and C. Streb, Eur. J. Inorg. Chem., 2016, 2016, 1425 CrossRef.
- (a) J. V. Lauritsen, S. Helveg, E. Lægsgaard, I. Stensgaard, B. S. Clausen, H. Topsøe and F. Besenbacher, J. Catal., 2001, 197, 1 CrossRef; (b) M. Daage and R. R. Chianelli, J. Catal., 1994, 149, 414 CrossRef; (c) W. Bensch, in Comprehensive Inorganic Chemistry II, Elsevier, 2013 Search PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308 CrossRef PubMed.
- G. Li, D. Zhang, Q. Qiao, Y. Yu, D. Peterson, A. Zafar, R. Kumar, S. Curtarolo, F. Hunte, S. Shannon, Y. Zhu, W. Yang and L. Cao, J. Am. Chem. Soc., 2016, 138, 16632 CrossRef PubMed.
- (a) P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, J. Catal., 2000, 189, 129 CrossRef; (b) P. Lazar and M. Otyepka, Chem.–Eur. J., 2017, 23, 4863 CrossRef PubMed; (c) J. V. Lauritsen, J. Kibsgaard, S. Helveg, H. Topsøe, B. S. Clausen, E. Lægsgaard and F. Besenbacher, Nat. Nanotechnol., 2007, 2, 53 CrossRef PubMed.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671 CrossRef PubMed.
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254 CrossRef.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef PubMed.
- H. Vrubel and X. Hu, ACS Catal., 2013, 3, 2002 CrossRef.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957 CrossRef.
- (a) D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC; (b) Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef PubMed.
- T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nat. Commun., 2016, 7, 10902 CrossRef PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef PubMed.
- Iron-Sulfur Clusters in Chemistry and Biology, ed. T. A. Rouault, De Gruyter, Berlin, 2014 Search PubMed.
- R. Bjornsson, F. Neese, R. R. Schrock, O. Einsle and S. DeBeer, J. Biol. Inorg Chem., 2015, 20, 447 CrossRef PubMed.
Footnote |
| † The first two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |