
Carbon nanotube/PVA aerogels impregnated with PEI: solid adsorbents for CO2 capture
A. V.
Gromov
 a,
A.
Kulur
a,
J. A. A.
Gibson
b,
E.
Mangano
b,
S.
Brandani
b and
E. E. B.
Campbell
*ac
a,
A.
Kulur
a,
J. A. A.
Gibson
b,
E.
Mangano
b,
S.
Brandani
b and
E. E. B.
Campbell
*ac
aEastCHEM and School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, Scotland, UK. E-mail: Eleanor.Campbell@ed.ac.uk
bSchool of Engineering, University of Edinburgh, Edinburgh, EH9 3FB, Scotland, UK
cDivision of Quantum Phases and Devices, School of Physics, Konkuk University, Seoul, 143-701 South Korea
First published on 9th May 2018
Abstract
A series of ultra-light aerogels made of oxidized carbon nanotubes and cross-linked polyvinyl alcohol has been prepared by freeze drying of hydrogels, characterised, and tested as amine impregnated solid supports for CO2 capture. The prepared spongy aerogels have demonstrated mechanical, chemical and thermal stabilities, and are electrically conducting. Polyethyleneimine impregnated aerogels with an amine content of 75–83% demonstrated CO2 capacity values ≥3.3 mmol g−1 in a dilute gas stream, which makes the prepared aerogels highly promising supports for amine impregnation in carbon capture applications.
1. Introduction
The concentration of CO2 in the atmosphere has increased from 340 ppm in 1980 to 402.8 ppm in 2016.1,2 According to generally accepted climate change scenarios, very serious environmental consequences can be expected if, as forecast, the atmospheric level of CO2 continues to rise above 450 ppm.3,4 An overview of the global carbon budget indicates that anthropogenic CO2 emission from fossil fuels, currently at ca. 9.9 pg C per year,1,5 continues to be the main source of emissions. The total level of emissions, including land-use emissions (1.3 ± 0.7 pg C per year), far exceeds the capacity of natural CO2 sinks of the planet in the ocean (2.4 ± 0.5 pg C per year) and on land (3.0 ± 0.8 pg C per year).1 According to international efforts to address climate change consolidated by the Tokyo Protocol (1997) and the Paris agreement (2016), carbon capture and storage/utilization (CCS) is one of the important mitigation strategies for limiting and/or reducing the levels of atmospheric CO2 in the medium-term.The current technologies for CCS, mainly amine scrubbers, can be efficiently implemented at large point sources of CO2 emission. Pre-combustion capture is typically associated with coal-fired integrated gasification combined cycle plants, i.e. conversion of coal to H2, CO and CO2, where the concentration of CO2 is ∼30%. For existing pulverized-coal, oil or gas fired plants, post-combustion CO2 capture technologies are required, where CO2 is to be removed from the typically dilute (<15% by volume) flue gas. Among the current trends in CCS there is a shift from absorption processes of CO2 capture by liquid (aqueous) amines to CO2 adsorption by solid porous materials. Separation by solid adsorbents may provide an energy benefit of up to 50% on the adsorbent regeneration with respect to the energy penalty in the regeneration step in the liquid amine based process.6–8 It is considered that for solid sorbents to be competitive with the existing MEA scrubbing system, the CO2 working capacity must be in the range of 3–4 mmol of CO2 per gram of sorbent.8,9
Solid porous materials for CCS that have been tested within the last decade can be classed into two large groups: (i) physisorbents (zeolites, porous carbons, and metal–organic frameworks) and (ii) porous supports grafted/impregnated with various bases (mainly various polyethyleneimines). Impregnation of porous carbon supports with polyamines completely shifts the CO2 adsorption mechanism to chemisorption.10 Due to the increased heat of CO2 adsorption on amine impregnated or functionalised porous materials, these materials demonstrate significantly higher selectivity11–14 with respect to CO2 when compared to physisorbents. This, however, also implies that the heat of desorption needed to release the CO2 and regenerate the sorbents is also high. Porous carbon supports have advantages over the more extensively investigated silica supports due to the possibility of incorporating an electrical swing process, making use of the electrical properties of the materials. The adsorption capacity is typically proportional to the amount of loaded amine, although the adsorption efficiency with respect to the amount of amino groups present is highly dependent on the surface morphology, pore size and available pore volume, Vtot, of the porous substrate.10 In earlier work, we observed a significant increase of CO2 uptake, reaching a value of 2.3 mmol g−1 at a CO2 partial pressure of 0.1 bar,15 when the pore size and, in particular, the pore volume of mesoporous carbons impregnated with polyethyleneimine (PEI) increased.
Impregnation of porous substrates with liquid amines results in a decrease of the free pore volume of the adsorbents. Analysis of the literature data of amine impregnated porous species demonstrates that the CO2 capacity and efficiency of amine utilization drop when the volume of amine used for filling the pores approaches the total available pore volume, Vtot, of the original substrate10,12,16 as a result of limited diffusion of CO2 gas into the bulk of the adsorbent. Typical values of Vtot for porous carbon substrates produced via resorcinol–formaldehyde condensation are in the range of 2–3.5 cm3 g−1 (ref. 13, 17–19 and 20) (corresponding to a material porosity of 80–87%), although values of 5.35 cm3 g−1 (ref. 21) and 6 cm3 g−1 (ref. 22) (92% porosity) were also reported when special templating procedures were applied. This implies that for typical highly porous carbon substrates the maximum amount of amine which can be used for impregnation will not exceed 75–80% of the total weight of the adsorbent when all pores in the substrate are filled.
A promising class of materials which can provide larger values of internal volume available for filling with organic polyamines are aerogels.23 Aerogels are ultralight materials with very high values of internal pore volume. Sol–gel chemistry methods result in cross linked hydro(organo)gels which, after drying and thermal annealing, were reported to produce aerogels with hierarchical pore structures.24,25 Resorcinol and formaldehyde are commonly used precursors in the synthesis of all-carbon aerogels.24 Building on this synthetic approach recent studies have reported the development of highly porous composite aerogels that incorporate carbon nanotubes (CNTs) or graphene into the structure.26–29 Carbon nanotube based aerogels were reported27 to have an extremely high pore volume and shown to be effective for absorption of organic28,30 and inorganic31 liquid spills. Cross-linking between carbon nanotubes and graphene flakes (GrOx) can occur via gelation of the carbon material with polyethylene oxide (PEO), PEI, surfactants, metal salts etc.,32 hydrothermal treatment33 or even without additional cross-linking species just using carbon nanotubes as spacers during lyophilisation.28 It has been shown that the gelation of carbon nanotubes/carbon nanofibres and graphene oxide may be assisted by small amounts of cross-linked PVA, which serves as a scaffold and provides a larger pore size and available pore volume in the resultant gel.30
These materials provide considerable scope for optimizing the pore structure to maximize the efficiency of CO2 uptake as well as being possible to synthesize in the form of monoliths. The advantages of using CNTs/graphene in such structures can include enhanced internal pore volumes, optimized electrical properties and a structure with greater mechanical stability.27
In this work we provide the first study, to our knowledge, of the application of carbon nanotube/PVA aerogels as low weight solid supports for incorporating liquid amines in order to increase CO2 uptake from dilute gas streams. Aerogels were prepared from various ratios of oxidised CNTs and PVA by freeze drying. Their mechanical and electrical properties were determined and their stability with respect to organic and inorganic solvents was tested. The effect of impregnating aerogels with polyethyleneimine was studied and the CO2 uptake was determined under dry conditions at a 0.1 bar CO2 partial pressure which is in the range of partial pressures found in the flue gas of fossil fuel power stations. The material is shown to be competitive with the best CO2 capture materials under conditions of atmospheric pressure and low CO2 partial pressure with the additional advantage of allowing the development of an electrical swing desorption process due to the possibility of rapid ohmic heating.
2. Experimental details
2.1. Aerogel synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1.
:2:1.
Analytical grade methanol, sulfuric acid (>95%) and nitric acid (70%) were purchased from Fisher Scientific.
:1 v/v ratio. The oxidised carbon nanotubes were then isolated by vacuum filtration, and washed in sequence with dist. water, 3% NaOH, water, 3% HCl, water and methanol. As a result of oxidation a weight loss of 3–4% was observed. XPS analysis of the oxidised carbon nanotubes showed 9 at% of oxygen in the material in comparison to 0.5 at% O in the purchased carbon nanotubes. The oxidation of carbon nanotubes was an essential step to ensure the formation of a stable aqueous dispersion.
:1, 1:2 and 1:3, i.e. 20 or 40 or 60 mg PVA were added to 20 mg of CNTs. The beaker was placed in an ultrasonic bath and sonicated for 30 minutes and then stirred for 15 min at room temperature; this sequence was repeated at least 3 times until a stable dispersion was formed. Glutaraldehyde (25% in H2O), 20 μl for each 20 mg of PVA, and HCl (2.5%), 35 μl for each 20 mg of PVA, were added to the mixtures, which were stirred at room temperature for an additional 30 minutes. The stir bars were removed and the CNT:PVA dispersions were left to gelate overnight at 50–55 °C on a hot plate. The prepared hydrogels were placed in a cold bath at −20 to −25 °C until complete freezing, and then kept at a temperature of −20 °C for an additional 30 minutes. The frozen gels were dried under vacuum, keeping the temperature at −20 °C for the first 15 minutes and then allowing to it to rise to room temperature; the complete removal of water and formation of aerogels was considered to have occurred when the residual pressure was 2–3 × 10−1 mbar and required overnight pumping.
Under these synthesis conditions a small reduction of the aerogel volume with respect to the original volume of the slurry before freezing was observed for all series of produced aerogels. The final aerogel volumes were measured to be 4.25 ± 0.35 ml for all CNT:PVA ratios for the ‘5 ml’ series and 2.36 ± 0.15 ml for the ‘3 ml’ series of samples. There was no systematic effect of CNT:PVA ratio on the aerogel volume.
The density of the aerogels was extremely low with calculated densities ranging from ∼10 mg cm−3 (CNT-to-PVA ratio 1:1) to ∼25 mg cm−3 (CNT-to-PVA ratio 1:3) for ‘5 ml’ samples, and from 20 to 35 mg cm−3 for ‘3 ml’ samples respectively.
The physical characteristics of the obtained aerogels are presented in Table 1.
| Series | CNT:PVA wt ratio |
Volume (ml) | Weight (mg) | Density (mg ml−1) | Porosity, % (est.b) |
|---|---|---|---|---|---|
| a The presented data show average values with standard deviation tolerance obtained from 3 to 5 samples of each kind. b Porosity was estimated from the average measured aerogel monolith volume and volumes of CNTs (density 1.9 g ml−1)34 and PVA (density 1.19 g ml−1). | |||||
| ‘5 ml’ | 1:1a |
4.2 ± 0.3 | 43 ± 3 | 10 ± 1 | 99.35 ± 0.05 |
| 1:2a |
4.3 ± 0.4 | 66 ± 5 | 15 ± 3 | 99.0 ± 0.1 | |
| 1:3a |
4.3 ± 0.3 | 87 ± 7 | 20 ± 3 | 98.6 ± 0.07 | |
| ‘3 ml’ | 1:1a |
2.3 ± 0.1 | 46 ± 1 | 20.3 ± 0.6 | 98.8 ± 0.03 |
| 1:2 |
2.4 | 66 | 28 | 98 | |
| 1:3 |
2.5 | 84 | 34 | 97.5 | |
2.2. Aerogel characterisation
 | ||
| Fig. 1 Photograph of a typical 1:1 CNT:PVA aerogel and the set-up used to determine mechanical stability and compressibility of aerogel materials: left – aerogel monolith before the test, centre – aerogel monolith under a load of 50 g and right – aerogel 5 min after removing the load. | ||
The pore volume available for impregnation was also estimated by filling the aerogels with methanol. The aerogels were placed on a PTFE platform in a test tube filled with excess of methanol, so that the methanol level was slightly below the platform. The capped test tube was inclined until the sample on the platform was in contact with the methanol. When it was visually determined that aerogels were completely filled with methanol, the test tube was returned to the vertical position and gently shaken to remove any excess methanol from the outside of the aerogel and the sample was removed from the test tube. The amount of the absorbed methanol was measured by the test tube weight difference before and after the soaking procedure. The available internal volume of the aerogel was considered to be equal to the volume of methanol absorbed by the aerogel.
3. Results and discussion
There have been many reports on the use of mesoporous substrates, mostly highly porous carbon and silica materials, impregnated with organic polyamines.19–21,39–42 The amount of amine that can be incorporated into such substrates is limited by the total pore volume. The typical measured values of total pore volume for porous carbons prepared by a templating method or by formaldehyde–resorcinol sol–gel condensation (‘carbon aerogels’) are in the range of 1–3.5 ml g−1,13,18,43 although pore volumes as high as 5.3 ml g−1 (ref. 21) and 6 ml g−1 (ref. 22) have been reported.Ultra-light aerogels (foams) made of gelated organic polymers with a carbon filler, such as the materials of interest here, show much higher values of total internal volume, with reported porosity values >98%.27,30,44 For the purposes of selective carbon capture, requiring elevated temperatures of adsorption/desorption within the range 75 to 90 °C, the ultra-light aerogels (sponges) should exhibit good thermal stability. In addition, they require high chemical stability towards the organic amines used for impregnation and high mechanical stability to enable the use of monolithic materials to reduce the pressure drop in a practical carbon capture process. The materials synthesised from PVA/CNT mixtures reported here satisfy all these requirements: (i) crosslinked PVA forms stable hydrogels in aqueous solutions, (ii) PVA has a high melting temperature (185 °C) and (iii) PVA does not react with amines. Additionally, the presence of hydroxyl groups in the composite was considered to provide good wetting with amines and also could facilitate the reaction of amines with CO2via the formation of alkyl carbonate salts45,46 or carbamate esters.47
The lyophilisation technique makes it possible to tune the resultant density of the aerogel by varying the initial concentrations of CNTs and PVA, i.e. by changing the amount of water in the hydrogel. During synthesis, the mass of CNTs was kept close to 20 mg whilst the quantity of PVA was varied in order to synthesise aerogels with the desired CNT:PVA ratio. The dispersions of CNTs in PVA solutions formed gels after 5–10 h of heating at 50–55 °C with negligible phase separation. The synthesized aerogels were mechanically stable. The aerogels with 1:1, 1:2 and 1:3 wt ratios of CNT:PVA had dry masses of approximately 40, 60 and 80 mg respectively, i.e. freeze drying removed practically all the water from the hydrogel.
The presence of CNTs is important for hydrogel formation. Pure PVA solutions at equivalent concentrations of 0.4–1.6% (20–80 mg PVA in 5 ml), which is below the critical PVA gelation concentration in water,48–51 after adding the cross-linking agent and heating at 50–55 °C for 24 h, became cloudy, but did not form bulk hydrogels. Freeze drying of solutions of pure PVA after cross-linking did result in stable aerogels, in agreement with the literature.49,52,53 All aerogel samples made of pure PVA underwent significant shrinkage during freeze-drying, the final volumes being 2.6 ml for the 20 mg sample (density 7.6 mg ml−1), 2.9 ml for the 40 mg sample (density 15.6 mg ml−1), 3.3 ml for the 60 mg sample (density 20.6 mg ml−1) and 3.05 ml for the 80 mg sample (density 26.8 mg ml−1).
An attempt to carbonise the CNT:PVA aerogels at 1000 °C in Ar flow did not produce rigid all-carbon aerogels and resulted in decomposition of the PVA component of the aerogel monolith and recovery of decarboxylated carbon nanotube powder.
3.1. Aerogel appearance
The prepared aerogel monoliths are black spongy materials in cylindrical forms with a diameter of ∼1.8 cm (corresponding to the inner diameter of the slurry container) and a height of 1.5–1.9 cm for an initial slurry volume of 5 ml. Fig. 1 shows a photograph of a typical aerogel sample prepared from a gelated 5 ml slurry.The internal pore structure of the materials was investigated by scanning electron microscopy (SEM). A selection of images is presented in Fig. 2. From the low magnification images (top row), a well-defined macroporous structure was observed, with large micrometre sized pores (up to a few tens of μm) distributed across the surface. At higher levels of magnification (bottom row), it is clearly seen that CNTs were well distributed throughout the polymer phase. From an examination of the SEM images it is also possible to conclude that the increase of the polymer fraction in the aerogel from 50% (1:1 CNT:PVA ratio) to 75% (1:3 ratio) resulted in a denser local distribution of the material and some growth of the size of internal voids.
 | ||
| Fig. 2 SEM images of aerogel monoliths prepared with CNT:PVA wt ratios of 1:1 (left column), 1:2 (middle) and 1:3 (right). The scale bar for the top images is 200 μm and for the bottom image the scale bar is 200 nm. | ||
3.2. Characterisation of the aerogel pore structure
Liquid nitrogen adsorption isotherms of aerogels prepared with different PVA contents are shown in Fig. 3. The isotherm curves are all similar and correspond to type III isotherms according to the IUPAC classification,54 typical for macroporous materials with pore sizes close to 100 nm.55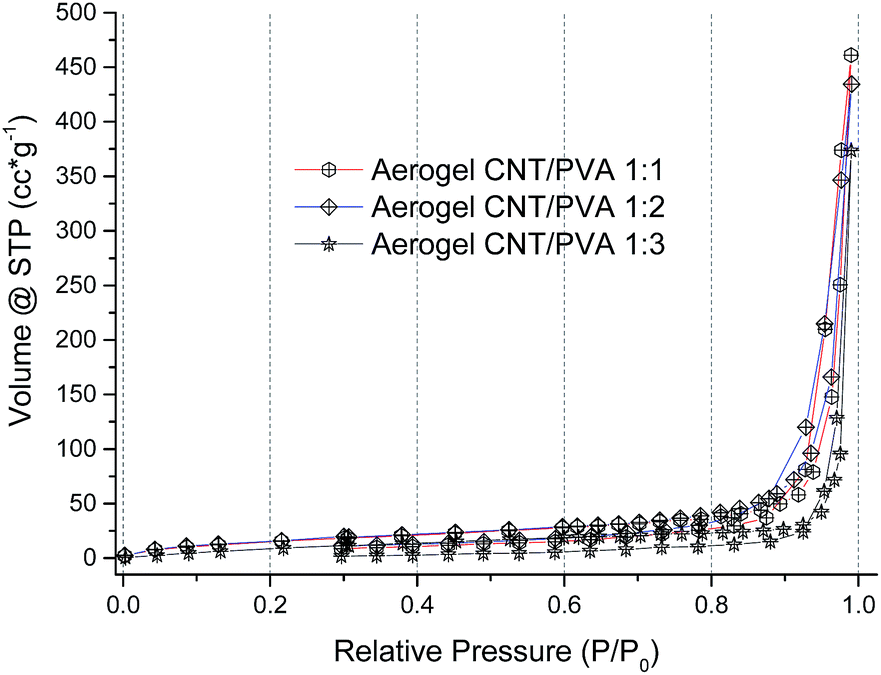 | ||
| Fig. 3 N2 adsorption isotherms at 77 K of aerogels with CNT:PVA weight ratios of 1:1, 1:2 and 1:3. | ||
The surface area of aerogels estimated from the N2 adsorption isotherms was 59 m2 g−1 for 1:1 species, 43 m2 g−1 for 1:2 species and 62 m2 g−1 for 1:3 species. It is known that gas sorption isotherms (Brunauer–Emmett–Teller method) are useful for determining the total specific surface areas of powders and porous materials and provide a reasonable estimate of pore volumes and pore size distributions for micro and mesoporous materials.56–58 However this technique is not suitable for the characterisation of the pore structure and pore size distribution in macroporous materials.59 The volume of macropores in the aerogel samples was estimated by measuring the amount of methanol that could be absorbed by the aerogel when it was put in contact with the liquid. The volume of the absorbed methanol was found to be ∼75% of the measured volume of aerogel cylindrical monoliths with 1:1 and 1:3 CNT:PVA ratios, and ∼70% for 1:2 species, i.e. an aerogel monolith of 4 ml absorbs ca. 3 ml of methanol. Thus, the volume available for filling with liquid in the prepared CNT:PVA aerogel sponges ranged from 35 ml g−1 for species with a CNT:PVA ratio of 1:3 to >70 ml g−1 for 1:1 species.
3.3. Compression test
A simple setup for testing the mechanical properties of CNT:PVA aerogels is shown in Fig. 1. After removing the weight, all tested aerogels recovered to >90% of their original size within 5 minutes. The results of the compression test are presented in Fig. 4. The stiffness of the aerogels grows with an increase of the PVA content in the material, corresponding to an increase of the material density. These simple experiments provided an estimation of the compressive Young's modulus values of 5 ± 0.4 kPa for aerogels with a CNT:PVA wt ratio of 1:1 and density ∼10 mg ml−1, 7.3 ± 1.1 kPa for samples with a ratio of 1:2 and density ∼15.5 mg ml−1, and 14.3 ± 1.9 kPa with a CNT/PVA wt ratio of 1:3 and density ∼21 mg ml−1. These values are consistent with the published Young's modulus data for other ultra-light foams, e.g. 33 kPa for a polyurethane foam with a density of 16 mg ml−1 (ref. 60) and >80 kPa for polyurethane foams with a density of 90 mg ml−1.61
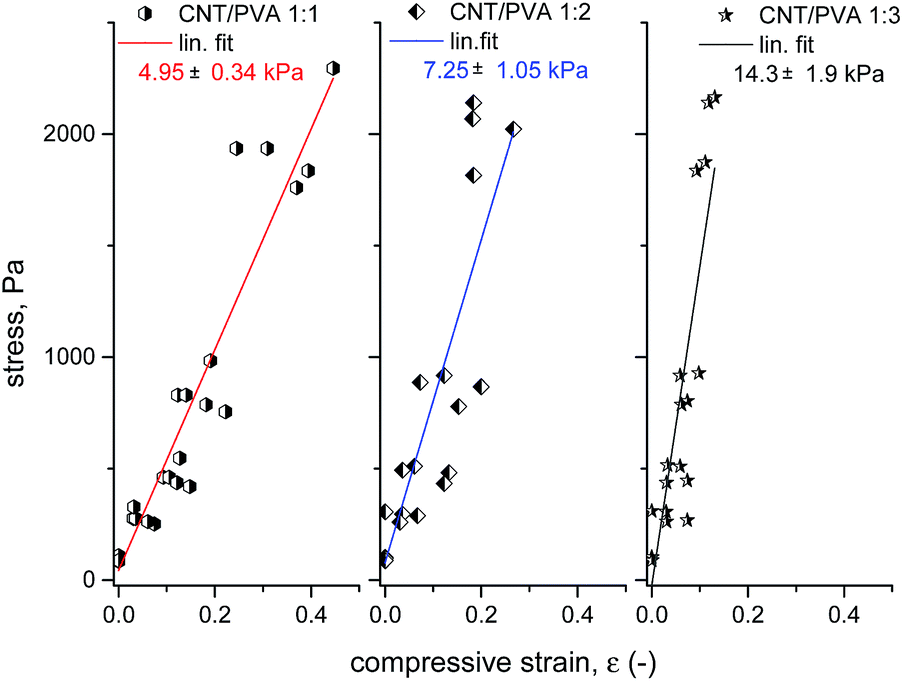 | ||
| Fig. 4 The results of the CNT:PVA compression test showing stress–strain (-) data points and their linear fit (assuming elastic behaviour of the material). Left – CNT:PVA ratio 1:1, centre – CNT:PVA ratio 1:2 and right – CNT:PVA ratio 1:3. The Young's modulus of the samples was derived from the linear fit slope; fit correlation coefficients (Pearson's r) for the presented data are in the range of 0.85–0.95. | ||
3.4. Electrical measurements
In contrast to pure PVA aerogels, all CNT containing aerogels were electrically conducting. The I–V characteristics of aerogels were measured using a 4-probe scheme (Fig. 5a) and electrical resistivity was calculated using the formula:38where ρ is the bulk resistivity of the material in Ω cm, V is the measured voltage, I is the electrical current, A is the surface cross-section and d is the distance between probe leads.
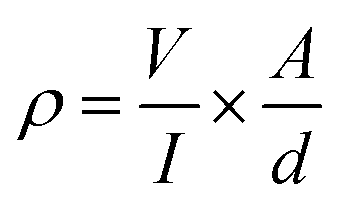
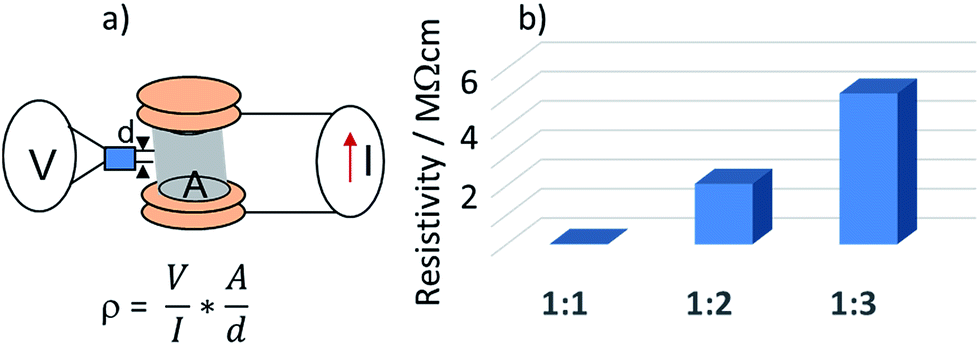 | ||
| Fig. 5 (a) schematic drawing of the experimental set-up for measuring electric resistivity of aerogel moulds; (b) resistivity of the aerogels with different CNT:PVA weight ratios. | ||
The resistivity was measured in the low power region; when the power applied to the aerogel grew beyond the values where the materials were subjected to ohmic heating (>0.5 W g−1 for all species), the resistivity exhibited a non-linear behaviour. The summary plot with resistivity of the prepared CNT:PVA aerogels is shown in Fig. 5b.
As expected, the resistivity of the aerogels dramatically increased with a decrease of the CNT content, ranging from 19 kΩ cm for materials with a CNT:PVA ratio of 1:1; 2 MΩ cm for a CNT:PVA ratio of 1:2 and 5 MΩ cm for species with a CNT:PVA ratio of 1:3.
Non-impregnated aerogel samples with CNT:PVA ratios of 1:1 and 1:2 could easily be heated to temperatures above 70 °C, and demonstrated a resistive heating dependence of 100–145 K g W−1. Aerogels with a CNT:PVA ratio of 1:3 demonstrated a heating rate of 280–370 K g W−1, although due to instrument limitations (max. compliance voltage of 200 V) we were unable to reach temperatures above 40 °C for these samples.
3.5. Impregnation and CO2 capture
In order to investigate the behaviour of aerogels during the wet impregnation procedure10,16,62 as well as to provide an estimate of the available internal volume, the uptake of methanol by the aerogels was investigated (Section 3.2).The wet impregnation procedure, typically used for porous carbons10,18 and silicas,62 where the porous material powder is mixed with the calculated amount of amine dissolved in an arbitrary amount of solvent followed by solvent evaporation, cannot be applied directly to aerogel sponges because a uniform distribution of amine inside the aerogel cannot be achieved. Therefore, the impregnation method applied here consisted of complete absorption of a measured volume of amine solution into the aerogel sponge, so that the total volume of the absorbed liquid was roughly equal to 85% of the aerogel pore volume as estimated by methanol uptake. This amount of liquid was found to provide complete wetting of the aerogel throughout the bulk, but avoided excessive amine concentration on the surface of the monolith during the evaporation step.
The applied vacuum drying scheme with slow methanol evaporation allowed significant shrinkage of the impregnated aerogels to be avoided. Fig. 6 shows the aerogel species before and after impregnation with 3-fold, 5-fold and 10-fold amounts of PEI600; the dimensions of impregnated aerogel species do not change significantly.
 | ||
| Fig. 6 Photographs of CNT:PVA (1:3 wt ratio): left – pieces of aerogel cut from the monolith before impregnation and right – the same pieces of aerogel after impregnation with 3-fold, 5-fold and 10-fold amounts of PEI600. | ||
This approach allowed the amount of added amine to be controlled within a few percent and provided uniform distribution of amine in the substrate. Fig. 7 shows SEM images of aerogels impregnated with various amounts of PEI. It is obvious that species containing 75% (×3) and 83.3% (×5) of amine still retain the 3D morphology inherent to the initial aerogel. Further increase of the amine content in the material (amine to substrate ratio ×10 and amine content in the sorbent 91%) resulted in the formation of continuous layers of amine inside the porous substrate.
 | ||
| Fig. 7 SEM images of aerogel (CNT:PVA ratio 1:1) (a) before impregnation and (b) after impregnation with a 3-fold amount of PEI (×3), (c) 5-fold amount of PEI (×5) and (d) 10-fold amount of PEI (×10). | ||
The volume available for impregnation in CNT:PVA aerogels (35–70 ml g−1) significantly exceeds the values of 2.5–6 ml g−1 that have been reported for typical porous materials used as supports for liquid amines.13,20,22
This implies that, contrary to typical highly porous carbon substrates with complete pore filling (blocking) at 70–80% of the amine content, leading to a decrease in amine utilization efficiency,15,18 the maximum amine loading in the impregnated aerogels can be higher than 85% wt without blocking the pores, thus, providing gas diffusion through the bulk of the adsorbent.
Note that the theoretical capacity for CO2 uptake with polyethyleneimine of the general formula H–(NH–CH2CH2)nNH2 with n ≈ 13.6 (PEI600) under dry conditions with the formation of the carbamate salt according to the generally accepted mechanism63 (Scheme 1) is ca. 12.2 mmol g−1.
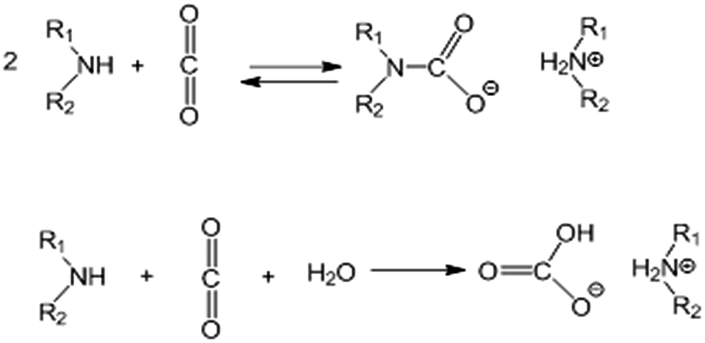 | ||
| Scheme 1 The generally accepted interaction mechanism between CO2 and amino groups: top – carbamate salt formation under dry conditions; bottom – competitive carbonate formation in the presence of water. | ||
For materials with a substrate impregnated with a 3-fold wt amount of amine (corresponding to the amine content of 75% in the material ) the theoretical CO2 capacity is 9.15 mmol g−1, for ×5 impregnation (83.3%) the theoretical CO2 capacity is 10.17 mmol g−1 and for ×10 impregnation (91%) it is 11.1 mmol g−1 (Fig. 8), assuming 100% utilization of the NH units, i.e. without taking into account the reverse reaction at 75 °C.
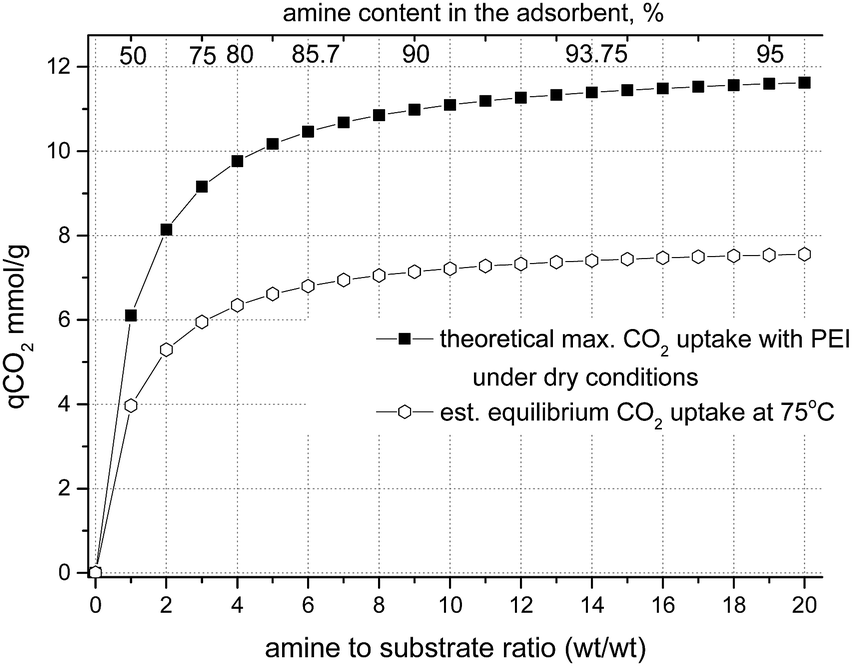 | ||
| Fig. 8 Theoretical maximum CO2 uptake (solid squares) and estimated equilibrium CO2 uptake expected at 75 °C (hollow circles) by porous species impregnated with PEI600 under dry conditions as a function of amine to substrate wt ratio. | ||
Therefore, a threefold increase of the amount of amine in the substrate (from ×3 to ×10) may result only in an ∼20% gain in CO2 uptake under dry conditions. It was therefore decided to focus on the adsorption properties of aerogels impregnated with 3- and 5-fold amounts of polyethyleneimine. It is necessary to take into account that the reaction of the formation of the carbamate salt under dry conditions is reversible and at a temperature of 75 °C the real uptake values will not reach the predicted theoretical uptake.
For example, for solid ammonium carbamate the rates of formation and decomposition reactions are the same at ∼70 °C (ref. 64) (Gibbs energy change of the reaction, ΔG = 0). Fernandes et al.65 investigated the formation of carbamate salts for various primary and secondary amines in solution by 1H NMR, measured the equilibrium stability (formation) constants and determined the standard molar enthalpy and entropy values for this reaction. We calculated equilibrium constants for carbamate salt formation at 75 °C using data from Fernandes et al.65 and obtained the values of Kcarbamate = 5.2 for monoethanolamine (MEA), ∼1.5 for secondary amines (morpholine and piperazine), and 0.5 for ammonia. These values correspond well to the analysis of Gupta et al.,66 who showed stability constants at 345 K of 2.7 and 1.55 for carbamate salts of MEA and diethanolamine respectively, and a decomposition equilibrium constant of ∼1 for ammonium carbamate at 70 °C.64 These equilibrium constants correspond to conversion to carbamates of ∼60% of secondary amines and ∼75% of primary amines under equilibrium conditions. The expected CO2 equilibrium uptake by a substrate impregnated with polyethyleneimine with ∼25% of primary amino groups and ∼50% of secondary amino groups will be ∼65–70% of the theoretical value (Fig. 8). Under adsorption conditions with excess of CO2, the carbamate salt formation reaction (Scheme 1) will be shifted to the products, and, therefore, the real uptake values will be in between the theoretical maximum uptake and the equilibrium uptake values.
CO2 capacities of aerogels with different CNT:PVA ratios, impregnated with 3-fold and 5-fold amounts of PEI600, measured by TGA at 75 °C, 0.1 bar CO2 under dry conditions, are presented in Table 2.
:PVA aerogels loaded with PEI600, measured at 75 °C, 0.1 bar dry CO2. The efficiency of the amine utilization is calculated as the ratio of the moles of CO2 taken up by the adsorbent to the number of moles of amino groups available for adsorption, q(CO2): [0.5 × mol(N)]
| CNT:PVA ratio in substrate |
PEI600 load M amine/M substrate | Av. qCO2@4 h mmol g−1 | q(CO2)/[0.5 × mol(N)] |
|---|---|---|---|
| 1:1 |
×5 | 3.3 ± 0.3 | 0.33 ± 0.03 |
| 1:1 |
×3 | 3.25 ± 0.3 | 0.36 ± 0.03 |
| 1:2 |
×5 | 2.6 ± 0.4 | 0.26 ± 0.04 |
| 1:3 |
×5 | 2.6 ± 0.4 | 0.26 ± 0.04 |
The CO2 capacities of PEI impregnated CNT:PVA aerogel monoliths are very competitive with respect to those of other porous supports impregnated with branched PEI600 measured with the same method (Table 3). Impregnated aerogels with a CNT:PVA wt ratio of 1:1 demonstrated on average better values of CO2 capacity per gram than more dense species with a higher PVA content in the aerogel (3.3 mmol g−1vs. 2.6 mmol g−1). The volumetric uptake at ca. 0.3 mmol CO2 per ml of sorbent is a about a factor of two or three less than that reported for impregnated carbon black materials at a partial pressure of 1 bar.16 The efficiency of utilization of amino groups under dry conditions for 1:1 samples was 33–36%, which was significantly higher than ≤25% efficiency that we observed earlier for mesoporous carbon impregnated with PEI600.10 CO2 uptake curves for aerogels impregnated with polyethyleneimine at 75 °C, 0.1 bar CO2 show fast initial sorption followed by a second, slower process (Fig. 9a), which is typical for porous substrates impregnated with organic amines (see references given in Table 3).
| Porous support | PEI 600 content, % | P CO2, bar | T, °C | q CO2, mmol g−1 | Ref. |
|---|---|---|---|---|---|
| Hexagonal mesoporous silica | 65 | 1 | 75 | 4.18 | Chen67 |
| Silica microcapsules | 83 | 1 | 75 | >5 | Qi68 |
| Silica foam | 70 | 0.1 | 75 | 2.3 | Subagyono69 |
| MCM-48 | 70 | 1 | 80 | 3.1 | Sharma12 |
| Carbon black | 50 | 1 | 75 | 3.1 | D. Wang16 |
| Hierarchical porous silica | 70 | 1 | 75 | 4.1 | J. Wang18 |
| Silica gel | 50 | 0.1 | 75 | 2.9 | Zhang70 |
| Mesoporous carbon spheres | 50 | 1 | 75 | 2.9 | M. Wang20 |
| Mesoporous carbon | 73 | 0.1 | 75 | 2.3 | Gibson15 |
| CNT:PVA aerogel sponge |
75, 83 | 0.1 | 75 | 3.25 ± 0.3, 3.3 ± 0.3 | This work |
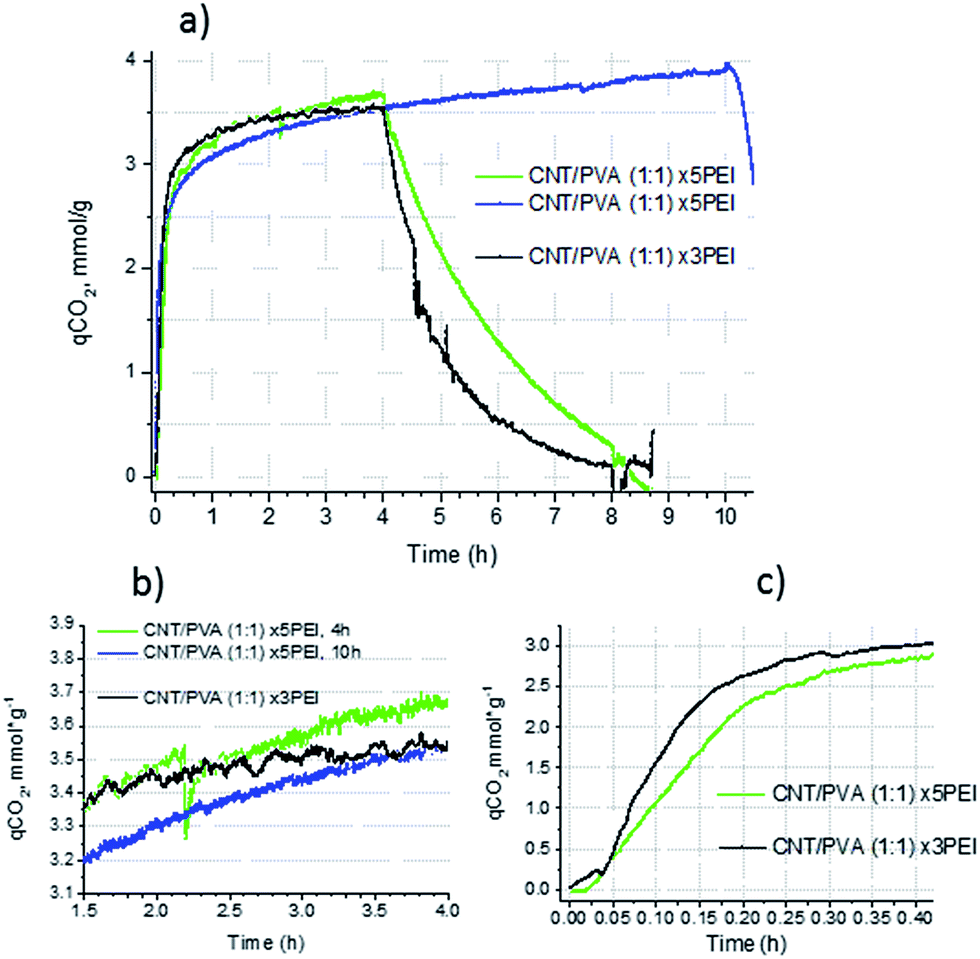 | ||
| Fig. 9 (a) CO2 uptake TGA curves of CNT–PVA aerogels with PEI600 loadings ×3 (black), ×5 (green), adsorption time 4 h at 75 °C, 0.1 bar CO2 and ×5 (blue), adsorption time 10 h; (b) the extended region demonstrating adsorption behavior in the range 1.5–4 h, and (c) the extended region demonstrating an uptake curve behavior in the initial fast sorption region. | ||
The second, slow sorption stage is associated with slow diffusion of CO2 through the amine and the formation of reaction products. This could be potentially improved by further optimising the pore structure and thickness of the amine layer or by using other schemes to increase the CO2 diffusivity through the layer of PEI, e.g. by using various additives.36,70,71
The CO2 uptake behaviour of aerogels loaded with PEI600 naturally depends on the amount of amine in the substrate. Fig. 9a shows CO2 uptake curves for aerogels with a CNT:PVA ratio of 1:1 loaded with 3- and 5-fold weight amounts of PEI600. Although the CO2 capacity values after 4 h of sorption are fairly close (3.66 and 3.54 mmol g−1 for ×5 samples vs. 3.55 mmol g−1 for ×3 sample) the slope of the curve for the ×5 sample is steeper indicating that the process is still far from equilibrium after 4 h.
When the CO2 uptake for a ×5 sample (different batches) was measured for 10 h the slow sorption step after 3 h of experiment exhibited a behaviour close to linear with a CO2 uptake rate of 0.075 mmol g−1 h−1. Fig. 9b shows the extended region of the CO2 uptake TGA curves. For both ×5 samples, despite a slightly different total CO2 capacity, the sorption behaviour is similar, while for the ×3 sample the slope beyond 3 h is much lower, indicating that the stock of the unreacted amino groups is small and the reaction of CO2 with amino groups may be close to equilibrium at 75 °C.
At a reaction time of 4 h the amine utilization efficiency is slightly higher for the ×3 PEI sample (0.36 vs. 0.33 for the ×5 PEI sample). This difference is significantly higher for the initial fast step and after the first 10 min (0.167 h) of sorption (Fig. 9c), where the ×3 PEI sample demonstrated a CO2 capacity of 2.46 mmol g−1 with an amine utilization efficiency of 27%, i.e. 0.2 g PEI per g of the sorbent reacted with CO2, compared to 1.91 mmol g−1 with an efficiency of 19% for the ×5 PEI sample (0.16 g PEI per g of the sorbent). Assuming a uniform distribution of amine on the surface of the aerogel with a surface area of 60 m2 g−1, the PEI layer should have a thickness of 50 nm for the ×3 PEI species and 83.5 nm for the ×5 PEI species. In the case of 100% utilisation of amino groups for the reaction with CO2, it is possible to estimate that in the ×3 PEI sample after 10 min of exposure to CO2, only the first 14 nm of the amine layer is involved in the reaction. The access to PEI which is deeper below the surface is hindered by diffusion of CO2 through the layer of liquid reaction products. For ×5 PEI such an estimate gives a similar value of 15.5 nm of the PEI thickness involved in the reaction during the fast initial step.
