
A bifunctional electrolyte additive for H2O/HF scavenging and enhanced graphite/LiNi0.5Co0.2Mn0.3O2 cell performance at a high voltage†
 *a
*a
Abstract
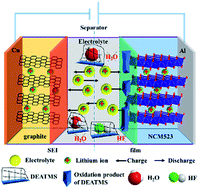
A novel strategy for selecting an additive based on understanding the influence of different functional groups on electrochemical characteristics is adopted. N,N-Diethylamino trimethylsilane (DEATMS) is tested as a bifunctional electrolyte additive to scavenge H2O and neutralize HF in the electrolyte and enhance the behaviour of graphite/LiNi0.5Co0.2Mn0.3O2 cells operating at a high voltage (4.5 V vs. Li/Li+) or elevated temperature (55 °C). With 2% DEATMS, no LiPF6 hydrolysis species is found after storage at 55 °C for 6 days, even with a moisture content of 2000 ppm in the electrolyte. In addition, the graphite/LiNi0.5Co0.2Mn0.3O2 cells containing DEATMS show cycling performances superior to those of the cells without DEATMS upon cycling for 100 cycles at a high voltage (85.5% vs. 72.0%) or high temperature (81.2% vs. 70.6%). The sacrificial oxidation of DEATMS on the cathode surface restricts direct contact between the electrolyte and cathode, improving the electrochemical performance. The ex situ surface analysis of the electrodes after cycling indicates that both the electrolyte decomposition and Mn dissolution are inhibited by the deposition of DEATMS oxidation products.
- This article is part of the themed collection: 2018 Sustainable Energy and Fuels HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

