
A hybrid molecular photoanode for efficient light-induced water oxidation†
Sergi
Grau‡
a,
Serena
Berardi‡
 *b,
Alicia
Moya
c,
Roc
Matheu
a,
Vito
Cristino
b,
Juan José
Vilatela
c,
Carlo A.
Bignozzi
b,
Stefano
Caramori
b,
Carolina
Gimbert-Suriñach
*a and
Antoni
Llobet
*a
*b,
Alicia
Moya
c,
Roc
Matheu
a,
Vito
Cristino
b,
Juan José
Vilatela
c,
Carlo A.
Bignozzi
b,
Stefano
Caramori
b,
Carolina
Gimbert-Suriñach
*a and
Antoni
Llobet
*a
aInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), Avda. Països Catalans 16, 43007 Tarragona, Spain. E-mail: cgimbert@iciq.cat; allobet@iciq.cat
bDept. of Chemistry and Pharmaceutical Sciences, University of Ferrara, Via di Mortara 17, 44121, Ferrara, Italy. E-mail: serena.berardi@unife.it
cIMDEA Materials Institute, C. Eric Kandel 2, 28906, Getafe, Madrid, Spain
First published on 17th May 2018
Abstract
A hybrid photoanode comprising a multilayered heterostructured WO3/BiVO4 semiconductor and a molecular water oxidation catalyst Ru(tda)(py-pyr)2 (Ru-WOC, where tda is [2,2′:6′,2′′-terpyridine]-6,6′′-dicarboxylato and py-pyr is 4-(pyren-1-yl)-N-(pyridin-4-ylmethyl)butanamide) is described. Both elements are linked by a highly conductive carbon nanotube fibre film (CNTf), which acts as charge transfer and anchoring platform, to which the catalyst is attached through π–π stacking interactions. Photoelectrochemical characterization of the resulting electrodes shows that the full photoanode WO3/BiVO4/CNTf/Ru-WOC outperforms the bare WO3/BiVO4 electrode in the potential range 0.3–0.8 V vs. NHE at pH 7, with current densities enhanced by 0.05–0.29 mA cm−2. Bulk electrolysis experiments and oxygen gas measurements show that the enhanced photocurrent is due to the catalytic water oxidation reaction. Detailed electrochemical impedance spectroscopy (EIS) analysis is used to investigate the roles of the multiple layers involved in the process. The CNTf/Ru–WOC interface is responsible for increasing charge accumulation and reducing recombination phenomena. The CNTf is able to hold the charge produced from light absorbed by the WO3/BiVO4 semiconductor, as shown by the high capacitive values observed for a WO3/BiVO4/CNTf electrode in the whole range of studied potentials (0.15–0.85 V vs. NHE). Furthermore, Ru-WOC transfers the charge to the solution through fast water oxidation catalysis. This is supported by the low resistivity shown by the full WO3/BiVO4/CNTf/Ru-WOC electrode at low potentials (E < 0.5 V vs. NHE). The robustness and high catalytic rate of Ru-WOC ensures the proper performance of the hybrid photoelectrode device. The latter is particularly important, as it provides opportunities to improve the performance of photoanodes for the water oxidation reaction based on the easy modification of ligands in the molecular catalyst to tune its structural, electronic, and catalytic properties. This is a unique advantage compared with commonly used catalysts based on metal oxides or oxy(hydroxides), which have limited tunability.
Introduction
The conversion of solar energy into a more manageable and sustainable energy source is a major focus of global scientific research. Although photovoltaic (PV) and solar thermal technologies have been realized, with solar panels used to generate a portion of global energy production, the conversion of solar energy into clean fuels has yet to be achieved on an industrial scale.1 The latter strategy offers a solution to the mismatch between sunlight intermittence and energy needs by converting solar energy into storable chemical bonds. The difficulty in finding efficient light absorbing materials to couple with active electrocatalysts, either through wires in a typical PV-electrochemical (PV-EC) configuration or via semiconductor–electrolyte interfaces, is among the problems hindering the implementation of such technology.1–3 These systems need to effectively absorb light and produce long-lived separated charges to facilitate fast electron and hole transfer to catalysts that drive the desired chemical reactions. The water oxidation reaction is one of the limiting processes within such complex systems, owing to the high requirements for efficient catalysts, which include a high thermodynamic potential and large kinetic complexity. Indeed, the latter complexity is a consequence of the need to transfer four electrons and four protons from two water molecules together with the generation of an O–O bond (Fig. 1).4–6 For this reason, much research effort is now devoted toward solar-to-fuel conversion, with the aim to find high-performance electroanodes and photoanodes for the water oxidation reaction.2,7–9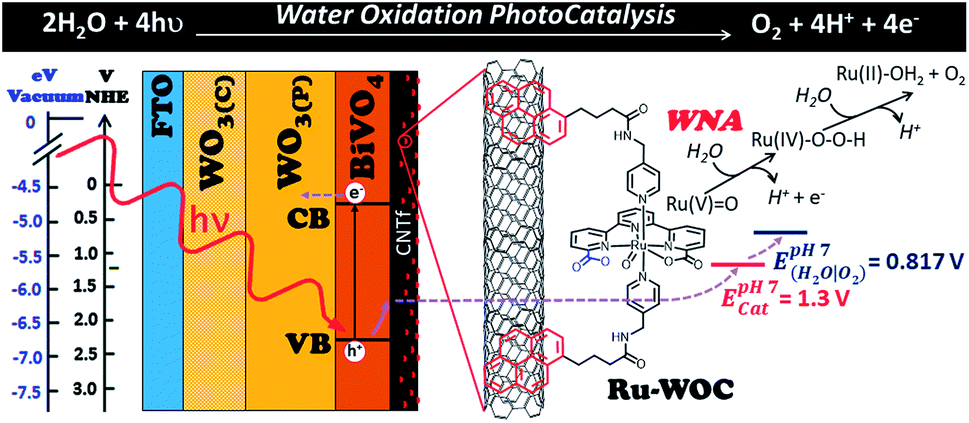 | ||
| Fig. 1 Hybrid photoanode design studied in this work. WO3(C) = compact tungsten oxide film, WO3(P) = mesoporous tungsten oxide layer, Ru-WOC = molecular ruthenium water oxidation catalyst with pyrene groups in red and dangling carboxylate group in blue, WNA = water nucleophilic attack, E(cat) = oxidation potential of catalyst to give Ru(V)-oxo active species, E(H2O/O2) = thermodynamic potential of water oxidation at pH 7. | ||
Tungsten oxide (WO3) and bismuth vanadate (BiVO4) are two promising semiconductor materials with band gaps and valence band positions suitable for photoinduced water oxidation.7,8 In particular, BiVO4 has shown excellent photocurrent densities at potentials close to and lower than the thermodynamic value. However, the performances of BiVO4 photoanodes are still far below their theoretical maximum capacities.7,8 This problem is associated with slow charge carrier mobility and the slow kinetics of the water oxidation reaction at the electrode surface. Four main strategies have been used to overcome these unfavourable phenomena: (i) doping with metallic elements (particularly good results obtained with W and Mo),10 (ii) tuning the morphology and nanostructure of the material,10–12 (iii) forming heterojunctions with other semiconductors (such as WO3)13,14 or conductive materials (such as graphene),7,15 and (iv) modifying the photoelectrode surface with suitable water oxidation catalysts (metal oxides or oxy(hydroxides)).16,17 While the first three strategies (i–iii) have been designed to enhance charge carrier mobility and charge separation, and/or avoid recombination, the latter strategy (iv) mainly concerns reducing the high kinetic barrier associated with the water oxidation reaction. Interestingly, recent work has indicated the poor activity of metal oxide derivatives as water oxidation catalysts.17,18 Instead, the enhanced photocurrent densities observed have been attributed to higher charge separation efficiencies obtained with metallic oxides deposited on the photoanode surface.17,18 These observations are in agreement with the intrinsic limited catalytic activity of metal oxides, such as cobalt oxide, which competes with BiVO4 catalysis. In contrast to metal oxide catalysts, molecular water oxidation catalysts (WOC) have seen tremendous improvements in catalytic rates, achieving maximum turnover frequencies three to four orders of magnitude higher than those achieved by the best oxide-based water oxidation catalysts. Indeed, molecular water oxidation catalysts outperform the naturally occurring oxygen evolving complex in photosystem II, responsible for oxygen release in the natural photosynthesis in plants, by two orders of magnitude.5,6,19 Recently, strategies to anchor these molecular catalysts on electrode surfaces have been developed, maintaining the high catalytic activity observed under homogeneous conditions and, in some cases, even increasing their performance.20–22
Herein, we have coupled a WO3/BiVO4 photoanode with a highly active molecular WOC with the aim to enhance the reaction kinetics, minimize recombination and oxide degradation, and improve the overall performance of the photoelectrode. A carbon nanotube fibre interlayer film between the photoactive material and catalyst is used as a highly conductive platform to facilitate anchoring of the functionalized molecular WOC and allows fast charge transfer between the two key photoelectrode components (semiconductor and catalyst) (Fig. 1).
Results
Fabrication of hybrid photoanode
The multilayered architecture of the WO3/BiVO4 photoactive material used in this study, as shown in Fig. 1, comprises three well-defined layers, namely, a compact tungsten oxide thin film layer (WO3(C)), a mesoporous layer of the same material (WO3(P)),23 and a top layer of porous BiVO4.16 The inner compact layer of WO3(C) is essential for isolating the back FTO contact from the electrolyte solution, avoiding dark currents associated with shunt phenomena. This effect is clearly shown in Fig. S5 (see ESI†), which shows that, in the absence of light, the electrode WO3(C)/WO3(P)/BiVO4 produces no current over the whole potential range, while the electrode without the WO3 compact underlayer (WO3(P)/BiVO4) shows significant dark currents from 0.75 V vs. NHE. On the other hand, the heterostructured WO3/BiVO4 porous material showed better performance than bare BiVO4 photoanodes.13,14 This improvement was attributed to good band alignment and synergistic effects between the two semiconductors, which lead to higher charge separation efficiencies.14 Detailed structural, electrochemical, and optical characterization of the photoanodes used in this work is shown in Fig. S1–S5 in the ESI.†Water oxidation catalyst Ru-WOC (Fig. 1) was selected for its high activity under both homogeneous and heterogeneous conditions.19,20,22 Ru-WOC is known to operate via an electrophilic Ru(V)-oxo species that suffers water nucleophilic attack (WNA) to form a hydroperoxo key derivative with a newly formed O–O bond. The high catalytic rate that characterizes this catalyst is attributed to the role of the dangling carboxylate group assisting O–O bond formation (Fig. 1, blue moiety in the Ru-WOC complex).24 Several reports have described the direct attachment of molecular catalysts to semiconducting materials, usually using phosphonate or carboxylate groups.25–27 In the present case, when the Ru-WOC was attached to the WO3/BiVO4 photoanode, no improvement in the electrode performance was observed. This result was attributed to deactivation of the dangling carboxylate group, which interacted directly with the metal oxide, hindering its activation.27 This could be avoided using a conductive support that isolates the catalyst from the photoelectrode surface. The support should guarantee fast charge transfer from the semiconductor to the catalyst, without altering the ruthenium catalytic centre. In this regard, carbon nanostructures, such as graphene and single or multiwalled carbon nanotubes, are good candidates owing to their unique properties, which include high surface areas, high thermal and electrical conductivities, and chemical inertness. In particular, carbon nanotube fibres (CNTf) are excellent conductive platforms. They are macroscopic arrays of CNTs with a combined surface area of around 250 m2 g−1, an electrical conductivity of 3.5 × 105 S m−1, and high electrochemical stability,28 and can be produced on a kilometre per day scale,29 for example, as free-form electrodes. Accordingly, the hybrid molecular photoanode device was prepared following a new strategy; a WO3/BiVO4 photoanode (approximately 1 × 1 cm2) was covered with a CNTf film (approximately 0.9 × 0.9 cm2; thickness, around 5 μm) estimated based on fibre linear density, winding rate, and spinning time.29 Next, the resulting WO3/BiVO4/CNTf structure was soaked in a methanolic solution (1 mM) of Ru-WOC precursor overnight to allow chemical attachment of the ruthenium complex on the surface of the electrode via π–π interactions between the complex pyrene groups and the graphitic structure of the CNTf (Fig. 1). After this treatment, the electrodes were rinsed with clean methanol, to ensure that excess homogeneous catalyst was removed, and carefully dried under an air flow. To estimate the catalyst loading on the electrode surface, analogous experiments were performed with non-photoactive ITO/CNTf electrodes. Analysis of the current underneath the Ru(III/II) wave in the cyclic voltammetry (CV) of the resulting ITO/CNTf/Ru-WOC electrodes allowed us to calculate the amount of catalyst precursor on the CNTf film, giving values up to Γ = 15 nmol cm−2, which were comparable to those observed in previous studies that used multiwalled carbon nanotubes (Fig. S9†).20,22 This confirmed that the free-standing format and high surface area of CNTf made them an attractive conducting support for heterogeneous catalysis.
Photoelectrochemical performance
The photoelectrochemical performance of the photoanodes was tested in a typical one-compartment three-electrode photoelectrochemical cell, as described in detail in the ESI.† As the CNTf was an extremely black layer that completely blocked incoming light, all photoelectrochemical results presented in this study are based on back illumination of the photoelectrodes. The photoelectrochemical behaviour of the bare WO3/BiVO4 photoanodes improved over consecutive linear sweep voltammetry (LSV) experiments, as shown in Fig. S8.† Therefore, all electrodes were electrochemically pretreated until a constant photocurrent response was achieved prior to any modification (usually 3–5 LSV cycles).Fig. 2 shows the chopped light LSV performances of a WO3/BiVO4 photoanode and a WO3/BiVO4/CNTf analogue in pH 7 buffered solution. A significant decrease in the photocurrent was observed when the CNTf was present (compare red and black traces). This result concurred with the CNTf film blocking the BiVO4 active centres responsible for water oxidation catalysis. Therefore, although the CNTf film was highly conductive, it was unable to perform the chemical reaction. In other words, the CNTf film did not act as a water oxidation catalyst under these conditions. In contrast, the presence of Ru-WOC on the surface of the electrode significantly improved the performance of the photocurrent, particularly in the range of 0.3 V to 0.8 V vs. NHE (compare blue trace with black and red traces in Fig. 2). The improvement increased from 0.05 mA cm−2 at 0.3 V to 0.29 mA cm−2 at 0.7 V vs. NHE. To ensure that this enhancement was due to the catalyst and not the methanolic solution treatment during catalyst deposition, analogous blank tests were conducted. Specifically, WO3/BiVO4/CNTf electrodes were soaked in clean methanol overnight and then dried. The resulting treated electrodes did not show a significant improvement in performance, confirming that the increased photocurrent observed in Fig. 2 was due to the presence of the catalyst (Fig. S10†).
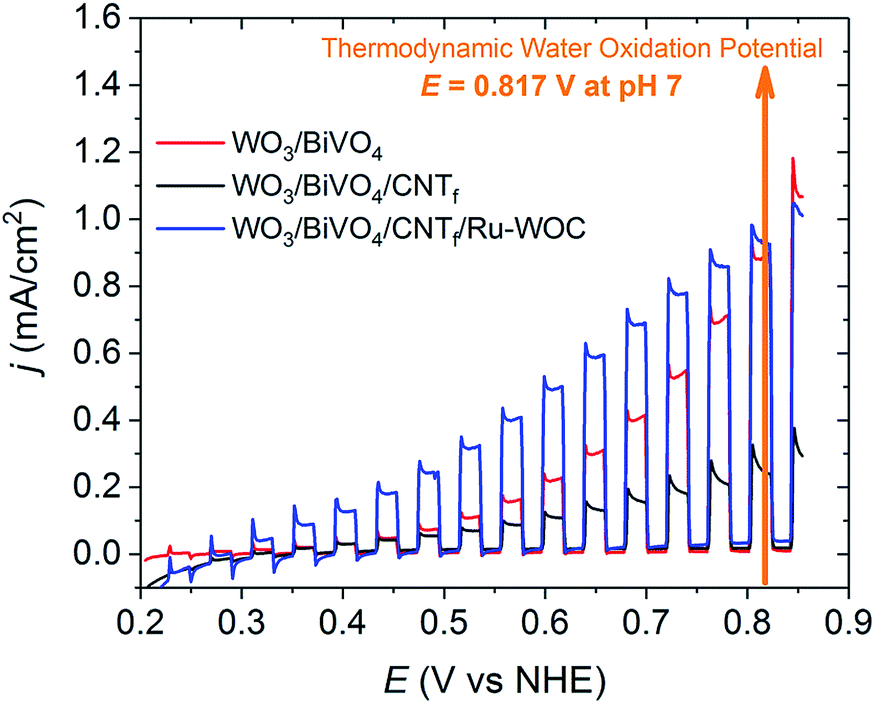 | ||
| Fig. 2 Chopped light LSV (5 mV s−1) of the different photoanodes in pH 7 phosphate buffer collected with IR drop compensation. WO3/BiVO4 (red), WO3/BiVO4/CNTf (black), and WO3/BiVO4/CNTf/Ru-WOC (blue). | ||
Bulk electrolysis experiments showed that the full electrode, WO3/BiVO4/CNTf/Ru-WOC, outperformed the bare WO3/BiVO4 electrode (Fig. 3, blue and red traces, respectively). This photocurrent enhancement was attributed to the higher catalytic activity of Ru-WOC compared with that of BiVO4. The integrity of the molecular water oxidation catalyst after bulk electrolysis experiments was tested by carefully removing the CNTf/Ru-WOC film from the surface of the WO3/BiVO4 electrode and placing it on a clean ITO electrode. As shown in Fig. S9,† the fingerprint of the ruthenium complex remained intact on the film.
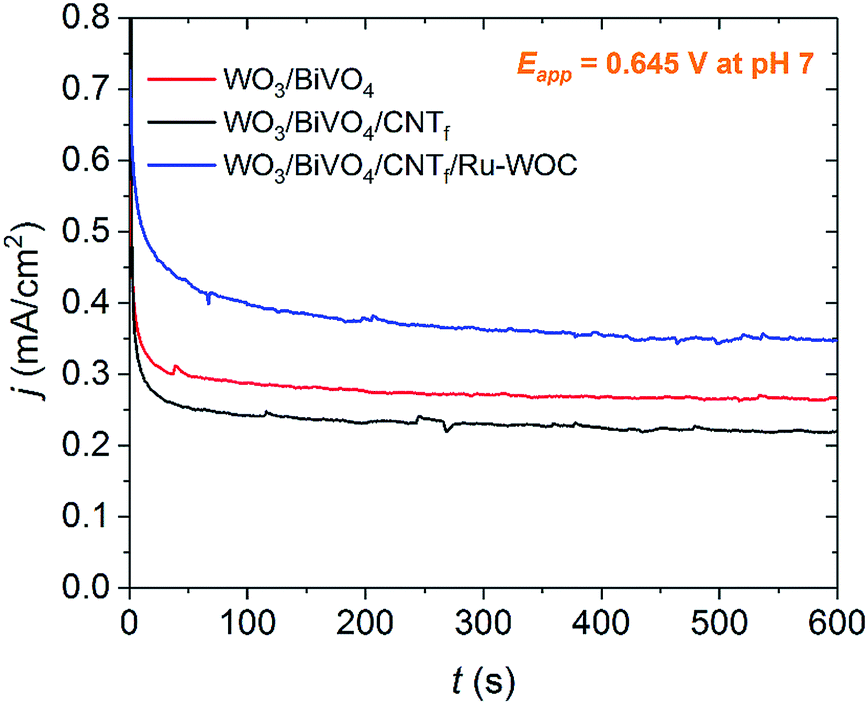 | ||
| Fig. 3 Bulk electrolysis experiment at applied potential Eapp = 0.645 vs. NHE in pH 7 phosphate buffer, IR drop was not compensated. WO3/BiVO4 (red), WO3/BiVO4/CNTf (black) and WO3/BiVO4/CNTf/Ru-WOC (blue). | ||
Oxygen detection experiments were performed to prove that the enhanced photocurrent obtained with the WO3/BiVO4/CNTf/Ru-WOC electrode was due to water oxidation catalysis. An adapted generator–collector device was used, as inspired by previously reported procedures designed for related photoelectrochemical systems (Fig. 4 and S11–S13 in the ESI†).30,31 In particular, a slow scan rate LSV is performed on the desired photoelectrode (generator) while a controlled potential electrolysis is run simultaneously on a closely positioned FTO electrode (collector). At the collector electrode, the oxygen generated at the photoanode is reduced after diffusion through the solution. Therefore, the current observed in the controlled potential electrolysis experiment is proportional to the O2 evolved by the water oxidation reaction on the photoelectrode. This method allows the oxygen gas to be measured in situ and the potential at which the chemical reactions starts to be controlled. As shown in Fig. 4 and S12,† the current flowing through the collector electrode sandwiched to the full hybrid electrode WO3/BiVO4/CNTf/Ru-WOC (blue line), attributed to the oxygen evolution reaction, started at a potential 140 mV lower (approx. 0.41 V) than that of the bare electrode WO3/BiVO4 (red line) (approx. 0.55 V). The delay in detecting the oxygen was attributed to the diffusion of oxygen gas from the generator electrode to the collector electrode. To rule out the formation of side-products derived from the oxidation of CNT fibres, a control experiment was performed with the WO3/BiVO4/CNTf electrode. Fig. S13† shows that no significant current was detected in the collector electrode in the full range of potentials. Therefore, the current of the collector electrode in Fig. 4 was attributed uniquely to the reduction of oxygen gas. In this scenario, the collector–generator methodology, when properly calibrated, allows accurate oxygen efficiency measurements to be conducted.31 Using this method, faradaic efficiencies of 94% for WO3/BiVO4/CNTf/Ru-WOC and 97% for WO3/BiVO4 were obtained (Fig. S13†).