4. Conclusions
A series of ultra-light aerogels made of oxidized carbon nanotubes and PVA has been prepared by freeze drying of hydrogels, characterised, and tested as amine impregnated solid supports for CO2 capture.The aerogel spongy materials were prepared with CNT:PVA wt ratios of 1:1, 1:2 and 1:3 with material bulk densities of ca. 10 mg ml−1, 15 mg ml−1 and 20 mg ml−1 respectively. The presence of carbon nanotubes was shown to be important for gelation of the CNT:PVA dispersions and for controlling the final density (volume) of aerogels. CNT:PVA aerogels have rather low surface areas (43–61 m2 g−1), but were shown to have very high values of internal volume available for filling (impregnation), 35 to >70 ml g−1, depending on the density of species.
Aerogels with all tested CNT:PVA weight ratios are mechanically stable due to good component homogeneity; the mechanical properties of the prepared aerogels are consistent with published data for other ultra-light foams and demonstrate the values of the compressive Young's modulus of 5 to 14.3 kPa.
CNT:PVA aerogels are electrically conducting; a decrease of the CNT content in the aerogel resulted in a dramatic increase of the material bulk resistivity from 19 kΩ cm for species with a CNT:PVA wt ratio of 1:1 to 5 MΩ cm for species with a CNT:PVA wt ratio of 1:3. Aerogels with CNT:PVA wt ratios of 1:1 and 1:2 are suitable for electrical swing adsorption (ohmic heating) but the 1:3 ratio materials are impractical as they require voltages >200 V for reaching temperatures feasible for solid sorbent regeneration.
Using a modified wet impregnation procedure, samples were prepared with a homogeneous distribution of amine (PEI600) within the aerogel monolith and precise dosing with an amine content of 75 to 91%, corresponding to a 3-fold (×3) to 10-fold (×10) amount of added amine with respect to the weight of the substrate. In ×3 and ×5 impregnated species the amine layer coats the initial surface texture inherent to the original aerogel, while in ×10 samples the amine layer is thick and forms a smooth and continuous surface.
PEI600 impregnated aerogels were tested under 10% CO2 partial pressure conditions in order to simulate the flue gas of a fossil fuel power plant. The obtained CO2 capacity values significantly exceed our previous results obtained with impregnated mesoporous carbons, and are competitive with the highest reported values for porous substrates impregnated with similar amines. This makes ultra-light CNT:PVA aerogel sponges very promising supports for amine impregnated solid sorbents for CO2 capture.
The value of the CO2 capacity was dependent on the structure of the carbon support; the species with the highest CNT content (CNT:PVA ratio 1:1) demonstrated on average the highest uptake values of ∼3.3 ± 0.3 mmol g−1 for ×3 and ×5 samples and an efficiency of amino group utilisation of >0.35. The analysis of TGA adsorption curves showed that the initial fast sorption step occurs during the first 10 minutes and involves the outer PEI600 layer to a depth of ≈15 nm, beyond which further CO2 uptake is limited by diffusion through the top layer of the products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed with financial support from the EPSRC FlexICCS project EP/N024613/1 which is gratefully acknowledged. To comply with RCUK requirements, the raw experimental data used in this paper can be found at DOI: 10.7488/ds/2346.Notes and references
- C. Le Quéré, R. M. Andrew, P. Friedlingstein, S. Sitch, J. Pongratz, A. C. Manning, J. I. Korsbakken, G. P. Peters, J. G. Canadell, R. B. Jackson, T. A. Boden, P. P. Tans, O. D. Andrews, V. K. Arora, D. C. E. Bakker, L. Barbero, M. Becker, R. A. Betts, L. Bopp, F. Chevallier, L. P. Chini, P. Ciais, C. E. Cosca, J. Cross, K. Currie, T. Gasser, I. Harris, J. Hauck, V. Haverd, R. A. Houghton, C. W. Hunt, G. Hurtt, T. Ilyina, A. K. Jain, E. Kato, M. Kautz, R. F. Keeling, K. Klein Goldewijk, A. Körtzinger, P. Landschützer, N. Lefèvre, A. Lenton, S. Lienert, I. Lima, D. Lombardozzi, N. Metzl, F. Millero, P. M. S. Monteiro, D. R. Munro, J. E. M. S. Nabel, S. I. Nakaoka, Y. Nojiri, X. A. Padín, A. Peregon, B. Pfeil, D. Pierrot, B. Poulter, G. Rehder, J. Reimer, C. Rödenbeck, J. Schwinger, R. Séférian, I. Skjelvan, B. D. Stocker, H. Tian, B. Tilbrook, I. T. van der Laan-Luijkx, G. R. van der Werf, S. van Heuven, N. Viovy, N. Vuichard, A. P. Walker, A. J. Watson, A. J. Wiltshire, S. Zaehle and D. Zhu, Earth Syst. Sci. Data Discuss., 2017, 2017, 1–79 Search PubMed.
- Earth's CO2, http://co2now.org/current-co2/co2-now/.
- M. Meinshausen, B. Hare, T. M. M. Wigley, D. Van Vuuren, M. G. J. Den Elzen and R. Swart, Clim. Change, 2006, 75, 151–194 CrossRef.
- J. Rockström, W. Steffen, K. Noone, Å. Persson, F. S. Chapin III, E. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. Schellnhuber, B. Nykvist, C. A. De Wit, T. Hughes, S. van der Leeuw, H. Rodhe, S. Sörlin, P. K. Snyder, R. Costanza, U. Svedin, M. Falkenmark, L. Karlberg, R. W. Corell, V. J. Fabry, J. Hansen, B. Walker, D. Liverman, K. Richardson, P. Crutzen and J. Foley, Ecol. Soc., 2009, 14(2), 32 CrossRef.
- C. Le Quéré, R. J. Andres, T. Boden, T. Conway, R. A. Houghton, J. I. House, G. Marland, G. P. Peters, G. R. van der Werf, A. Ahlström, R. M. Andrew, L. Bopp, J. G. Canadell, P. Ciais, S. C. Doney, C. Enright, P. Friedlingstein, C. Huntingford, A. K. Jain, C. Jourdain, E. Kato, R. F. Keeling, K. Klein Goldewijk, S. Levis, P. Levy, M. Lomas, B. Poulter, M. R. Raupach, J. Schwinger, S. Sitch, B. D. Stocker, N. Viovy, S. Zaehle and N. Zeng, Earth Syst. Sci. Data, 2013, 5, 165–185 CrossRef.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef PubMed.
- M. L. Gray, K. J. Champagne, D. Fauth, J. P. Baltrus and H. Pennline, Int. J. Greenhouse Gas Control, 2008, 2, 3–8 CrossRef.
- N. El Hadri, D. V. Quang, E. L. V. Goetheer and M. R. M. Abu Zahra, Appl. Energy, 2017, 185, 1433–1449 CrossRef.
- J. A. A. Gibson, A. V. Gromov, S. Brandani and E. E. B. Campbell, Microporous Mesoporous Mater., 2015, 208, 129–139 CrossRef.
- C. Chen, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2014, 31, 1919–1934 CrossRef.
- P. Sharma, I.-H. Baek, Y.-W. Park, S.-C. Nam, J.-H. Park, S.-D. Park and S. Y. Park, Korean J. Chem. Eng., 2012, 29, 249–262 CrossRef.
- J. Wang, M. Wang, B. Zhao, W. Qiao, D. Long and L. Ling, Ind. Eng. Chem. Res., 2013, 52, 5437–5444 CrossRef.
- J. A. A. Gibson, A. V. Gromov, S. Brandani and E. E. B. Campbell, J. Porous Mater., 2017, 24, 1473–1479 CrossRef.
- J. A. A. Gibson, E. Mangano, E. Shiko, A. G. Greenaway, A. V. Gromov, M. M. Lozinska, D. Friedrich, E. E. B. Campbell, P. A. Wright and S. Brandani, Ind. Eng. Chem. Res., 2016, 55, 3840–3851 CrossRef.
- D. Wang, X. Ma, C. Sentorun-Shalaby and C. Song, Ind. Eng. Chem. Res., 2012, 51, 3048–3057 CrossRef.
- L. Marques, P. Carrott and M. Carrott, Adsorpt. Sci. Technol., 2013, 31, 223–232 CrossRef.
- J. Wang, H. Chen, H. Zhou, X. Liu, W. Qiao, D. Long and L. Ling, J. Environ. Sci., 2013, 25, 124–132 CrossRef.
- J. Wang, H. Huang, M. Wang, L. Yao, W. Qiao, D. Long and L. Ling, Ind. Eng. Chem. Res., 2015, 54, 5319–5327 CrossRef.
- M. Wang, L. Yao, J. Wang, Z. Zhang, W. Qiao, D. Long and L. Ling, Appl. Energy, 2016, 168, 282–290 CrossRef.
- S. Gadipelli, H. A. Patel and Z. Guo, Adv. Mater., 2015, 27, 4903–4909 CrossRef PubMed.
- Q. Chen, D. Long, L. Chen, X. Liu, X. Liang, W. Qiao and L. Ling, J. Non-Cryst. Solids, 2011, 357, 232–235 CrossRef.
- G. Reichenauer, Adv. Sci. Technol., 2014, 91, 54–63 CrossRef.
- A. M. ElKhatat and S. A. Al-Muhtaseb, Adv. Mater., 2011, 23, 2887–2903 CrossRef PubMed.
- M. Antonietti, N. Fechler and T.-P. Fellinger, Chem. Mater., 2014, 26, 196–210 CrossRef.
- M. B. Bryning, D. E. Milkie, M. F. Islam, L. A. Hough, J. M. Kikkawa and A. G. Yodh, Adv. Mater., 2007, 19, 661–664 CrossRef.
- S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794–830 RSC.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef PubMed.
- L. Dong, Q. Yang, C. Xu, Y. Li, D. Yang, F. Hou, H. Yin and F. Kang, Carbon, 2015, 90, 164–171 CrossRef.
- Q. Zheng, Z. Cai and S. Gong, J. Mater. Chem. A, 2014, 2, 3110–3118 Search PubMed.
- M. Zhang, B. Gao, X. Cao and L. Yang, RSC Adv., 2013, 3, 21099–21105 RSC.
- H. Bai, C. Li, X. Wang and G. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 Search PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef PubMed.
- S. H. Kim, G. W. Mulholland and M. R. Zachariah, Carbon, 2009, 47, 1297–1302 CrossRef.
- W.-J. Son, J.-S. Choi and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef.
- J. Wang, D. Long, H. Zhou, Q. Chen, X. Liu and L. Ling, Energy Environ. Sci., 2012, 5, 5742–5749 Search PubMed.
- Z. Chen, S. Deng, H. Wei, B. Wang, J. Huang and G. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 6937–6945 Search PubMed.
- M. A. Tupta, Electronic Engineering Times Europe, 2011, pp. 21–24 Search PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 Search PubMed.
- E. S. Sanz-Pérez, A. Arencibia, R. Sanz and G. Calleja, Adsorption, 2016, 22, 609–619 CrossRef.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. J., 2014, 251, 293–303 CrossRef.
- M. U. Thi Le, S.-Y. Lee and S.-J. Park, Int. J. Hydrogen Energy, 2014, 39, 12340–12346 CrossRef.
- Y. Zhu, H. Hu, W. Li and X. Zhang, Carbon, 2007, 45, 160–165 CrossRef.
- R. R. Kohlmeyer, M. Lor, J. Deng, H. Liu and J. Chen, Carbon, 2011, 49, 2352–2361 CrossRef.
- A. L. S. Ethier, R. Jackson, A. C. Rumple, W. Medina-Ramos, Z. Li, J. Fisk, B. Holden, L. Gelbaum, P. Pollet, C. A. Eckert and C. L. Liotta, Processes, 2015, 3, 497–513 CrossRef.
- D. J. Heldebrant, P. K. Koech, J. E. Rainbolt, F. Zheng, T. Smurthwaite, C. J. Freeman, M. Oss and I. Leito, Chem. Eng. J., 2011, 171, 794–800 CrossRef.
- A. Ion, C. V. Doorslaer, V. Parvulescu, P. Jacobs and D. D. Vos, Green Chem., 2008, 10, 111–116 RSC.
- A.-L. Kjøniksen and B. Nyström, Macromolecules, 1996, 29, 5215–5222 CrossRef.
- F. Auriemma, C. De Rosa and R. Triolo, Macromolecules, 2006, 39, 9429–9434 CrossRef.
- M. Bercea, S. Morariu and D. Rusu, Soft Matter, 2013, 9, 1244–1253 RSC.
- C. Tang, C. D. Saquing, J. R. Harding and S. A. Khan, Macromolecules, 2010, 43, 630–637 CrossRef.
- T. Hatakeyema, J. Uno, C. Yamada, A. Kishi and H. Hatakeyama, Thermochim. Acta, 2005, 431, 144–148 CrossRef.
- E. Otsuka and A. Suzuki, J. Appl. Polym. Sci., 2009, 114, 10–16 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef.
- H.-J. Kuppers, B. Hirthe and M. Thommes, GIT Lab. J., 2001, 5, 110–113 Search PubMed.
- K. Sing, Colloids Surf., A, 2001, 187–188, 3–9 CrossRef.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef.
- J. Rouquerol, G. V. Baron, R. Denoyel, H. Giesche, J. Groen, P. Klobes, P. Levitz, A. V. Neimark, S. Rigby, R. Skudas, K. Sing, M. Thommes and K. Unger, Microporous Mesoporous Mater., 2012, 154, 2–6 CrossRef.
- W. Witkiewicz and A. Zieliński, Adv. Mater. Sci., 2006, 6, 35–51 Search PubMed.
- P. S. Patel, D. E. Shepherd and D. W. Hukins, BMC Musculoskeletal Disord., 2008, 9, 137 CrossRef PubMed.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29–45 CrossRef.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2011, 51, 1438–1463 CrossRef.
- R. N. Bennett, P. D. Ritchie, D. Roxburgh and J. Thomson, Trans. Faraday Soc., 1953, 49, 925–929 RSC.
- D. Fernandes, W. Conway, R. Burns, G. Lawrance, M. Maeder and G. Puxty, J. Chem. Thermodyn., 2012, 54, 183–191 CrossRef.
- M. Gupta, E. F. d. Silva and H. F. Svendsen, Energy Procedia, 2014, 51, 161–168 CrossRef.
- C. Chen, W.-J. Son, K.-S. You, J.-W. Ahn and W.-S. Ahn, Chem. Eng. J., 2010, 161, 46–52 CrossRef.
- G. Qi, Y. Wang, L. Estevez, X. Duan, N. Anako, A.-H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 Search PubMed.
- D. J. N. Subagyono, Z. Liang, G. P. Knowles and A. L. Chaffee, Chem. Eng. Res. Des., 2011, 89, 1647–1657 CrossRef.
- Z. Zhang, B. Wang, Q. Sun and X. Ma, Energy Procedia, 2013, 37, 205–210 CrossRef.
- M. A. Sakwa-Novak, S. Tan and C. W. Jones, ACS Appl. Mater. Interfaces, 2015, 7, 24748–24759 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |