Displacement reduction routed Au–Pt bimetallic nanoparticles: a highly durable electrocatalyst for methanol oxidation and oxygen reduction†
Received
23rd November 2017
, Accepted 8th May 2018
First published on 10th May 2018
1. Introduction
Architecting two nanoparticles as bimetallic systems in different forms like alloys, core–shell and cluster in cluster not only increases the catalytic activity but also improves the electronic and optical properties mainly due to the synergistic effect between the bimetallic nanoparticles (BMNPs).1–4 The properties of BMNPs mainly depend on the size and composition of the individual NPs in the bimetallic assembly.3–5 Especially, tuning the composition of the NPs in a bimetallic system to enhance the catalytic activity is a paramount task to researchers. Among the bimetallic systems reported in the literature, Au–PtNPs have received huge interest in energy applications including oxidation of methanol,6–10 ethanol,10–12 formic acid13,14 and glucose14,15 and reduction of dioxygen5,8,16 because of their high stability and composition dependent catalytic activity.
Direct alkaline alcohol fuel cells have been investigated over the past decade due to their faster kinetics for alcohol oxidation and the oxygen reduction reaction (ORR) in alkaline solutions, higher power generation efficiency, easy handling of liquid fuels and low emission of pollutants.6–12,17 Similarly, methanol fuel cells are promising energy conversion devices because of their high energy density, low operating temperature, low pollutant emission, easy storage and transportation of fuels, and high conversion efficiency.6–10,17–20 Even though Pt catalysts are well known for methanol oxidation due to their surface binding properties, there are a few critical issues faced by them which include sluggish kinetics and poor stability besides high cost.18–20 Since CO is very sensitive to the surface binding properties, it strongly binds with the active sites of the Pt catalyst completely poisoning the active surface.21 To remove CO from the surface of PtNPs, the formation of Pt–OH is very essential (eqn (1) and (2)), but it occurs only at high potential.22
| | | Pt–CH2O → Pt–CO + 2H+ + 2e− | (1) |
| | | Pt–CO + Pt–OH → 2Pt + CO2 + H+ + e− | (2) |
Hence, designing an alternate catalyst or modifying the Pt catalyst is highly essential to sort out the above problems. Moreover, it is essential to reduce the cost of the Pt catalyst for a sustainable commercialization of fuel cells. Combining Au with Pt as BMNPs can overcome the disadvantages of the Pt catalyst, and the presence of Au provides a platform for the oxidation of poisonous intermediates such as CO by changing the electronic band structure and activation energy.6–10 Further, the Au–OH formation is facile in the Au-based catalyst in contrast to the Pt catalyst.8–10,23 It has already been reported that Au–PtNPs have higher catalytic activity than Pt but they depend on the composition of Pt and Au.6–10,24 The availability of the free Pt surface in the Au–Pt assembly would be expected to enhance the oxidation of methanol.
It has already been shown that Au–PtNPs serve as a strong platform for methanol oxidation.6–10,24 Several methods have been reported in the literature for the synthesis of Au–PtNPs which include co-reduction,25 sonication, wet chemical synthesis,26 seed mediated synthesis,9,27 two phase synthesis28 and the galvanic displacement reaction (GDR).29,30 Among these methods, the GDR method gains considerable attention due to its ease of handling, cost effectiveness, high throughput and simple experimental set up.29,30 The electroless mechanism is the basis of the GDR in which the reduction of metal ions to metal NPs is favoured without a reducing agent as well as external electric current.16,29–31 Very recently, Engelbrekt et al. prepared Au@PtNPs by GDR-like synthesis. They first prepared AuNPs by the chemical reduction of HAuCl4 in buffered glucose-starch solution at 95 °C and then added various concentrations of H2PtCl6 to prepare AuNPs to form Au@PtNPs. The resultant Au@PtNPs were mixed with graphitized carbon black for electrocatalytic applications.32 The present study provides a facile approach for the synthesis of Au@PtNPs in an aqueous solution as well as their fabrication on an electrode surface for electrocatalytic applications.
In the present study, the synthesis of Au–PtNPs in the solution phase by a galvanic displacement reaction and their application as a catalyst for methanol oxidation and oxygen reduction were described. Citrate capped PtNPs were initially prepared by a wet chemical method, and then various concentrations of HAuCl4 were added to them to form Au–PtNPs. The formation of Au–PtNPs follows the GDR between Pt atoms and AuCl4− ions which is mainly due to the difference in the thermodynamic potential. The as prepared Au–PtNPs were characterized by UV-vis spectroscopy, high resolution transmission electron microscopy (HR-TEM), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). Further, Au–PtNPs were fabricated on a glassy carbon (GC) electrode using 1,6-hexanediamine (HDA) as a linker for the electrochemical oxidation of methanol and the reduction of dioxygen.
2. Experimental section
2.1 Chemicals
Hydrogen tetrachloroaurate (HAuCl4), platinic acid (H2PtCl6·6H2O) and sodium borohydride (NaBH4) were purchased from Sigma Aldrich. Trisodium citrate (Na3C6H5O7) (TSC), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O), disodium hydrogen phosphate dihydrate (Na2HPO4·2H2O) and methanol were purchased from Merck, India and were used as received. 1,6-Hexanediamine (HDA), the commercial Pt/C catalyst (Pt 40%) and glassy carbon (GC) plates were purchased from Alfa-Aesar. Na2HPO4 and Na2H2PO4 were used to prepare phosphate buffer (PB) solution (pH 7). Double distilled water was used to prepare the solutions used in the present work. All glassware used in the preparation of Au–PtNPs was cleaned with freshly prepared aqua regia and rinsed thoroughly with water.
2.2 Instrumentation
Absorption spectra were measured using a Perkin-Elmer lambda 35 and a JASCO V-550 UV-visible spectrophotometer. Electrochemical measurements were carried out with a CHI model 643B (Austin, TX, USA) electrochemical analyzer. The measurements were performed in a conventional two-compartment three electrode cell with a 3 mm polished GC electrode as a working electrode, a platinum wire as a counter electrode and a NaCl saturated Ag/AgCl electrode as a reference electrode. All the electrochemical experiments were carried out under a nitrogen atmosphere except the ORR experiment at room temperature. HR-TEM images of Au–PtNPs were obtained using a JEOL JEM 3010 operating at 200 kV. The samples were prepared by dropping 2 μL of Au–Pt solution on a carbon-coated copper grid. XRD analysis was carried out with a Rigaku X-ray diffraction unit using Ni-filtered Cu Kα (λ = 1.5406) radiation. A large volume (500 ml) of Au–PtNPs was synthesized and centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), and then the particles were separated. They were repeatedly washed with water and dried in a vacuum. The dried Au–PtNPs powder was used for XRD measurements. Scanning electron microscopy (SEM) measurements were carried out on a VEGA3 TESCAN, USA. Energy-dispersive X-ray spectroscopy (EDS) and line scan analysis were carried out using a Bruker Nano, Germany. For SEM, XRD, EDS and line scanning measurements, ITO plates were used as substrates with a similar modification to that used for the GC electrode.
000 rpm), and then the particles were separated. They were repeatedly washed with water and dried in a vacuum. The dried Au–PtNPs powder was used for XRD measurements. Scanning electron microscopy (SEM) measurements were carried out on a VEGA3 TESCAN, USA. Energy-dispersive X-ray spectroscopy (EDS) and line scan analysis were carried out using a Bruker Nano, Germany. For SEM, XRD, EDS and line scanning measurements, ITO plates were used as substrates with a similar modification to that used for the GC electrode.
2.3 Synthesis of citrate capped PtNPs
Citrate capped PtNPs were prepared according to the reported procedure.33 0.8 mL of 0.2% H2PtCl6 was added to 9.2 ml of boiling water. After a minute, 0.2 mL each of 1% TSC and 0.05% citric acid were added, and half a minute later 0.1 mL of 0.08% freshly prepared NaBH4 was quickly added with constant stirring. The color of the solution turned to brown from light yellow on the addition of NaBH4 confirming the formation of PtNPs. The product was cooled down to room temperature after 10 min.
2.4 Synthesis of citrate capped AuNPs
Citrate capped AuNPs were prepared according to the following procedure.31 0.1 ml of 31.7 mM HAuCl4·3H2O and 0.2 ml of 38.8 mM TSC were added to 9.5 ml of water with constant stirring for 20 min. To this solution, 0.2 mL of ice cold 20 mM NaBH4 was added drop by drop, and the stirring was continued for another 20 min. The color of the solution turns from light yellow to wine red immediately after the final addition, indicating the formation of citrate capped AuNPs.
2.5 Synthesis of bimetallic Au–PtNPs
100–900 μL of 0.03 mM HAuCl4 was added drop by drop to the cit-PtNPs with constant stirring. The color of the solution changed from light brown to reddish brown gradually by the addition of various concentrations of HAuCl4, indicating the formation of Au–PtNPs, and the stirring was continued for another 20 min. The preparation and plausible mechanism for the formation of Au–PtNPs are schematically shown in Scheme 1.
 |
| | Scheme 1 Synthesis and plausible mechanism of the formation of Au@PtNPs. | |
2.6 Fabrication of Au–PtNPs on the GC electrode
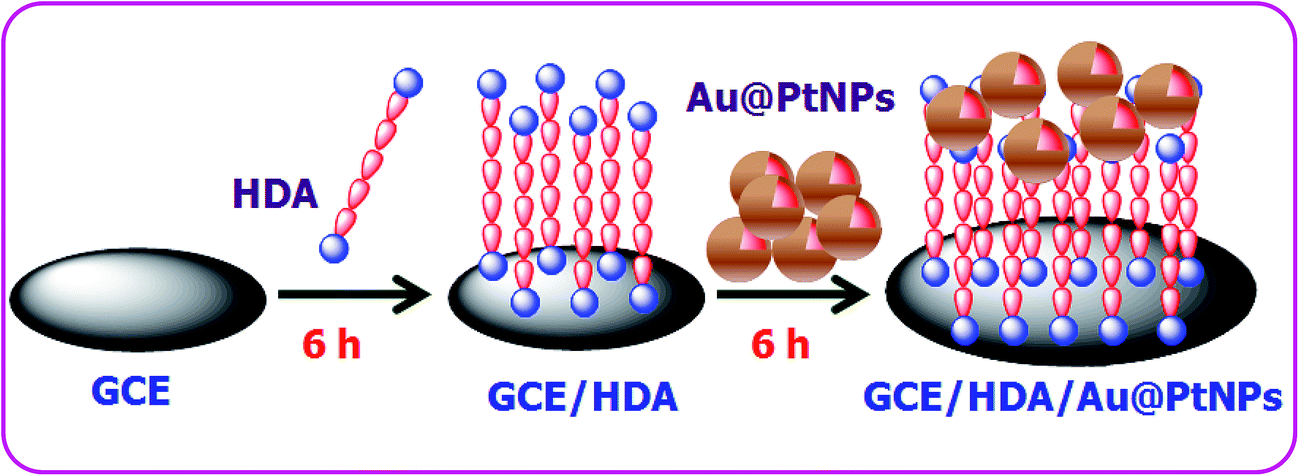
The GC electrode was polished with 0.05 μm alumina powder and sonicated in double distilled water for 5 min. The HDA linker was used to attach the Au–PtNPs on the GC electrode by immersing the well cleaned GC electrode in an aqueous solution of HDA for 6 h. The attachment of amine on the GC electrode follows Michael's nucleophilic addition of amine groups on the olefinic bond present in the GC electrode.34 The HDA modified electrode was then immersed in the colloidal solution of Au–PtNPs for 6 h. The resultant electrode was washed with double distilled water. This electrode is termed the GC/HDA/Au–PtNPs electrode. Scheme 2 illustrates the attachment of Au–PtNPs on the GC electrode.
 |
| | Scheme 2 Fabrication of Au@PtNPs on a GCE. | |
3. Results and discussion
3.1 Characterization of Au–PtNPs by UV-visible spectroscopy
Electronic spectroscopy is the most vital technique to characterize metal NPs because they exhibit characteristic absorption bands due to the surface plasmon resonance (SPR) in the visible region. Fig. S1A† shows the absorption spectra recorded for platinic acid and citrate capped PtNPs. Platinic acid shows an absorption band at 280 nm corresponding to ligand to metal charge transfer (Fig. S1A,† curve a) whereas cit-PtNPs do not show any absorption band in the visible region (Fig. S1A,† curve b) due to the very weak d–d interband transition.34 On the other hand, the cit-AuNPs show an absorption maximum at 520 nm (Fig. S1B†) corresponding to the SPR band of AuNPs. These results confirmed the successful formation of PtNPs and AuNPs. The color of the formed PtNPs and AuNPs was found to be brownish yellow and wine red, respectively (insets, Fig. S1A and B†). While adding HAuCl4 to PtNPs, the brownish yellow color of the solution turned to reddish brown, indicating the formation of Au–PtNPs. Further, Au–PtNPs with various mole ratios were prepared, and the formation of Au–PtNPs was monitored by UV-vis spectroscopy. Fig. 1 shows the absorption spectra recorded for the Au–PtNPs with different mole ratios of Pt and Au. The Pt:Au mole ratio of 1:0 does not show any absorbance band in the visible region (Fig. 1, curve a) whereas on changing the mole ratio to 1:0.03, a new band appeared at 545 nm corresponding to the AuNPs suggesting the formation of Au–PtNPs (Fig. 1, curve b). On further increasing the mole ratios to 1:0.06 and 1:0.09, the SPR band intensity increased concomitantly (Fig. 1, curves c and d) and attained saturation at a 1:0.09 ratio, and further increment in the mole ratio to 1:0.12 does not affect the SPR (Fig. 1, curve e). In addition, the color of the solution gradually turns to reddish brown from brownish yellow upon the addition of HAuCl4 to the PtNP solution (Fig. 1, inset). The obtained spectral and color changes clearly suggest the successful formation of Au–PtNPs.
 |
| | Fig. 1 UV-vis absorption spectra obtained for (a) PtNPs and (b–e) Au–PtNPs with different Pt:Au mole ratios of (b) 1:0.03, (c) 1:0.06, (d) 1:0.09 and (e) 1:0.12 and their corresponding photographs (inset). | |
3.2 Mechanism of the formation of Au–PtNPs
The formation of Au–PtNPs follows the galvanic displacement reaction (GDR), which is driven by an electroless mechanism involving spontaneous reduction of Au3+ ions to Au(0) by Pt(0) in the absence of external sources of electric current. The GDR between Au3+ ions and Pt(0) to form the Au–PtNPs was induced by the difference in their standard reduction potentials: Pt2+/Pt (+0.73 V vs. SHE) and Au3+/Au (+0.99 V vs. SHE). The surface of PtNP seeds serves as a reducing agent and electron source for the reduction of AuCl4−.30,31 When the active site of the Pt seeds undergoes the GDR, Pt was dissolved by releasing electrons and holes were generated on the facets (anodic reaction). The released electrons can easily migrate to the surface of the nanoparticle to reduce AuCl4− ions to Au(0) (cathodic reaction).30,31 The shell deposition can continue as long as Pt ions can permeate and electrons can transfer through the shell. The general stoichiometric reaction between the Au3+ and Pt(0) is given below (3).| | | 3Pt(s) + 2AuCl4− → 2Au(s) + 3Pt2+ + 8Cl− | (3) |
3.3 Characterization of Au–PtNPs by HR-TEM and XRD
The size and morphology of the Au–PtNPs were examined by HR-TEM. Fig. 2 shows the HR-TEM images of PtNPs and Au–PtNPs with a Pt:Au mole ratio of 1:0.09. The HR-TEM image of the PtNPs shows that the formed PtNPs were spherical in shape, and they were aligned to form chain like structures (Fig. 2A). The average particle size of the formed PtNPs was found to be ∼4 nm (Fig. S2A†). On the other hand, the HR-TEM image of Au–PtNPs shows elongated spherical particles (Fig. 2B) with an average size of ∼11 nm (Fig. S2B†). The increase in the particle size suggests the formation of Au–PtNPs. Further, lattice images of a single crystal are shown with different magnifications (Fig. 2C and D), clearly suggesting that the formed Au–PtNPs were in the form of core–shell NPs. Further, they show a hollow structure which was covered with another layer, maybe the holes generated by the PtNPs, and the reduced AuNPs may be placed inside the holes to form core–shell Au–PtNPs (Au@Pt core@shell NPs). The selected area electron diffraction patterns of Pt and Au–PtNPs show discrete dots, illustrating the crystalline nature of Pt and Au–PtNPs (Fig. S3A and B†). The obtained features again confirmed that the crystallinity of the PtNPs was not affected after the formation of Au–PtNPs.
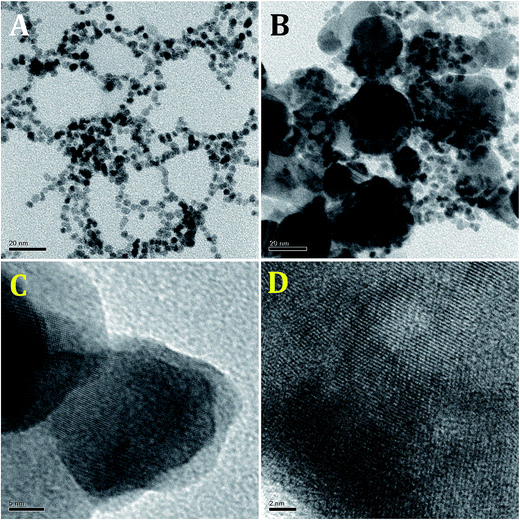 |
| | Fig. 2 HR-TEM images obtained for (A) PtNPs and (B–D) Au–PtNPs with different magnifications. | |
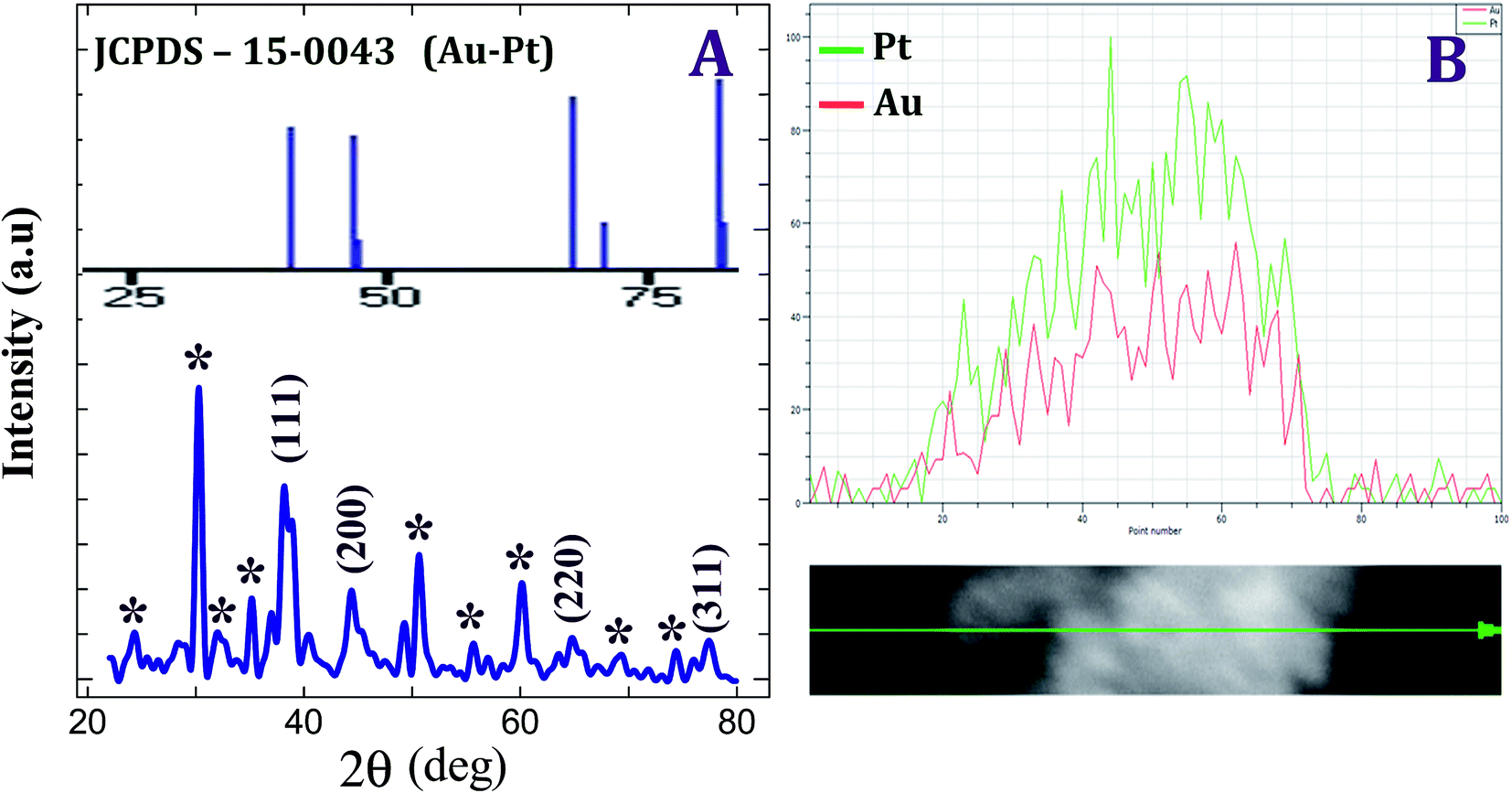
Fig. 3 shows the XRD pattern of Au–PtNPs with a Pt:Au mole ratio of 1:0.09. It illustrates diffraction features at 38.17°, 44.34°, 64.52°, 77.48° and 82.10° corresponding to (111), (200), (220), (311) and (222) planes, respectively (JCPDS card no. 03-065-2870). Among these, the peak corresponding to the (111) plane is more intense than those corresponding to the other planes. The ratio between the intensity of the (200) and (111) diffraction peaks is much lower, indicating that the (111) plane is a predominant orientation. The width of the (111) peak was employed to calculate the average crystallite size of the Au–PtNPs using the Scherrer equation.35 The calculated average size of Au–PtNPs is 14 nm, which closely matches with the particle size obtained from HR-TEM. Besides, compared with Au (111), the peak intensity of Pt (111) is much higher, implying the Au (core)–Pt (shell) orientation.
 |
| | Fig. 3 XRD pattern obtained for Au–PtNPs. | |
3.4 Characterization of Au–PtNPs by XPS
The as-prepared Au–PtNPs were further characterized by XPS, which provides information about the oxidation states of Au and Pt present in the Au–PtNPs. Fig. 4A shows the XPS survey spectrum of Au–PtNPs. It shows three major peaks in the range of 70–90 eV and at 280 and 530 eV corresponding to Au 4f and Pt 4f and C 1s and O 1s regions, respectively. The appearance of C 1s and O 1s peaks is mainly from the capping ligand, TSC. Fig. 4B shows the deconvoluted XPS profile of the Au and Pt region from the Au–PtNPs. It shows four peaks at 71.16, 74.54, 83.89 and 87.58 eV. The obtained 71.16 and 74.54 eV with a difference of 3.7 eV are the characteristic peaks of 4f7/2 and 4f5/2 of the zero valent nature of Pt, respectively. The peaks at 83.89 and 87.58 eV with a difference of 3.7 eV correspond to the 4f7/2 and 4f5/2 of Au, respectively, which is consistent with the zero valent nature of Au.16 This once again reveals the successful formation of Au–PtNPs and the zero valent nature of Au and Pt in the Au–PtNPs system. Further, it is noted that the Pt layer which covered the AuNPs exhibits more intense peaks than the Au core in XRD and XPS analyses. Hence, based on the results obtained from HR-TEM, XRD and XPS, it is proposed that the Au–Pt bimetallic nanoparticles exist in the form of a core–shell structure.
 |
| | Fig. 4 XPS obtained for Au–PtNPs: (A) survey spectrum and (B) deconvoluted Au 4f and Pt 4f regions of Au–PtNPs (inset A: enlarged survey spectrum). | |
3.5 Characterization of the Au–PtNPs modified electrode by SEM, XRD and EDS
The ultimate aim of the present work is to utilize the Au–PtNPs for electrocatalytic applications, and hence, the colloidal Au–PtNPs were attached on the electrode surface. The modification of Au–PtNPs on the electrode surface was confirmed by SEM, XRD and EDS techniques.
The morphology of the Au–PtNPs modified substrate with a Pt:Au mole ratio of 1:0.09 was characterized by SEM. Fig. S4† shows the SEM image obtained for the Au–PtNPs modified substrate. It shows that the attached Au–PtNPs were spherical in shape, and their size was found to be ∼20 nm. It also illustrates that the particles were distributed throughout the surface uniformly. Further, the Au–PtNPs modified substrate was characterized by XRD. Fig. 5A shows the XRD pattern obtained for substrates modified with Au–PtNPs with a Pt:Au mole ratio of 1:0.09. It shows diffraction features at 38.17°, 44.34°, 64.52° and 77.48° corresponding to the (111), (200), (220) and (311) planes of Au–PtNPs (JCPDS card no. 03-065-2870) (Fig. 5A, inset). It was found that there is no change in the crystalline planes. This suggests that the crystalline nature of the formed Au–PtNPs remains the same after being fabricated on the substrate. Among these, the peak corresponding to the (111) plane is more intense than those corresponding to the other planes. The ratio between the intensity of the (200) and (111) diffraction peaks was much less, indicating that the (111) plane is a predominant orientation. In addition, four characteristic peaks for Au–PtNPs marked by their indices ((111), (200), (220), and (311)) indicated that the resultant bimetallic nanoparticles have a face centered cubic (fcc) structure.30,31
 |
| | Fig. 5 (A) XRD obtained for the Au–PtNPs modified substrate (* peaks correspond to the ITO substrate) (inset: JCPDS file of Au–PtNPs) and (B) EDS line spectra of the Au–PtNPs modified substrate. | |
Fig. S5† shows the EDAX spectra of PtNPs, AuNPs and Au–PtNPs with a Pt:Au mole ratio of 1:0.09. The EDAX spectra of Pt and AuNP modified substrates show peaks at 2.050 and 9.442 keV corresponding to Pt and 2.21 and 9.71 keV corresponding to Au along with carbon, nitrogen and oxygen at 0.27, 0.39 and 0.53 keV, respectively (Fig. S5A and B†). On the other hand, the Au–PtNPs modified substrate shows both Pt and Au peaks, which confirms the successful modification of the Au–PtNPs (Fig. S5C†). It was found that the Pt and Au weight percentages of Au–PtNPs with a Pt:Au ratio of 1:0.09 are 7.23 and 4.17%, respectively. The Au–PtNPs with a Pt:Au mole ratio of 1:0.09 were further characterized by line scanning analysis.
Qualitative information on elemental distributions can be obtained by line-scanning analysis on the EDS analyzer, in which the diffusion profile of elements at an interface can be plotted as the number of X-ray quanta being counted vs. the spatial location along a line. Further, the normalised intensity scales simply make comparison of major and minor elements. Hence, it is a very much useful tool for the characterization of the bimetallic system.31Fig. 5B shows the line spectra of the Au–PtNPs modified substrate with a Pt:Au mole ratio of 1:0.09. It confirms that the particles present on the substrate were Au–PtNPs, and the intensity of PtNPs (Fig. 5B green line) was higher than that of AuNPs (Fig. 5B red line), implying that the AuNPs were covered with PtNPs. This clearly demonstrates that the PtNPs were in the outer shell, and the AuNPs were in the inner core. Hence, it is proposed that the formed Au–PtNPs were in the form of the Au(core)@Pt(shell) structure.
3.6 Characterization of the Au–PtNPs modified electrode by cyclic voltammetry and electrical impedance spectroscopy
Fig. S6† shows the cyclic voltammograms (CVs) obtained for GC/HDA/PtNP and GC/HDA/AuNP electrodes in 0.5 M H2SO4 at a scan rate of 10 mV s−1. The PtNP modified electrode shows an oxidation peak at +0.75 V and a reduction peak at +0.37 V, corresponding to the formation and the subsequent reduction of Pt oxide along with hydrogen adsorption and desorption peaks at −0.25 V (Fig. S7A†), whereas the AuNP modified electrode shows an Au oxidation peak at +1.40 V and its subsequent reduction peak at +0.90 V (Fig. S7B†), confirming the successful fabrication of PtNPs and AuNPs on the electrode surface.21 On the other hand, the Au–PtNPs modified GC electrode exhibits a characteristic hydrogen adsorption–desorption peak at −0.25 V along with Pt and Au oxidation and reduction peaks (Fig. 6),16,32 illustrating the successful modification of Au–PtNPs on the GC electrode. It was found that while increasing the ratio of Au in the Au–Pt system, the Au oxide and reduction peak currents increased linearly (Fig. 6, curves b–d) and attained saturation at a 1:0.09 ratio.
 |
| | Fig. 6 CVs obtained for (a) GC/HDA/PtNPs, (b–d) GC/HDA/Au–PtNPs with Pt:Au mole ratios of (b) 1:0.06, (c) 1:0.09 and (d) 1:0.12 in 0.5 M H2SO4 at a scan rate of 10 mV s−1. Insets: enlarged view of formation and reduction of (i) Pt-oxide and (ii) Au-oxide. | |
In order to investigate the conducting nature of the Au–PtNPs electrode, an EIS study was carried out. Fig. 7 shows the Nyquist plots obtained for electrodes modified with GC/HDA/PtNPs, GC/HDA/AuNPs and GC/HDA/Au–PtNPs with a Pt:Au mole ratio of 1:0.09 in 1 mM K3[Fe(CN)6] containing 0.2 M PB solution (pH 7) at scanning frequencies from 0.01 to 100000 Hz. A Randles circuit model [RS(CPE − RCT)] (Fig. 7, inset) was used to fit the impedance spectral data where RS refers to the solution resistance and CPE refers to the constant phase element. The charge transfer resistance (RCT) can be calculated from the semicircle obtained in the Nyquist plot, and it can control the interfacial electron-transfer rate of the redox probe between the solution and the electrode. The bare GC electrode shows a charge transfer resistance (RCT) value of 37.77 kΩ (curve a), whereas the AuNP and PtNP modified GC electrodes exhibit 25.89 and 21.47 kΩ, respectively (curves b and c). On the other hand, the Au–PtNPs modified electrode drastically reduced the RCT value to 16.34 kΩ (curve d). The increase in the conductivity of the Au–PtNPs modified electrode is mainly due to the higher surface homogeneity of the bimetallic system when compared to its respective monometallic counterparts. The obtained RS, C, and RCT values are given in Table S1.† The heterogeneous electron-transfer rate constant (ket) was calculated using eqn (4).31
| |  | (4) |
where
R,
T and
F have their usual representations,
A is the electrode area (0.07 cm
2),
C0 is the concentration of the redox couple (1 mM) and
n is the number of electrons transferred per molecule of the redox probe (
n = 1 for the K
3[Fe(CN)
6] probe). The calculated
ket values are 1.01 × 10
−4, 1.47 × 10
−4, 1.77 × 10
−4 and 4.12 × 10
−3 cm s
−1 for bare GC, Au and PtNP and Au–PtNPs modified GC electrodes, respectively. The obtained higher
ket value for the Au–PtNPs modified electrode indicates that the electron transfer reaction was faster at this electrode when compared to bare GC, Pt and AuNP modified electrodes.
 |
| | Fig. 7 Nyquist plots obtained for (a) bare GC, (b) GC/HDA/AuNP, (c) GC/HDA/PtNP and (d) Au–PtNPs electrodes in 1 mM K3[Fe(CN)6] in 0.2 M PB solution at scanning frequencies from 0.01 to 100000 Hz. Inset: equivalent electrical circuit used for fitting the impedance spectra. | |
3.7 Electrochemical oxidation of methanol at the Au–PtNPs modified GC electrode
Inspired by the composition dependent formation of Au–PtNPs, the electrocatalytic activity of the Au–PtNPs modified electrode was examined towards the oxidation of methanol by cyclic voltammetry. Fig. 8 shows the CVs obtained for 1.0 M methanol at GC electrodes modified with PtNPs and Au–PtNPs with different Pt:Au mole ratios in 1.0 M KOH at a scan rate of 50 mV s−1. The Au–PtNPs modified electrode does not show any response in the absence of methanol (Fig. 8, dotted line), indicating that this electrode is inert in this potential window. In the presence of methanol, the PtNP modified electrode shows an oxidation peak at −0.15 V (Fig. 8, curve a). When the AuNPs were blended with PtNPs in the form of Au–PtNPs with a Pt:Au mole ratio of 1:0.06, the oxidation peak current of methanol was dramatically enhanced by 6-fold, suggesting that Au–PtNPs act as a better catalyst when compared to PtNPs alone (Fig. 8, curve b). Moreover, it exhibits two oxidation peaks on the forward as well as the reverse scan. It is well known that the sharp current ramp in the forward scan can be ascribed to the characteristic methanol oxidation on the electrode surface, forming adsorbed carbonaceous intermediates such as CO and HCO−, which will be oxidized at higher potentials due to the formation of Au–OH species.6–10,26,27 In the reverse scan, desorption of OH-ads or reduction of Au-oxides regenerates the active metallic Au surface which allows further oxidation of methanol molecules at lower potentials. Further varying the Pt:Au mole ratio to 1:0.09, the oxidation peak current was enhanced (Fig. 8, curve c), and it decreased at a ratio of 1:0.12 (Fig. 8, curve d), indicating that the Pt:Au ratio of 1:0.09 is a suitable composition for the oxidation of methanol.
 |
| | Fig. 8 CVs obtained for 1 M methanol in 1 M KOH at (a) GC/HDA/PtNPs and (b–d) GC/HDA/Au–PtNPs with a Pt:Au mole ratio of (b) 1:0.06, (c) 1:0.09 and (d) 1:0.12 electrodes at a scan rate of 50 mV s−1 (dotted line: in the absence of methanol). | |
The forward peak current densities of the PtNP and Au–PtNPs catalysts with different mole ratios are shown in Fig. S7.† It can be seen that the current density (A m−2) of the Au–Pt catalyst increases with the increase of the Pt:Au ratio from 1:0 to 1:0.09 and decreased on further increasing the Au content in the Au–PtNPs assembly. The obtained maximum electrocatalytic activity of the Au–PtNPs catalyst (6.52 A m−2) with a Pt:Au ratio of 1:0.09 is about 7-fold higher than that of the PtNP catalyst (1.12 A m−2). The order of the catalytic activity of the prepared PtNPs and Au–PtNPs towards methanol oxidation is as follows: Au–PtNPs (Pt:Au = 1:0.09) > Au–PtNPs (Pt:Au = 1:0.06) > Au–PtNPs (Pt:Au = 1:0.12) > PtNPs (Pt:Au = 1:0).
The catalytic activity of the Au–PtNPs (Pt:Au = 1:0.09) modified electrode was further compared with that of the commercial Pt40/C catalyst. For this, a large volume (500 ml) of Au–PtNPs (Pt:Au = 1:0.09) was synthesized, centrifuged (10000 rpm), washed with water and then dried in a vacuum. The dried Au–PtNPs powder was dispersed in water (1 mg/0.5 mL), and 30 μL of the solution was drop-cast on the electrode surface and dried overnight. The same amount of commercial Pt/C (30 μL of 1 mg/0.5 mL) was coated on the GC electrode with a similar procedure. The Au–PtNPs loading level on the GC electrode is 0.86 mg cm−2. Fig. 9A shows the CVs obtained for 1.0 M methanol in 1.0 M KOH at commercial Pt40/C and Au–PtNPs (1:0.09) modified GC electrodes at a scan rate of 50 mV s−1. The Pt/C modified electrode shows methanol oxidation at −0.20 V (curve a) whereas the Au–PtNPs modified electrode shows its oxidation at +0.23 V with an 8-fold higher oxidation current (curve b). The electrochemically active surface area (ECSA) of the Pt/C and Au–PtNPs modified electrodes can be calculated from the Coulomb charge for hydrogen adsorption and desorption (QH) in the negative-going potential scan, carried out at a scan rate of 10 mV s−1 using the following equation:7,8
| | 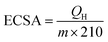 | (5) |
where,
m is the loading amount of the catalyst in the electrode (mg),
QH is the charge obtained from hydrogen desorption (μC) and 210 is the charge required to oxidize a monolayer of H
2 on bright Pt (μC cm
−2). The ECSA values were found to be 170.26 and 73.58 m
2 g
−1 for Au–PtNPs and Pt/C catalysts, respectively. The largest ECSA for the Au–PtNPs catalyst is most likely because of the high specific surface area, high tolerance of poisoning and surface homogeneity of the Au–PtNPs, which may influence the electrocatalytic activity and stability during methanol oxidation. The above results suggest that the Au–PtNPs modified electrode can be used as an effective catalyst for methanol oxidation. Further, the mass activity of the catalyst was calculated by normalizing the methanol oxidation current densities with metal loadings on the electrode surface. The mass activity of the Pt/C catalyst was found to be 29 mA mg Pt
−1. On the other hand, the present Au–PtNPs modified electrode exhibits higher mass activity (∼132 mA mg Pt
−1) than the commercial Pt/C catalyst, indicating the excellent electrocatalytic activity of the Au–PtNPs catalyst towards methanol oxidation when compared to the Pt/C catalyst.
 |
| | Fig. 9 (A) CVs obtained for 1 M methanol in 1 M KOH at (a) commercial Pt/C and (b) Au–PtNPs(1:0.09) modified GC electrodes at a scan rate of 50 mV s−1. (B) i–t curves obtained for 1 M methanol at (a) commercial Pt/C and (b) Au–PtNPs(1:0.09) modified GC electrodes in 1.0 M KOH (Eapp = +0.60 V). | |
The transportation characteristics of methanol at the Au–Pt (Pt:Au–1:0.09) modified electrode were investigated by varying the scan rate in 1.0 M KOH, and the results are shown in Fig. S8.† While increasing the scan rate from 10–100 mV s−1, the anodic peak current increased. The relationship between the anodic peak current and the square root of scan rate is linear with a correlation coefficient of 0.9991 (Fig. S8,† inset), indicating that the electrochemical oxidation of methanol at the GC/Au–PtNPs electrode is a diffusion controlled process.
The obtained electrocatalytic activity of the PtNP and Au–PtNPs modified electrodes towards methanol oxidation can be explained as follows.26,27,32 The advantage of the core–shell structure is the possibility of coating the Au layer with the electrochemically active Pt thin layer. The efficiency of the electrocatalytic reaction strongly depends on the proximity of the reactant and the catalyst employed in the reaction. When the Pt content is high in the Au–Pt catalyst or completely Pt, it is difficult to form Pt–OH at lower potentials, and hence the electrocatalytic activity will be affected. Moreover, a thicker Pt layer will not allow methanol into the inner part of the material, and hence the formation of Au–OH is not possible. On the other hand, the AuNPs present in the Au–PtNPs assembly can catalyze poisonous intermediates such as CO that were absorbed on the active sites of PtNPs significantly, which results in the increased electrocatalytic activity of Au–PtNPs with the increase of Pt:Au mole ratio. These factors make the Au–PtNPs a suitable candidate for the electrochemical oxidation of methanol. On the other hand, AuNPs are not able to catalyze methanol, and thus, the electrocatalytic activity decreases at a higher Au content.
Further, the long-term stability of the prepared Au–PtNPs catalyst was examined towards the oxidation of methanol. Fig. 9B shows the amperometric i–t curve obtained for the oxidation of 1.0 M methanol at PtNP and Au–PtNPs modified electrodes in 1.0 M KOH at an applied potential of +0.6 V for 4000 s. It can be seen that the oxidation current of methanol at the Au–PtNPs modified electrode decreases gradually with the extension of time. After 100 s, the oxidation current remains unchanged, suggesting that the prepared Au–PtNPs modified electrode is highly stable towards the oxidation of methanol. It also exhibits higher stability and electrocatalytic activity than the Pt/C catalyst (Fig. 9B). The stability of the Au–PtNPs catalyst was also examined using a CV accelerated test. Fig. S9† shows CVs obtained for 1.0 M methanol at Pt/C and Au–PtNPs modified GC electrodes in 1.0 M KOH at a scan rate of 50 mV s−1. The Pt/C catalyst exhibits a stable electrochemical response towards methanol even after 100 cycles (Fig. S9,† curves a and a′). Even though, the prepared Au–PtNPs catalyst showed a slight shift in the oxidation potential of methanol, it exhibits higher oxidation current than the Pt/C catalyst (Fig. S9,† curves b and b′). The onset potential of the Au–PtNPs catalyst shifts positively by only 20 mV after the CV accelerated test, which is insignificant. The highest peak current density after potential cycling indicates that the catalyst not only possesses enhanced catalytic activity but also reveals considerable stability in the MOR.
The performance of the present modified electrode towards methanol oxidation was compared with those of the reported Au–Pt catalyst modified electrodes (Table 1).7,9,27,32,36 It can be seen from Table 1 that the Au–PtNPs prepared in the present study showed an 8-fold higher oxidation current with a less positive potential (+0.23 V) for methanol oxidation when compared to the commercially available Pt/C catalyst. Even though the Pt–Au string bead nanochain network modified GC electrode showed higher oxidation current for methanol oxidation its oxidation occurs at a more positive potential (+0.86 V). The excellent long-term stability of the Au–PtNPs catalyst further signifies that they can act as a suitable candidate in direct methanol fuel cells (DMFCs).
Table 1 Comparison of the performance of various Au–PtNPs modified electrodes towards methanol oxidation
| S. no. |
Electrodes |
Oxidation potential (vs. SCE) |
Medium |
Mass activity (mA mg Pt−1) |
Comparison with commercial Pt/C (oxidation current) |
Ref. |
|
Multi-walled carbon nanotubes.
Potential is converted into a SCE potential.
|
| 1 |
Au–PtNPs/ITO |
+1.03 V |
0.5 M H2SO4 |
— |
2.5-Fold higher |
7
|
| 2 |
Au–PtNPs/GCE |
+0.68 V |
0.5 M H2SO4 |
161.6 |
6-Fold higher |
9
|
| 3 |
Au–Pt honeycomb/GCE |
+0.45 V |
0.5 M H2SO4 |
148.5 |
1.6-Fold higher |
27
|
| 4 |
Au@PtNPs/GCE |
+0.54 V |
0.1 M H2SO4 |
219 |
2-Fold higher |
32
|
| 5 |
Au@PtNPs/MWCNTsa/GCE |
−0.32 V |
0.5 M KOH |
— |
3.5-Fold higher |
36
|
| 6 |
Au@PtNPs/GCE |
+0.18 Vb |
1.0 M KOH |
132 |
8-Fold higher |
This work |
3.8 Electrocatalytic reduction of dioxygen
The present study also aims to utilize the Au–PtNPs modified electrode towards the oxygen reduction reaction (ORR). The ORR is a relatively sluggish process in a fuel cell, governing the overall performance of the fuel cell.5,8,16,37,38 It has been well established in the literature that Pt based nanomaterials were widely used as ORR catalysts. Alloying Pt with Au reduces the cost of the catalyst to become economically viable. The generalized mechanism for the ORR is shown below:16,38–40| | | H2O2 + 2H+ + 2e− → 2H2O | (8) |
Fig. 10 shows the CVs obtained for the ORR at bare GC, GC/HDA/PtNP and GC/HDA/Au–PtNPs electrodes with different mole ratios in oxygen saturated 0.5 M H2SO4 at a scan rate of 50 mV s−1. At the bare GC electrode, the ORR occurs at −0.46 V (Fig. 10, curve a). For the PtNP modified GC electrode, the ORR was obtained at −0.05 V which is a 410 mV more positive potential shift in the reduction potential when compared to the bare GC electrode (Fig. 10, curve b). On the other hand, when the Au–PtNPs with a Pt:Au mole ratio of 1:0.06 were modified on the GC electrode, the ORR current increased with a 100 mV more positive potential shift (Fig. 10, curve c). Interestingly, the Au–PtNPs with a Pt:Au mole ratio of 1:0.09 drastically enhanced the ORR current with a 150 mV more positive potential shift compared to the PtNP catalyst and a 660 mV more positive potential shift compared to the bare GC electrode (Fig. 10, curve d). Further, with increasing the Au content in the Au–PtNPs assembly to 1:0.12 mole ratio of Pt:Au, the ORR current decreased (Fig. 10, curve e). The obtained results suggest that the Au–PtNPs act as a suitable catalyst for the ORR, and the catalytic activity depends on the Au content.
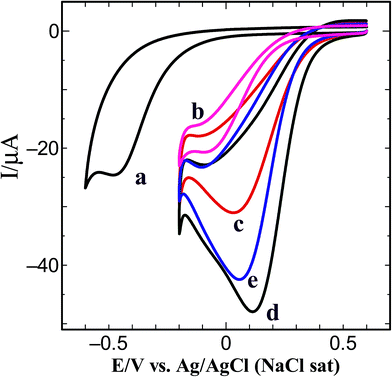 |
| | Fig. 10 CVs obtained for the ORR at (a) bare GC and (b) GC/HDA/PtNPs and (c–e) GC/HDA/Au–PtNPs with a Pt:Au mole ratio of (c) 1:0.06, (d) 1:0.09 and (e) 1:0.12 electrodes in O2 saturated 0.5 M H2SO4 at a scan rate of 50 mV s−1. | |
The ORR was also carried out at the Au–PtNPs modified electrode using a rotating disc electrode (RDE). Fig. 11A shows linear sweep voltammograms obtained for the Au–PtNPs modified electrode in O2 saturated 0.5 M H2SO4 with the electrode rotation rates from 100 to 1600 rpm at a scan rate of 5 mV s−1. A gradual increase in the limiting current was observed while increasing the rotation rate owing to the increase in the mass transport to the electrode surface (Fig. 11A). The observed current density is normalized to the geometric area of the electrode in the respective curves. According to the Koutecky–Levich equation (eqn (9)), the inverse of current density could be given as the sum of the inverses of the above current components. It could also be given in terms of jk, B and ω expressed in revolutions per minute (rpm).
where
j is the current density and
jk is the kinetic current density.
B is evaluated using
eqn (9a).
| | | B = 0.2nFCO2(DO2)2/3ν−1/6 | (9a) |
where 0.2 is a constant,
n is the number of electrons transferred per mole of oxygen,
F is the Faraday constant (96
485 C mol
−1), CO
2 is the bulk concentration of oxygen (1.1 × 10
−6 mol cm
−3), DO
2 is the diffusion coefficient of oxygen in sulfuric acid (1.4 × 10
−5 cm
2 s
−1) and
ν is the kinematic viscosity of sulfuric acid (1.0 × 10
−2 cm
−2 s
−1).
41,42 The Au–PtNPs modified electrode (
Fig. 11B) shows a linear relationship of
j−1vs. ω−1/2. The slope (
B) of the straight line of the
K–
L plot could be used to calculate the number of electrons involved in the ORR. The Au–PtNPs modified electrode shows a
B value of 12.39 × 10
−2 mA
−1 cm
−2 rpm
−1/2, and its corresponding
n value is 4.3. The theoretical
B value calculated for the 4-electron transfer process is 10.6 × 10
−2 mA
−1 cm
−2 rpm
−1/2.
41 The experimental
B and
n values closely match with the theoretical values. This confirms that the Au–PtNPs modified electrode follows a 4-electron pathway for the reduction of oxygen as shown in
eqn (6).
 |
| | Fig. 11 (A) LSVs obtained for the ORR at the GC/Au@PtNP electrode in O2 saturated 0.5 M H2SO4 at the rotation rates of 100, 200, 400, 800 and 1600 rpm and at a scan rate of 5 mV s−1. (B) The corresponding K–L plot. | |
4. Conclusions
In this paper, we have demonstrated the facile synthesis of Au–PtNPs by a galvanic displacement reaction and fabricated them on a GC electrode for methanol oxidation and dioxygen reduction. The composition of the Au–PtNPs was tuned by varying the mole ratio of Pt:Au, and the formed Au–PtNPs were characterized by UV-vis spectroscopy. The newly appeared SPR band at 545 nm confirmed the formation of Au–PtNPs by the GDR. HR-TEM, XRD, XPS and line spectra results confirmed that the formed Au–PtNPs were in the form of the Au(core)–Pt(shell) nanostructure. The as prepared Au–PtNPs were fabricated on a GC electrode using HDA as a linker, and the modification was confirmed by SEM, XRD, EDS, CV and EIS. The Au oxide formation and reduction and Pt oxide formation and reduction peaks in the cyclic voltammograms again confirmed the successful fabrication of Au–PtNPs on the electrode surface. Further, the Au–PtNPs modified electrode exhibited composition dependent electrocatalytic activity towards methanol oxidation and the ORR. The Au–PtNPs modified electrode with a Pt:Au mole ratio of 1:0.09 showed higher electrocatalytic activity by showing 7-fold higher oxidation and 2.5-fold higher reduction currents towards methanol and the ORR, respectively, than the electrodes modified with bare GC, PtNPs and Au–PtNPs with other mole ratios. The Au–PtNPs prepared from this work showed methanol oxidation at +0.23 V with an 8-fold higher oxidation current when compared to the commercially available Pt/C catalyst besides long term stability. Thus, Au–PtNPs can be used as a suitable catalyst for methanol oxidation and the ORR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Department of Science and Technology-SERB (EMR/2016/002898), New Delhi for the financial support. B. Sinduja thanks the Department of Science and Technology (DST), New Delhi, India for the award of a DST-INSPIRE Senior Research Fellowship (IF-150600(2015)).
Notes and references
- M. Shankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- R. Shi, J. Shao, X. Zhu and X. Lu, J. Phys. Chem. C, 2011, 115, 2961–2968 Search PubMed.
- H.-L. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- F. Tao, Chem. Soc. Rev., 2012, 41, 7977–7979 RSC.
- S. Cao, F. Tao, Y. Tang, Y. Li and J. Yu, Chem. Soc. Rev., 2016, 45, 4747–4765 RSC.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef PubMed.
- W. Ye, H. Kou, Q. Liu, J. Yan, F. Zhou and C. Wang, Int. J. Hydrogen Energy, 2012, 37, 4088–4097 CrossRef.
- L.-L. He, J.-N. Zheng, P. Song, S.-X. Zhong, A.-J. Wang, Z. Chen and J.-J. Feng, J. Power Sources, 2015, 276, 357–364 CrossRef.
- M. Y. Duan, R. Liang, N. Tian, Y.-J. Li and E. S. Yeung, Electrochim. Acta, 2013, 87, 432–437 CrossRef.
- L. Zhao, J. P. Thomas, N. F. Heinig, M. Abd-Ellah, X. Wang and K. T. Leung, J. Mater. Chem. C, 2014, 2, 2707–2714 RSC.
- H. M. Song, D. H. Anjum, R. Sougrat, M. N. Hedhili and N. M. Khashab, J. Mater. Chem., 2012, 22, 25003–25010 RSC.
- X. Han, D. Wang, D. Liu, J. Huang and T. You, J. Colloid Interface Sci., 2012, 367, 342–347 CrossRef PubMed.
- S. Wan, X. Wang and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 6883–6891 RSC.
- Y. Holade, A. Lehoux, H. Remita, K. B. Kokoh and T. W. Napporn, J. Phys. Chem. C, 2015, 119, 27529–27539 Search PubMed.
- Y. J. Lee and J. Y. Park, Sens. Actuators, B, 2011, 155, 134–139 CrossRef.
- N. S. K. Gowthaman and S. A. John, RSC Adv., 2015, 5, 42369–42375 RSC.
- E. H. Yu, X. Wang, U. Krewer, L. Li and K. Scott, Energy Environ. Sci., 2012, 5, 5668–5680 Search PubMed.
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 Search PubMed.
- H. Kunitomo, H. Ishitobi and N. Nakagawa, J. Power Sources, 2015, 297, 400–407 CrossRef.
- R. B. Kurtz, B. Braunschweig, P. Mukherjee, R. L. Behrens, D. D. Dlott and A. Wieckowski, J. Catal., 2011, 278, 181–188 CrossRef.
- H. Wang, Z. Jusys and R. J. Behm, Fuel Cells, 2004, 4, 113–125 CrossRef.
- L. D. Burke, J. A. Collins, M. A. Horgan, L. M. Hurley and A. P. O'Mullane, Electrochim. Acta, 2000, 45, 4127–4134 CrossRef.
- J. H. Mokkath and U. Schwingenschlogl, RSC Adv., 2013, 3, 15350–15353 RSC.
- Y. Zhao, C. Ye, W. Liu, R. Chen and X. Jiang, Angew. Chem., Int. Ed., 2014, 53, 8127–8131 CrossRef PubMed.
- L. Yang, J. Chen, X. Zhong, K. Cui, Y. Xu and Y. Kuang, Colloids Surf., A, 2007, 295, 21–26 CrossRef.
- Z. Guo, X. Zhang, H. Sun, X. Dai, Y. Yang, X. Li and T. Meng, Electrochim. Acta, 2014, 134, 411–417 CrossRef.
- D. Mott, J. Luo, P. N. Njoki, Y. Lin, L. Wang and C. J. Zhong, Catal. Today, 2007, 122, 378–385 CrossRef.
- Y. Song, K. Liu and S. Chen, Langmuir, 2012, 28, 17143–17152 CrossRef PubMed.
- D. A. Ferrer, L. A. Diaz-Torres, S. Wu and M.-J. Yacaman, Catal. Today, 2009, 147, 211–216 CrossRef.
- N. S. K. Gowthaman, B. Sinduja and S. A. John, RSC Adv., 2016, 6, 63433–63444 RSC.
- C. Engelbrekt, N. Seselj, R. Poreddy, A. Riisager, J. Ulstrupa and J. Zhang, J. Mater. Chem. A, 2016, 4, 3278–3286 Search PubMed.
- N. C. Bigall, T. Hartling, M. Klose, P. Simon, L. M. Eng and A. Eychmuller, Nano Lett., 2008, 8, 4588–4592 CrossRef PubMed.
- I. Gallardo, J. Pinson and N. Vila, J. Phys. Chem. B, 2006, 110, 19521–19529 CrossRef PubMed.
-
B. D. Cullity, Elements of X-ray diffraction, Indiana, 2nd edn, 1978 Search PubMed.
- Y. Xu, Y. Dong, J. Shi, M. Xu, Z. Zhang and X. Yang, Catal. Commun., 2011, 13, 54–58 CrossRef.
- A. T. E. Vilian, S. K. Hwang, C. H. Kwak, S. Y. Oh, C.-Y. Kim, G.-W. Lee, J. B. Lee, Y. S. Huh and Y. K. Han, Synth. Met., 2016, 219, 52–59 CrossRef.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef PubMed.
- S.-P. Lin, K.-W. Wang, C.-W. Liu, H.-S. Chen and J.-H. Wang, J. Phys. Chem. C, 2015, 119, 15224–15231 Search PubMed.
- N. S. K. Gowthaman, S. Shankar and S. A. John, J. Electroanal. Chem., 2018, 812, 37–44 CrossRef.
- J. J. Salvador-Pascual, S. Citalan-Cigarroa and O. SolorzaFeria, J. Power Sources, 2007, 172, 229–234 CrossRef.
- J. Zeng, S. Liao, J. Y. Lee and Z. Liang, Int. J. Hydrogen Energy, 2010, 35, 942–948 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV-vis spectra of Au and PtNPs, histograms of HR-TEM images, SAED patterns, SEM image, EDAX spectra and CVs obtained for Au–PtNPs and methanol oxidation comparison and impedance spectral data of Au–PtNPs. See DOI: 10.1039/c7se00565b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*