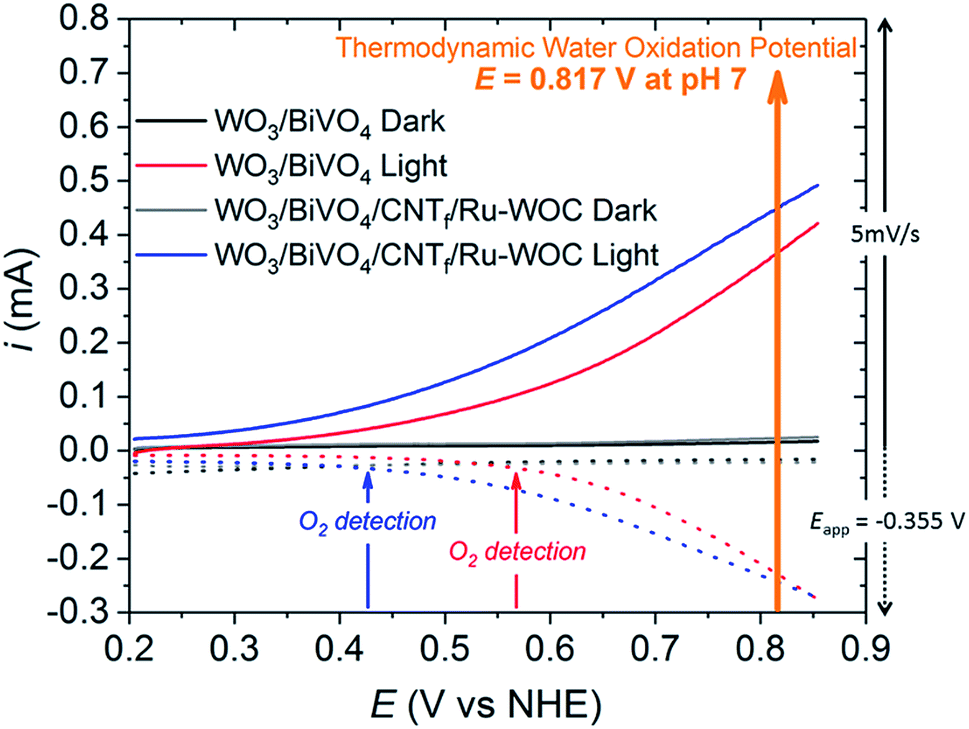 | ||
| Fig. 4 Oxygen gas detection experiments using an adapted generator–collector method performed in pH 7.30,31 Solid lines correspond to LSV (5 mV s−1) performed on the photoanode electrode (generator). The dotted lines correspond to the in situ current observed on the collector electrode due to O2 reduction (controlled potential electrolysis at Eapp = −0.355 V vs. NHE, see details in the ESI†). WO3/BiVO4 in the dark (black), WO3/BiVO4 under 1 sun illumination (red), WO3/BiVO4/CNTf/Ru-WOC in the dark (grey), and WO3/BiVO4/CNTf/Ru-WOC under 1 sun illumination (blue). | ||
Electrochemical impedance spectroscopic analysis
To get further insight into the role of the different interfaces involved in photocurrent generation, electrochemical impedance spectroscopy (EIS) experiments were conducted under illumination and different applied biases. The results have been fitted using a circuital model adapted from reported works (Fig. 5A).14,32,33 Briefly, it consists of serial resistance of the electrochemical cell (R1) in series with a R2-CPE2 mesh and an extended element (TL). The latter is commonly used to represent the transmission line of mesoporous films (WO3/BiVO4 in our case), through which the photogenerated carriers travel, overcoming the transport resistance (Rtr) between the fused nanoparticles. An approximation of a negligible capacitance contribution between the particles is generally used. Furthermore, the model includes a resistive contribution (Rct) due to charge carrier recombination (which eventually influences charge transfer to the electrolyte), and a capacitive contribution accounting for the accumulation of photogenerated charges in the film (CPEfilm). Furthermore, the R2-CPE2 mesh is introduced to account for the FTO/WO3 interface or the CNTf/electrolyte interface when the CNTf is present.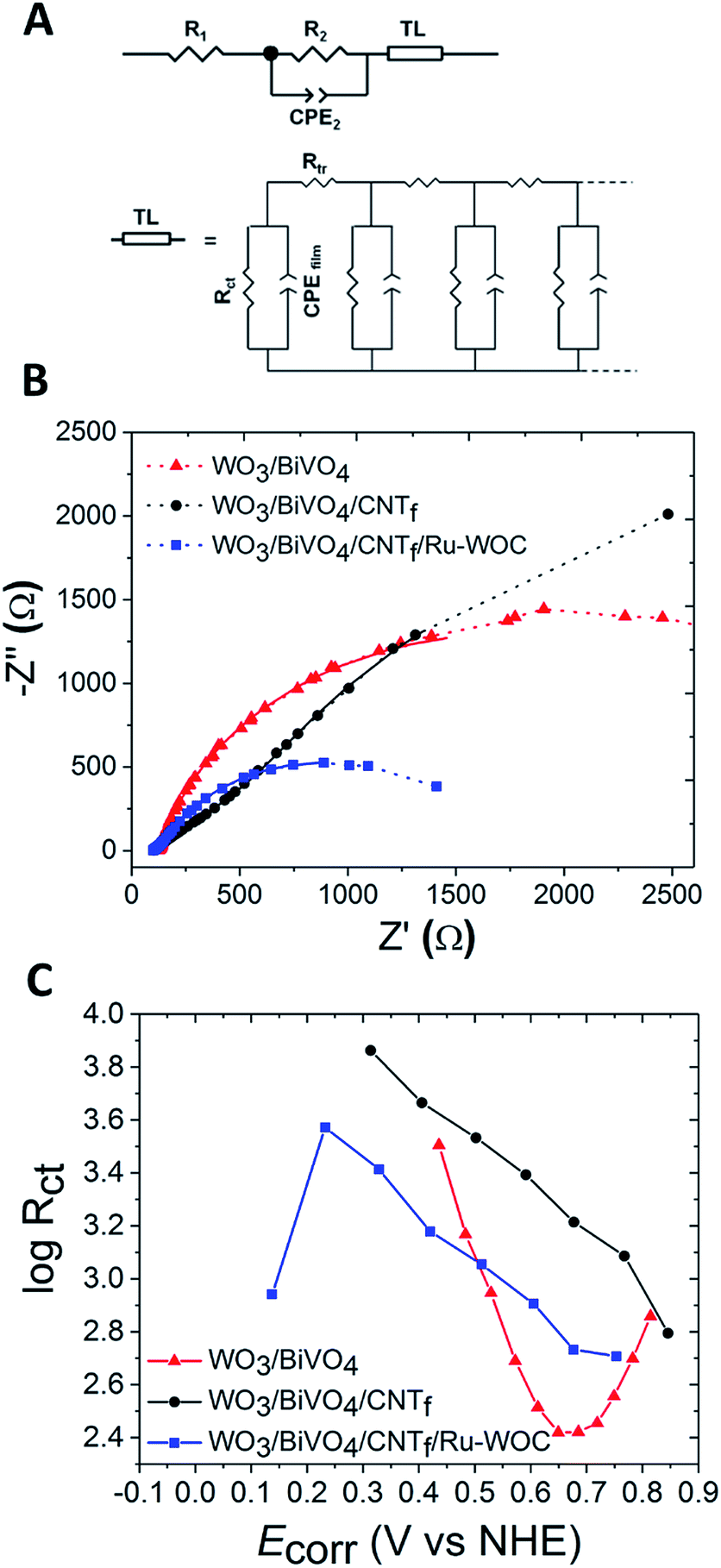 | ||
Fig. 5 (A) Equivalent circuit used to fit the EIS data of the photoanodes. (B) Complex plane Nyquist plots for the different electrodes measured under 1 sun illumination in pH 7 phosphate buffer at 0.440 V vs. NHE. The corresponding fits are also reported as thick solid lines. (C) log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Rctvs. potential. Legend code: WO3/BiVO4 (red), WO3/BiVO4/CNTf (black), and WO3/BiVO4/CNTf/Ru-WOC (blue). Rctvs. potential. Legend code: WO3/BiVO4 (red), WO3/BiVO4/CNTf (black), and WO3/BiVO4/CNTf/Ru-WOC (blue). | ||
The experimental Nyquist plots of different photoanodes, and the corresponding fittings, are reported in Fig. 5B and S14,† showing the good agreement provided by the selected circuital model. The major contribution of total resistance is given by Rct, which has the same dependence as the applied bias of the inverse of the differential resistance (Fig. S15†). The exact values, obtained for different resistive and capacitive contributions, are shown in Tables S1 and S2,† respectively.
In the model, Rct represents recombination through the nanostructured film, so its contribution is expected to decrease for systems that can effectively transfer charge to the electrolyte and perform fast water oxidation catalysis. Fig. 5C reports the bias dependence of the logarithm of Rct and gives a quantification of this recombination phenomenon. The presence of Ru-WOC coupled to the CNTf reduces the Rct value of the corresponding photoanode approx. three-fold over the whole potential range (compare blue and black traces). The charge transfer rate to the aqueous electrolyte is indeed expected to be lower for the WO3/BiVO4/CNTf electrodes without the molecular catalyst, due to the hydrophobicity of the CNTf layer and blockage of the active catalytic sites of BiVO4. Regarding the bare WO3/BiVO4 electrode, the charge transfer by Ru-WOC was only improved at low potentials, as evidenced by the crossing point of the blue and red traces in Fig. 5C. Furthermore, the WO3/BiVO4 electrode showed bell-shaped behaviour (red trace in Fig. 5C) with a minimum at around 0.69 V vs. NHE, which is the inflection point of the LSV curve of the photoelectrode (Fig. S14†). For other resistive contributions, the resistance associated with the inert FTO/WO3 interface of bare WO3/BiVO4 was < 15 Ω and independent of the applied bias, as expected when the interface is not directly involved in the photocurrent generation mechanism.14 In contrast, for the WO3/BiVO4/CNTf electrodes, the R2 values were much higher (in the range 330–625 Ω) and bias-dependent. Therefore, in this case, the R2-CPE2 mesh has been assigned to the CNTf/electrolyte interface, whose higher resistive contribution covers that due to the FTO/WO3 interface, that was considered in the first place. This characteristic can be seen in the Nyquist plots, resulting in an additional arc for the WO3/BiVO4/CNTf electrode (Fig. 5B, black trace). When the latter is coupled with Ru-WOC, the R2 values (assigned by analogy to the functionalized CNTf/electrolyte interface) decreased by at least one order of magnitude (<25 Ω) with respect to non-catalytic WO3/BiVO4/CNTf. This was expected from the improved surface kinetics in the presence of the catalyst, which also induces wetting of the porous CNTf layer. Furthermore, the R2 values were bias dependent for WO3/BiVO4/CNTf/Ru-WOC, with a minimum at 0.64 V vs. NHE, close to the inflection point of the LSV curve (Fig. S14 and S15E†). More information on the nanofibres/electrolyte interface can be extracted from the capacitance values given by CPE2, which follows a similar potential dependence for both WO3/BiVO4/CNTf and WO3/BiVO4/CNTf/Ru-WOC (Fig. S16†). These results support the hypothesis of reduced electron recombination with surface trapped holes (that may translate into WO3/BiVO4 defects, oxidized CNTf, or oxidized catalyst) as the potential increases.
In the case of WO3/BiVO4 electrodes, the logarithm of the capacitance associated with charge accumulation in the film (CPEfilm) displayed a linear dependence on the applied bias after the surface states had been emptied by the voltage at approximately 0.54 V vs. NHE (Fig. S17†). This behaviour was typical of a chemical capacitor, as expected for this kind of electrode.34 A higher CPEfilm value has been observed for WO3/BiVO4/CNTf/Ru-WOC photoanodes, confirming that, in the presence of the catalyst, a higher charge can be accumulated and then transported to the electrolyte. Notably, the higher CPEfilm values for WO3/BiVO4/CNTf/Ru-WOC corresponded to lower Rct values in the range 0.15–0.50 V vs. NHE, where the photocurrent response is anticipated with respect to the bare WO3/BiVO4 electrode (compare red and blue traces in Fig. 5C and S17†). Furthermore, we observed that the CPEfilm followed the same bias dependence as WO3/BiVO4/CNTf/Ru-WOC and WO3/BiVO4 electrodes at high potentials (>0.59 V vs. NHE). However, at lower values, the results were essentially constant, as also observed in the case of WO3/BiVO4/CNTf. The higher CPEfilm values obtained in the latter case suggested significant charge accumulation on the CNTf layer (or on BiVO4 states covered by CNTf), which can be followed by good charge transfer to the electrolyte only when coupled with the WOC.
Discussion
Fig. 1 shows the potential values required to oxidize the molecular catalyst attached to the surface of the WO3/BiVO4/CNTf/Ru-WOC photoanode and generate the Ru(V)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O active species (E(RuV/IV) = 1.3 V vs. NHE). This potential should be easily overcome by the oxidative power of the low BiVO4 valence band upon illumination (EVB,pH 7 = 2.2–2.4 V vs. NHE).35 Once the Ru(V)O species is formed, the water oxidation catalytic process is triggered and occurs at a maximum rate of almost 8000 cycles per second.19,20 Therefore, upon illumination, the WO3/BiVO4/CNTf/Ru-WOC electrode should be able to perform oxygen evolution catalysis. Fig. 2 shows that the WO3/BiVO4/CNTf/Ru-WOC electrode indeed produced an enhanced photocurrent at a potential lower than the WO3/BiVO4 and WO3/BiVO4/CNTf electrodes (compare blue with red and black traces). Bulk electrolysis and oxygen measurement experiments confirmed that the enhanced photocurrent was sustained over several minutes, and that the current was due to oxygen gas formation (Fig. 3 and 4). Furthermore, oxygen gas was detected at approximately 0.41 V vs. NHE when using the catalyst-modified electrode WO3/BiVO4/CNTf/Ru-WOC, which was 140 mV earlier than for the bare photoanode WO3/BiVO4 (Fig. 4 and S12†) with faradaic efficiencies close to 100%. These results not only prove that Ru-WOC performed the reaction, but also showed that the charge was efficiently transferred through the entire multilayered architecture of the photoanode, from the inner semiconductor to the CNT fibres and, finally, the catalyst. Accordingly, the CNT fibres form an effective and stable interlayer with excellent charge transfer abilities. Furthermore, their work function being above the conduction band of BiVO4 might contribute to charge separation by preventing recombination phenomena.36 The CNTf layer also constitutes an excellent platform for the easy anchoring of molecular catalysts.
O active species (E(RuV/IV) = 1.3 V vs. NHE). This potential should be easily overcome by the oxidative power of the low BiVO4 valence band upon illumination (EVB,pH 7 = 2.2–2.4 V vs. NHE).35 Once the Ru(V)O species is formed, the water oxidation catalytic process is triggered and occurs at a maximum rate of almost 8000 cycles per second.19,20 Therefore, upon illumination, the WO3/BiVO4/CNTf/Ru-WOC electrode should be able to perform oxygen evolution catalysis. Fig. 2 shows that the WO3/BiVO4/CNTf/Ru-WOC electrode indeed produced an enhanced photocurrent at a potential lower than the WO3/BiVO4 and WO3/BiVO4/CNTf electrodes (compare blue with red and black traces). Bulk electrolysis and oxygen measurement experiments confirmed that the enhanced photocurrent was sustained over several minutes, and that the current was due to oxygen gas formation (Fig. 3 and 4). Furthermore, oxygen gas was detected at approximately 0.41 V vs. NHE when using the catalyst-modified electrode WO3/BiVO4/CNTf/Ru-WOC, which was 140 mV earlier than for the bare photoanode WO3/BiVO4 (Fig. 4 and S12†) with faradaic efficiencies close to 100%. These results not only prove that Ru-WOC performed the reaction, but also showed that the charge was efficiently transferred through the entire multilayered architecture of the photoanode, from the inner semiconductor to the CNT fibres and, finally, the catalyst. Accordingly, the CNT fibres form an effective and stable interlayer with excellent charge transfer abilities. Furthermore, their work function being above the conduction band of BiVO4 might contribute to charge separation by preventing recombination phenomena.36 The CNTf layer also constitutes an excellent platform for the easy anchoring of molecular catalysts.
EIS analysis allowed the two potential ranges in which the CNTf/Ru-WOC interface have distinct influence on the overall catalytic performance to be differentiated. At high potentials, although the charge accumulation capacity of the complete photoanode WO3/BiVO4/CNTf/Ru-WOC is higher than that of simple WO3/BiVO4, recombination is also higher, resulting in a similar performance (Fig. 5C and S17,†E > 0.5 V vs. NHE). In this potential range, water oxidation catalysis by Ru-WOC starts competing with catalysis by the BiVO4 active centres of the bare WO3/BiVO4 electrode, which are available in higher quantities in the absence of carbon nanotube fibres. In contrast, at low potentials, the full WO3/BiVO4/CNTf/Ru-WOC electrode outperformed the WO3/BiVO4 photoanode due to a higher charge accumulation capacity and lower recombination phenomena (Fig. 5C and S17,†E < 0.5 V vs. NHE). This was particularly remarkable in the 0.3–0.5 V vs. NHE range, where the WO3/BiVO4 photoanode has practically no activity (Fig. 2). This was a consequence of the high kinetic barrier of water oxidation catalysis at BiVO4 catalytic centres, which requires a higher potential to perform the reaction and avoid recombination phenomena. In sharp contrast, the specially designed Ru-WOC was able to perform the catalytic reaction at potentials as low as 0.3 V vs. NHE, which was around 300 mV higher than the semiconductor capacities (E = −0.05 V vs. NHE, Fig. S5†). This value was close to those reported for the best BiVO4-based photoanodes to date, with onset potentials for water oxidation catalysis in the range of −0.2 V to 0.2 V vs. NHE. Among these, the best performing examples contain metal oxides (ruthenium, iron, cobalt, or nickel) as water oxidation catalysts on the photoelectrode surface.11,12,17,18,37–39 Few examples of photoanodes modified with molecular catalysts with lower efficiency than that of the photoanode WO3/BiVO4/CNTf/Ru-WOC in this work have been reported.40–42 Importantly, the photoelectrode reported here can perform the reaction at 510 mV below the thermodynamic potential of the water oxidation reaction at pH 7, allowing for overlap with state-of-the-art photocathodes for the overall unassisted water splitting reaction.37,39 This work highlights the promising role of robust and fast true molecular water oxidation catalysts in enhancing photoanode performance.
Summary and conclusions
The preparation of a multilayer heterostructured WO3/BiVO4 photoanode containing two layers of WO3 (compact and mesoporous) and a porous BiVO4 film has been described. The resulting photoactive material was covered by a highly conductive carbon nanotube fibre (CNTf) that was able to accumulate charge, but unable to perform the water oxidation reaction. Finally, the surface of the CNTf was functionalized with a ruthenium complex (Ru-WOC) modified with pyrene groups to form π–π stacking bonds. The Ru-WOC takes the charge accumulated on the CNTf and rapidly transfers it to the solution by performing the water oxidation reaction. The system works at low potentials, where bare WO3/BiVO4 shows no activity in the oxygen evolution reaction. However, at high potentials, the beneficial influence of the CNTf/Ru-WOC interfaces is not sufficient to overcome the good performance of bare WO3/BiVO4. These results have been rationalized using EIS experiments, which showed the advantage of using a highly active molecular catalyst to trigger the water oxidation reaction at 510 mV below the thermodynamic potential of the reaction. Further improvements of the current molecule-based photoanode can be focused on increasing the catalyst loading, which should lead to enhanced current densities at low potentials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MINECO and FEDER (CTQ2016-80058-R, CTQ2015-73028-EXP, SEV 2013-0319, ENE2016-82025-REDT, CTQ2016-81923-REDC), and AGAUR (2017-SGR-1631), are gratefully acknowledged for providing financial support. The project leading to this application has received funding from the European Union’s Horizon 2020 research and innovation programmes under the ERC-STG 678565 and the Marie Skłodowska-Curie Grant Agreement No 705723. Dr Marcel Risch is gratefully acknowledged for providing and helping with experimental setup.Notes and references
- N. S. Lewis, Science, 2016, 351 Search PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2016, 16, 70 CrossRef PubMed.
- X. Sala, I. Romero, M. Rodríguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842 CrossRef PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088 RSC.
- H. L. Tan, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2017, 5, 16498 Search PubMed.
- S. S. M. Bhat and H. W. Jang, ChemSusChem, 2017, 10, 3001 CrossRef PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef PubMed.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044 RSC.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990 CrossRef PubMed.
- Y. Kuang, Q. Jia, H. Nishiyama, T. Yamada, A. Kudo and K. Domen, Adv. Energy Mater., 2016, 6, 1501645 CrossRef.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781 Search PubMed.
- X. Shi, I. Herraiz-Cardona, L. Bertoluzzi, P. Lopez-Varo, J. Bisquert, J. H. Park and S. Gimenez, Phys. Chem. Chem. Phys., 2016, 18, 9255 RSC.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240 RSC.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186 CrossRef PubMed.
- Y. Ma, A. Kafizas, S. R. Pendlebury, F. Le Formal and J. R. Durrant, Adv. Funct. Mater., 2016, 26, 4951 CrossRef.
- C. Zachaus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712 RSC.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786 CrossRef PubMed.
- J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard and A. Llobet, Angew. Chem., Int. Ed., 2016, 55, 15382 CrossRef PubMed.
- P. Garrido-Barros, C. Gimbert-Suriñach, D. Moonshiram, A. Picón, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907 CrossRef PubMed.
- R. Matheu, I. A. Moreno-Hernandez, X. Sala, H. B. Gray, B. S. Brunschwig, A. Llobet and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 11345 CrossRef PubMed.
- V. Cristino, S. Marinello, A. Molinari, S. Caramori, S. Carli, R. Boaretto, R. Argazzi, L. Meda and C. A. Bignozzi, J. Mater. Chem. A, 2016, 4, 2995 Search PubMed.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525 CrossRef.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef PubMed.
- M. Wang, Y. Yang, J. Shen, J. Jiang and L. Sun, Sustainable Energy Fuels, 2017, 1, 1641 Search PubMed.
- L. Francàs, C. Richmond, P. Garrido-Barros, N. Planas, S. Roeser, J. Benet-Buchholz, L. Escriche, X. Sala and A. Llobet, Chem.–Eur. J., 2016, 22, 5261 CrossRef PubMed.
- E. Senokos, V. Reguero, J. Palma, J. J. Vilatela and R. Marcilla, Nanoscale, 2016, 8, 3620 RSC.
- V. Reguero, B. Alemán, B. Mas and J. J. Vilatela, Chem. Mater., 2014, 26, 3550 CrossRef.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076–7082 CrossRef PubMed.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625 Search PubMed.
- J. Bisquert, Phys. Chem. Chem. Phys., 2000, 2, 4185 RSC.
- J. Bisquert, G. Garcia-Belmonte, F. Fabregat-Santiago and A. Compte, Electrochem. Commun., 1999, 1, 429 CrossRef.
- J. Bisquert, Phys. Chem. Chem. Phys., 2003, 5, 5360 RSC.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, P.-A. Glans, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, Chem. Mater., 2014, 26, 5365 CrossRef.
- A. Moya, N. Kemnade, M. R. Osorio, A. Cherevan, D. Granados, D. Eder and J. J. Vilatela, J. Mater. Chem. A, 2017, 5, 24695 Search PubMed.
- F. Jiang, Gunawan, T. Harada, Y. Kuang, T. Minegishi, K. Domen and S. Ikeda, J. Am. Chem. Soc., 2015, 137, 13691 CrossRef PubMed.
- F. Lin, D. Wang, Z. Jiang, Y. Ma, J. Li, R. Li and C. Li, Energy Environ. Sci., 2015, 5, 6400 Search PubMed.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. van de Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959 Search PubMed.
- K. S. Joya, N. Morlanés, E. Maloney, V. Rodionov and K. Takanabe, Chem. Commun., 2015, 51, 13481 RSC.
- W. Li, S. W. Sheehan, D. He, Y. He, X. Yao, R. L. Grimm, G. W. Brudvig and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 11428 CrossRef PubMed.
- B. Liu, J. Li, H.-L. Wu, W.-Q. Liu, X. Jiang, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 18577 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00146d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |