
Hybrid biopolymer electrodes for lithium- and sodium-ion batteries in organic electrolytes†
A. M.
Navarro-Suárez‡
 a,
J.
Carretero-González
b,
N.
Casado
c,
D.
Mecerreyes
cd,
T.
Rojo
*ae and
E.
Castillo-Martínez§
*a
a,
J.
Carretero-González
b,
N.
Casado
c,
D.
Mecerreyes
cd,
T.
Rojo
*ae and
E.
Castillo-Martínez§
*a
aCIC energiGUNE, Alava Technology Park, Miñano, Álava 01510, Spain. E-mail: trojo@cicenergigune.com; ilaisza@hotmail.com
bInstitute of Polymer Science and Technology, ICTP-CSIC, Juan de la Cierva 3, Madrid, 28006, Spain
cPOLYMAT, University of the Basque Country, UPV/EHU, Joxe Maria Korta Center, Avda. Tolosa 72, Donostia-San Sebastian 20018, Spain
dIkerbasque, Basque Foundation for Science, Bilbao E-48011, Spain
eInorganic Chemistry Department, University of the Basque Country, P.O. Box 644, 48080 Bilbao, Spain
First published on 25th January 2018
Abstract
The use of earth abundant and renewable materials is encouraging for the future development of environmentally clean, safe and affordable electrodes for lithium- and sodium-ion batteries. Biohybrid electrodes based on lignin and several conducting polymers have been studied mainly for supercapacitor applications. Here, we show that biohybrid electrodes containing natural lignin and a PEDOT conjugated polymer serve as electroactive materials for lithium- and sodium-ion batteries using liquid organic electrolytes. A reversible discharge capacity of 74 mA h g−1, at C/20 (4 mA g−1) rate, was achieved in the voltage range between 1 V and 4.5 V, with peak values of up to 159 mA h g−1. These properties make the natural lignin–PEDOT hybrid material a suitable organic positive electrode for Li- and Na-ion batteries.
Introduction
Electrochemical storage of energy in batteries is desirable because of their relatively high energy density, flexibility and scalability.1–3 Lithium ion battery technology has occupied practically the whole niche of the market for power electronics and portable devices because of its high energy density compared to other technologies.2 Research on sodium ion batteries is advancing fast4 towards lowering the cost of the batteries so that they can be applied for storing large amounts of energy from renewable sources. There is extensive interest due to the following reasons:5–8 (i) the high abundance and ubiquitous distribution of sodium; (ii) its similar energy density to lithium; (iii) the copper current collector can be replaced by lighter aluminium, partly overcoming the decrease in the gravimetric energy density on going from lithium to sodium; and (iv) the expected rising cost of lithium because of the increase in long-term demand.Numerous inorganic compounds, mainly oxides, phosphates, and alloys of Na with group IV and V elements, have been assessed for Na+ ion storage.9–11 Although most of them have shown gravimetric capacity and long cycle life comparable to those demonstrated by their lithium analogues, some issues related to kinetic limitations during Na+ ion intercalation12 as well as cost still remain unsettled. So, alternative chemistries and materials are needed for both lithium and sodium battery technologies so that they can achieve the required electrochemical performance at relatively low cost while meeting the safety and environmental protection requirements.13
Because large devices are needed for storing huge amounts of electricity, the most important properties of battery materials are not their energy density and efficiency, but more importantly a greatly prolonged cycle life and the use of safe and abundant materials.14 Electrodes based on redox active polymers prepared from renewable biomass appear to be one attractive solution.15–17 As a flexible macromolecular framework, organic polymers can accommodate large cations reversibly without much steric hindrance, therefore enhancing the kinetics for cation insertion and extraction reactions.18–21 Moreover, most of them do not contain costly and environmentally unfriendly metals and are abundant all over the Earth.22
Quinone-based materials have been studied as organic positive-electrode materials, since quinone moieties undergo a two-electron redox reaction which should lead to a high discharge capacity at relatively high voltages vs. Li or Na plating.23,24 Natural lignin is the most abundant aromatic polymer on Earth. In aqueous media, lignin can reversibly store two electrons and two protons quinone group present in its macromolecular structure.25,26 Interestingly, in non-aqueous solvents quinones can be reversibly reduced in two one-electron steps to its anion radical and dianion, respectively.27–29 Hybrid polymer electrodes containing quinone redox moieties and conjugated units have recently shown exceptional electrochemical proton storage properties in organic electrolyte.30 The strong ionic interaction between the reduced carbonyl functionality and small cations such as lithium (Li+), sodium (Na+), magnesium (Mg2+) and aluminium (Al3+) has been exploited to increase the gravimetric energy by largely modifying the voltage of the reaction.28 Primary lithium batteries based on hydrolysis lignin as the positive electrode material composite containing carbon and a polymeric binder have been developed.31 More recently, PEDOT/lignin composites have also been studied as cathode materials for sodium ion batteries with ionic liquid electrolytes, delivering a maximum capacity of 70 mA h g−1 at 25 mA g−1.32 Liquid electrolytes have much lower cost and should lead to more viable Na-ion batteries. Here, we study the possible storage mechanisms of natural lignin, PEDOT and lignin/PEDOT hybrids in a series of organic liquid electrolytes for lithium and sodium ion batteries. Given the insulating character of lignin, the electrochemical performance is greatly enhanced when it is hybridized with a conjugated polymer (PEDOT) into a biopolymer composite electrode material. To the best of our knowledge, this is the first report on the use of natural lignin as an electrode material for lithium and sodium ion batteries with liquid organic electrolytes.
Experimental
Lignin extraction
Natural lignin was isolated from black liquor after treatment at room temperature by adding 125 ml of 1 M sulfuric acid (H2SO4) to 20 ml of black liquor. After precipitation, the raw lignin was collected by centrifugation (4000 rpm for 5 minutes). The solid was dissolved with sodium hydroxide (NaOH, 0.1 M) and precipitated again with 1 M H2SO4 in a lignin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOH/H2SO4 volume ratio of 1:2; this procedure was repeated five times. The purified biopolymer was separated by centrifugation and dried at 80 °C under vacuum overnight.
:NaOH/H2SO4 volume ratio of 1:2; this procedure was repeated five times. The purified biopolymer was separated by centrifugation and dried at 80 °C under vacuum overnight.
Purification of lignin with NaOH was confirmed by analysing the polymer by EDX under a SEM. The results shown in Table S1† demonstrate that after purification, the percentages of sodium, oxygen and sulfur on the sample have largely decreased.
Synthesis of lignin/PEDOT polymers
Lignin/PEDOT polymers were synthesized via chemical oxidative polymerization of EDOT monomers in the presence of lignin, by using iron(III) chloride as the catalyst and sodium persulfate as the primary oxidant, at room temperature over a period of 8 hours, as explained elsewhere.33 The initial lignin:EDOT mass ratios used were 0:100, 20:80, 40:60 and 60:40, yielding the so-called PEDOT, Lig/PEDOT 20/80, Lig/PEDOT 40/60 and Lig/PEDOT 60/40 polymers, respectively. In order to confirm the polymerization of EDOT in the presence of lignin and the composite character of the final products, Fourier transform infrared (FTIR) and thermogravimetric (TG) analysis experiments were performed on the lignin, and the Lig/PEDOT polymers.
Characterization of lignin and lignin/PEDOT polymers
The chemical composition of natural lignin before and after purification was analyzed by using energy dispersive X-ray (EDX) spectroscopy in a Quanta 200 FEG (FEI) microscope.FTIR spectra were acquired at room temperature using a Thermo Scientific Model Nicolet 6700 FT-IR spectrometer, collecting 10 scans in transmission mode using KBr pellets.
TG analyses were performed on a TGA Q500 (TA Instruments). Measurements were carried out by heating around 3 mg of sample at 10 °C min−1 under a nitrogen atmosphere from room temperature to 800 °C.
Electrochemical testing
Electrochemical tests were conducted in 2032 coin cells, which were assembled in an argon filled dry box. The working electrodes were produced in powder form and tested as cathodes against lithium (Li) or sodium (Na) foil. The cathodes were composed of 4–5 mg of lignin_C65 (80 wt% lignin and 20 wt% Timcal Super C65 conductive carbon black), PEDOT or lignin/PEDOT polymers. When carbon was included, the powder electrodes were prepared by mixing the active polymer with Timcal Super C65 conductive carbon black by hand grinding in a mortar. The electrolytes used were lithium perchlorate (LiClO4), lithium hexafluorophosphate (LiPF6), sodium perchlorate (NaClO4), and sodium hexafluorophosphate (NaPF6) in a 50/50 vol% mixture of ethylene carbonate and dimethyl carbonate (EC/DMC). LiClO4 and LiPF6 were used with the Li anode, while NaClO4 and NaPF6 with the Na anode. Lignin, lignin/PEDOT polymers and carbon C65 were dried under vacuum at 80 °C overnight prior to battery assembly, while the metal anodes and electrolytes were kept inside the dry box. The battery cycling rate is reported in terms of C/n, where n is the number of hours needed to fully charge or discharge the battery based on the experimental capacity of lignin–polypyrrole hybrid bioelectrodes of 80 mA h g−1, reported by Milczarek and Inganäs.26 Then, our cells were cycled at C/20 (4 mA g−1), C/10 (8 mA g−1), and C/2 (40 mA g−1) for five cycles at each rate, and finally at C/20 (4 mA g−1) for 25 cycles. The capacities were calculated per gram of lignin/PEDOT in the electrode. All the experiments started with a reduction step as the PEDOT was already oxidized.Results and discussion
Lignin/PEDOT polymers were synthesized via oxidative polymerization of EDOT in the presence of lignin as previously reported.33Fig. 1 shows the schematic structure of the lignin/PEDOT hybrids. The thermal and chemical characterization of lignin, lignin/PEDOT and PEDOT is shown in Fig. S1.† Lignin/PEDOT hybrid biopolymers showed infrared bands characteristic of both lignin and PEDOT, as well as thermal properties intermediate between those of these materials, confirming the successful hybridization between PEDOT and lignin.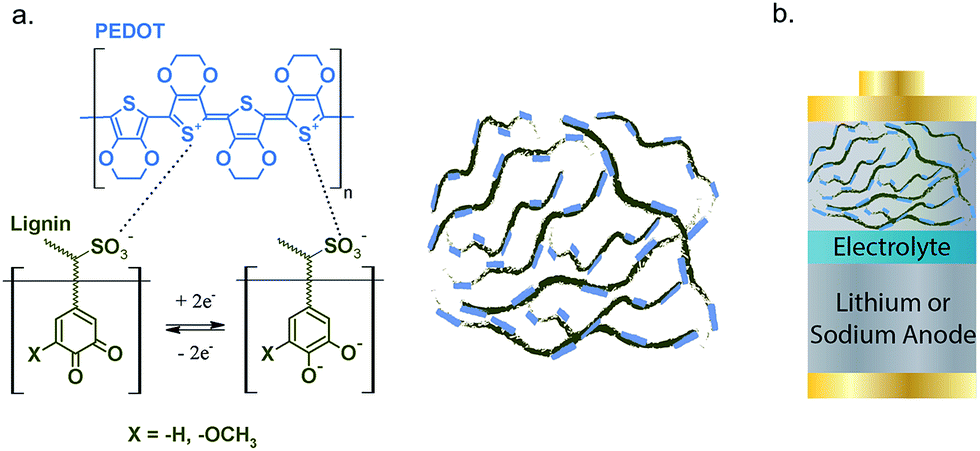 | ||
| Fig. 1 Schematic structure of PEDOT (blue), lignin (olive), and Lig/PEDOT (a) and of the assembled lignin/PEDOT batteries (b). | ||
Upon battery assembly, the OCV of all Lig/PEDOT hybrids varied between 2.5 and 3.0 V vs. Na+/Na or Li+/Li, suggesting the presence of oxidized species (either quinones or oxidized PEDOT or both). The first galvanostatic cycle for all the compounds is shown in Fig. S2† and their respective coulombic efficiencies are listed in Table S2.† Natural lignin stores cations most probably by ionic coupling with the reduced carbonyl functionalities of lignin, while PEDOT storage depends on the ingress/egress of the ions.27,32,34 The extra capacity of lignin_C65 during charging might be caused by the remnant sodium from the NaOH used during purification of the extracted biopolymer. The Lig/PEDOT hybrids showed capacities in the range of 5–35 mA h g−1 during the first reduction, likely due to Li or Na ion intercalation along with quinone reduction. They all showed much larger capacities during the subsequent first oxidation, although this was quite dependent not only on the PEDOT content, but also on the electrolyte, which suggests that most of this capacity must be related to anion insertion into the PEDOT/lignin. Although a contribution from electrolyte decomposition cannot be ruled out, in most cases the oxidation capacity was higher in the presence of LiPF6 than LiClO4, the latter being prone to oxidation at lower voltages, which suggests that this was not the main source of irreversible capacity.
The reversible capacities during the second (Fig. 2 and S3†) and subsequent cycles increased for all the hybrid biopolymers in each of the different electrolytes.
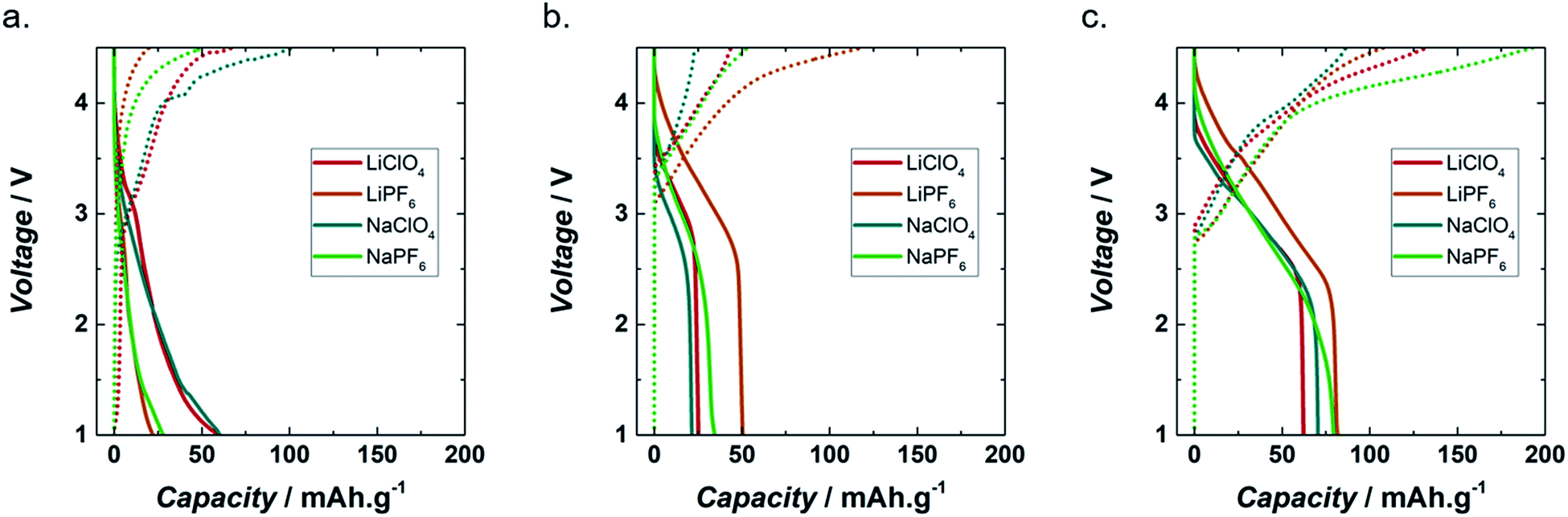 | ||
| Fig. 2 Capacity profiles of lignin_C65 (a), PEDOT (b) and Lig/PEDOT 20/80 (c) for the 2nd cycle at C/20 (4 mA g−1) in 1 M LiClO4 EC/DMC, 1 M LiPF6 EC/DMC, 1 M NaClO4 EC/DMC and 1 M NaPF6 EC/DMC. Continuous and discontinuous lines indicate discharge and charge, respectively. | ||
Fig. 2a shows the galvanostatic voltage profile of lignin_C65 with lithium and sodium perchlorate and hexafluorophosphate. All the discharge curves showed a change in slope at around 3 V where the reduction of lignin along with cation (i.e. Li+ or Na+) insertion started. The highest discharge capacity, 60 mA h g−1, was achieved in both LiClO4 and NaClO4 based organic electrolytes. In the case of the hexafluorophosphate electrolytes, the capacities achieved were only 22 and 28 mA h g−1, for LiPF6 and NaPF6, respectively.
Despite the observed trend in the discharge capacity, mainly depending on the electrolyte anion, the behaviour of the lignin_C65 electrode during charging varied mostly according to the cation. In LiClO4 and LiPF6, the charging redox process started at 1 and 1.1 V, respectively. Then, several changes in slope were observed starting at 2.7 and 3.4 V, respectively. The total charge capacity for LiClO4 and LiPF6 was 67 and 20 mA h g−1, respectively.
In NaClO4 and NaPF6, several changes in slope were observed, but only above 2.3 V for both electrolytes. The total charge capacity was approximately 103 and 51 mA h g−1, respectively. Generally, quinone-based materials in organic electrolytes present a series of asymmetric plateaux at around 2.5–3.5 V. The voltage of these plateaux depends on the chemical environment of the quinone moieties.24,35,36
The charge capacity was higher than that of discharge in LiClO4, NaClO4 and NaPF6, with extra capacities of 12, 72 and 82% respectively being achieved during charging. In LiPF6, there was an irreversible capacity of about 2 mA h g−1 or nearly 9% of the discharge.
Fig. 2b shows the galvanostatic discharge and charge profiles of PEDOT in each of the different electrolytes during the second cycle. In contrast to lignin_C65, which showed a steeper profile down to 1 V, all the redox processes in PEDOT seemed to occur at voltages above 2.5 V vs. A+/A (A = Li, Na), with negligible capacity below that voltage only in the case of NaPF6 containing electrolyte. The redox mechanism of conducting polymers has been reported to comprise the intercalation of both ions of the electrolyte.37 For PEDOT the charging capacities were about 44 and 23 mA h g−1 for LiClO4 and NaClO4, respectively. In LiPF6 the discharge and charge capacities were 50 and 120 mA h g−1, respectively. In NaPF6, the discharge and charge capacities were 34 and 53 mA h g−1, respectively.
Fig. 2c presents the electrochemical behaviour of Lig/PEDOT 20/80 during the second cycle. This biopolymer showed the highest capacity of all the mixed compositions, in LiClO4, LiPF6, NaClO4 and NaPF6. Data of Lig/PEDOT 40/60 and 60/40 are included in Fig. S3.† In general, the voltage profile of the discharge curves bore closer resemblance to that of pure PEDOT cathodes (Fig. 2b), which is the major component, than that of lignin (Fig. 2a), although the voltage range for cation insertion was extended to lower voltages. More importantly, there was a clear decrease in polarization, since the oxidation processes in Lig/PEDOT 20/80 started at voltages below 3 V vs. A+/A, while they only started at 3.1 to 3.4 V vs. A+/A in PEDOT. As a result, there was an increase in the capacity values, in comparison with bare PEDOT and bare lignin. This suggests a synergistic effect in the Lig/PEDOT 20/80 composite that might be attributed to the coupling of their redox processes, i.e. the swelling of PEDOT, which facilitates ion transportation, may assist in the electrochemical reaction of lignin. Moreover, the high content of conducting PEDOT in physical proximity with insulating lignin also aided the electronic transport of lignin.
The intercalation of the cations in Lig/PEDOT 20/80 depended on the anion present in the electrolyte. At around 2.4 V, Lig/PEDOT 20/80 stopped intercalating cations in LiClO4, LiPF6 and NaClO4, while in NaPF6 there was only a change of slope, after which the intercalation continued.
The discharge capacities for Lig/PEDOT 20/80 were 82, 79, 71 and 62 mA h g−1 in LiPF6, NaPF6, LiClO4, and NaClO4, respectively.
During charging, the de-intercalation of the cations was independent of the electrolyte and started at around 2.7 V.
In order to compare the evolution upon cycling, a more detailed evaluation of the galvanostatic curves at C/20, with the capacities normalized to their maximum values, is shown in Fig. 3 and S4.† In general, the intercalation of ions in PEDOT and Lig/PEDOT 20/80 started at higher voltages than for lignin_C65 likely due to better conductivity and lower resulting polarization, while the de-intercalation seemed to initiate first in lignin than in the other two polymers. As mentioned before, the galvanostatic curves of lignin did not show a clear plateau and the Lig/PEDOT 20/80 curves resembled the ones observed for PEDOT.
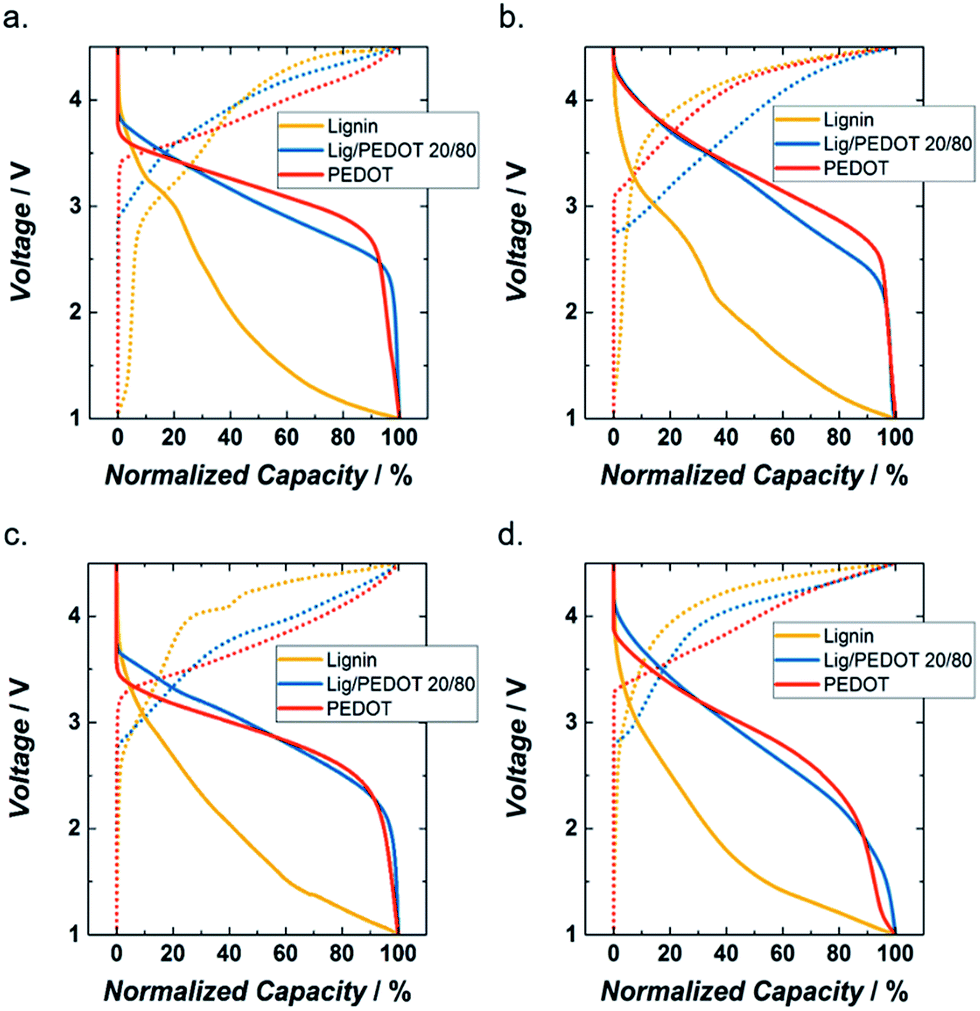 | ||
| Fig. 3 Normalized capacity profiles of lignin_C65, PEDOT and Lig/PEDOT 20/80 for the 2nd cycle at C/20 (4 mA g−1) in 1 M LiClO4 EC/DMC (a), 1 M LiPF6 EC/DMC (b), 1 M NaClO4 EC/DMC (c) and 1 M NaPF6 EC/DMC (d). Continuous and discontinuous lines indicate discharge and charge, respectively. | ||
PEDOT and Lig/PEDOT 20/80 achieved most of their discharge capacity at higher voltages than lignin_C65; this might be a consequence of a much slower cation transfer through lignin. By using Lig/PEDOT biopolymers as cathodes, we took advantage of the ability of both initial materials to store energy; however, an excess of lignin in the biopolymer might result in a decrease of the accessible electroactive sites.32
Fig. 4 shows the results of cycling lignin_C65, Lig/PEDOT 20/80 and PEDOT at different rates in each of the electrolytes. It can be seen that in LiClO4 (Fig. 4a), Lig/PEDOT 20/80 presented the most favourable combination of total capacity (62 mA h g−1 at C/20) and capacity retention (44% after 25 cycles at C/20). Lignin_C65 showed a similar performance, yet the capacity values were lower and less stable while PEDOT exhibited poor behaviour. The other Lig/PEDOT proportions did not intercalate lithium ions (Fig. S5a†) in this electrolyte. This behaviour proves that in this electrolyte, lignin requires either a conductive filler (C65) or a high proportion (80%) of electron conducting polymer.
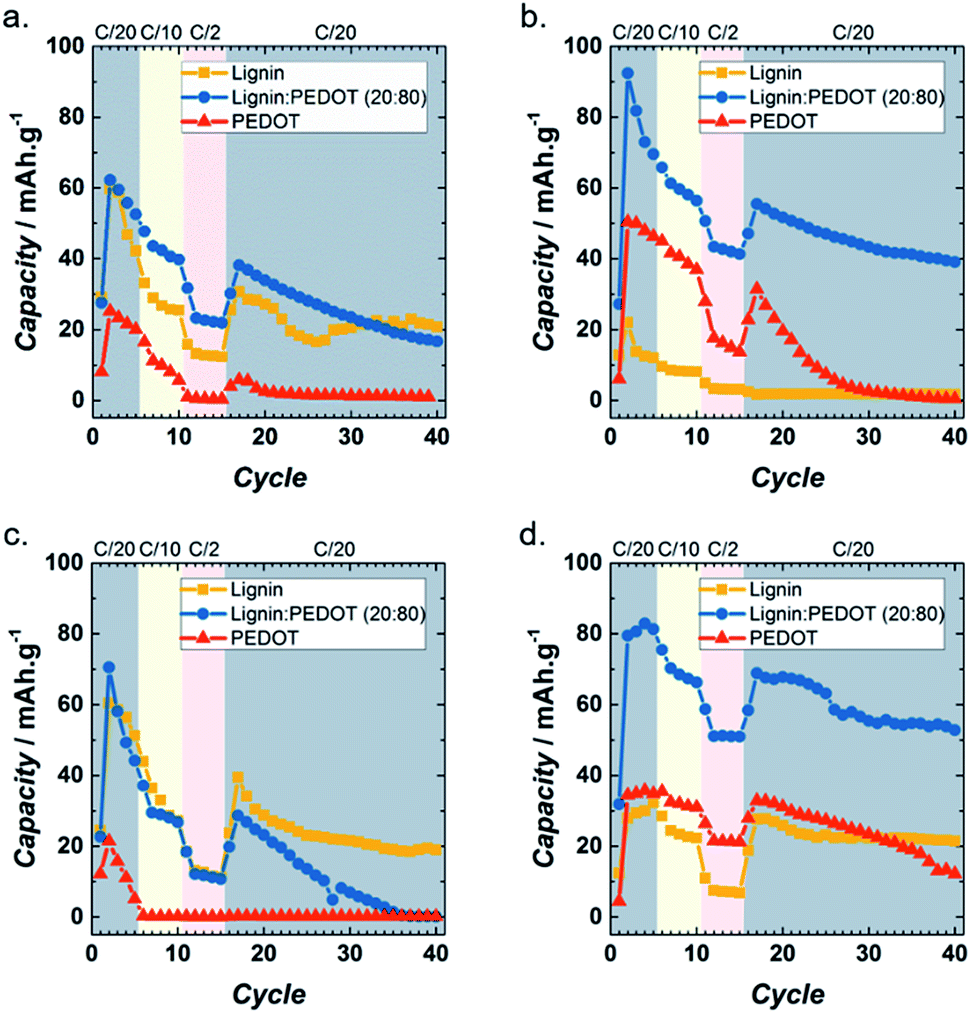 | ||
| Fig. 4 Rate capabilities of lignin_C65, PEDOT and Lig/PEDOT 20/80 at various current rates in 1 M LiClO4 EC/DMC (a), 1 M LiPF6 EC/DMC (b), 1 M NaClO4 EC/DMC (c) and 1 M NaPF6 EC/DMC (d). | ||
In LiPF6, Lig/PEDOT 20/80 exhibited the highest capacity of all the materials (Fig. 4b) and showed an improvement in cycle-life in comparison to the same material in LiClO4. Capacity values up to 92 mA h g−1 at C/20 and a capacity retention of 70% after 25 cycles at C/20 were achieved with this material. In contrast to the performance in LiClO4, all the other Lig/PEDOT proportions (Fig. S5b†) inserted lithium ions into the cathode, showing an improvement over the intercalation properties of lignin.
Quite different results were achieved when cycling with Na electrolytes vs. Na+/Na. In the case of NaClO4 (Fig. 4c and S5c†), the C65 conductive filler allowed the continuous insertion of ions into the lignin cathode, retaining 48% of its initial capacity. The hybridization of lignin and PEDOT worsened the Na+ intercalation abilities in NaClO4. Lig/PEDOT 20/80 achieved the highest capacity during the second discharge, but the capacity faded very fast.
A contrasting effect is observed in NaPF6 (Fig. 4d and S5d†), where all the lignin-derived materials presented electrochemical activity even after 40 cycles. Increasing the proportion of PEDOT enhanced the capacity values of lignin, with a maximum value of 83 mA h g−1 for Lig/PEDOT 20/80 at C/20.
In general, the behaviour of these cathode biohybrid materials seemed to be highly dependent on the salt anion. Indeed, comparing the plots on the left and the right of Fig. 4 and S5,† it is evident that the electrochemical process was enhanced in the Lig/PEDOT hybrids when PF6− was the anion counter-balancing the alkali metal cation.
Incremental capacity analysis (ICA) is a powerful technique used to obtain information on aging mechanisms. ICA is based on dQ/dV vs. V plots and therefore transforms voltage plateaus and inflection points in voltage curves into dQ/dV peaks.38 To investigate the aging mechanism of Lig/PEDOT 20/80 in the different electrolytes, ICA of different stages of cycling, i.e. the 2nd, 16th, and 40th cycles, is shown in Fig. 5. The 16th cycle corresponds to the first cycle after cycling at different current densities.
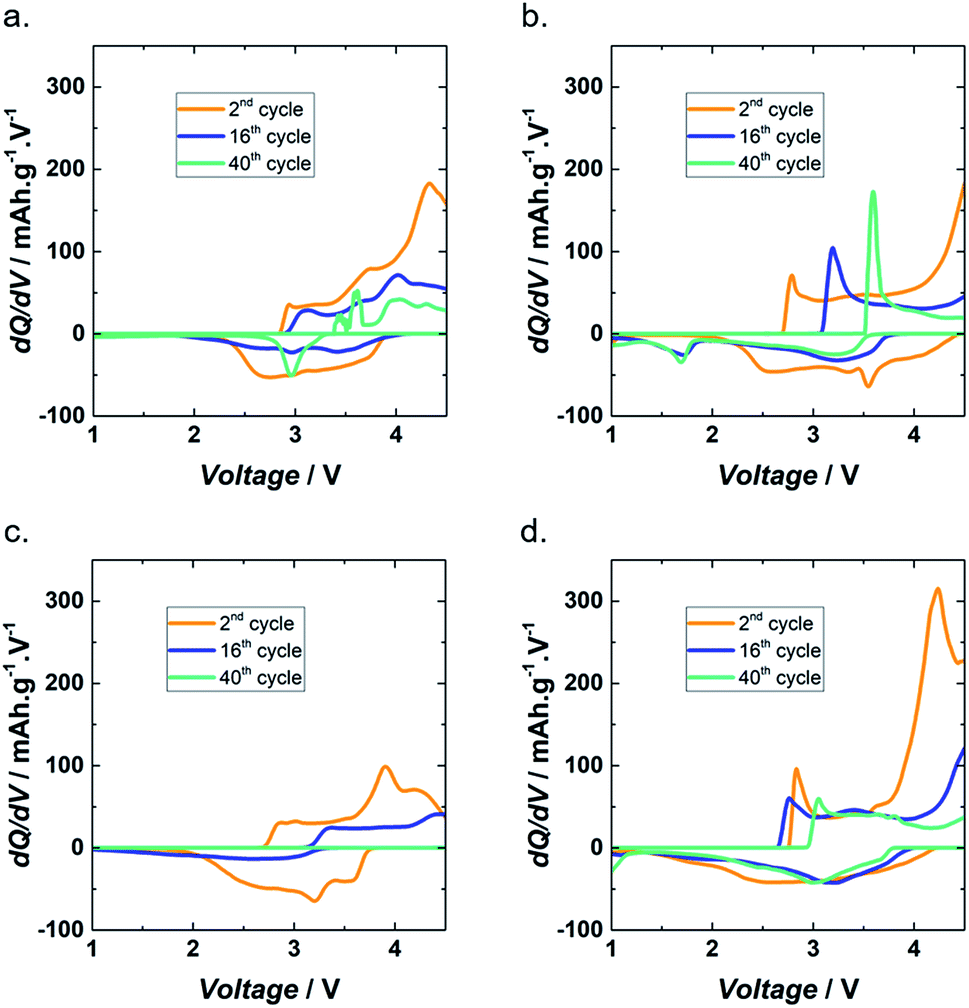 | ||
| Fig. 5 dQ/dV of Lig/PEDOT 20/80 at different cycles in 1 M LiClO4 EC/DMC (a), 1 M LiPF6 EC/DMC (b), 1 M NaClO4 EC/DMC (c) and 1 M NaPF6 EC/DMC (d). | ||
In general, the charging and discharging peaks of Lig/PEDOT 20/80 were stochastic. This might be caused by the resistivity of the electrodes and difficulty for Li+ and Na+ to intercalate.
During cycling, two common characteristics were observed in the dQ/dV plots in all the electrolytes. On the one hand, the diminishing peak intensity indicated loss of active material, implying that there was a loss of electrical contact coming from a percolation origin.39
On the other hand, the shift towards higher/lower voltages during discharge/charge could suggest an evolution of the composition and morphology of the electrode material or an indication of underdischarging and undercharging. The fact that the underdischarging and undercharging effect appeared during both discharge and charge with a similar magnitude indicated that this increase of polarization was ohmic in nature.38
In order to improve the electronic conductivity and cation diffusion, a composite made from 20 wt% Timcal Super C65 conductive carbon and 80 wt% Lig/PEDOT 20/80 (herein Lig/PEDOT 20/80 + C65) was tested as the cathode for Li- and Na-ion batteries. LiPF6 and NaPF6 were selected as the electrolytes, given the excellent performance of Lig/PEDOT 20/80 in them. Fig. 6 shows the cycle-life data of Lig/PEDOT 20/80 and Lig/PEDOT 20/80 + C65 in LiPF6 and NaPF6. The inclusion of C65 in the biopolymer electrode improved the discharge capacity values up to 119 mA h g−1 and 159 mA h g−1 at C/20 in LiPF6 and NaPF6, respectively. These values were calculated per gram of Lig/PEDOT.
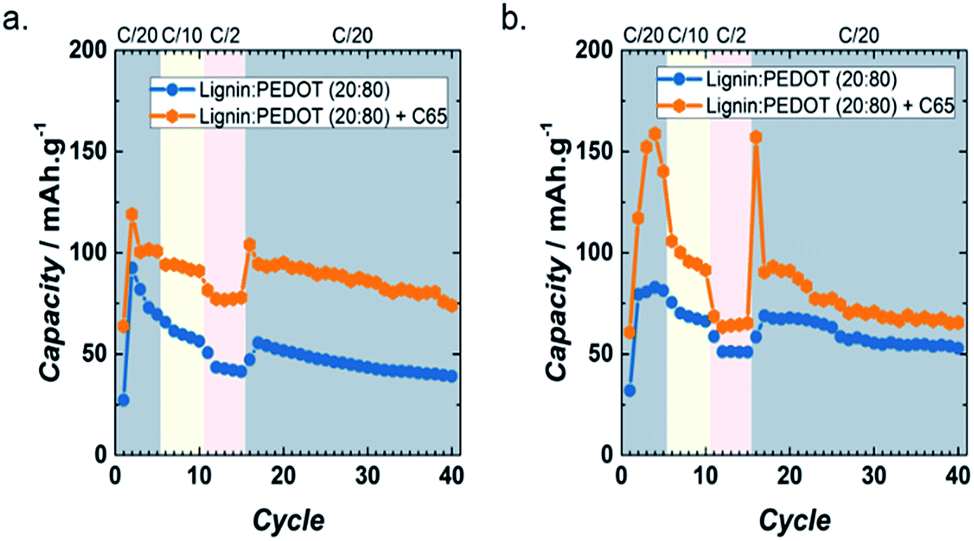 | ||
| Fig. 6 Rate capabilities of Lig/PEDOT 20/80 and Lig/PEDOT 20/80 + C65 at various current rates in 1 M LiPF6 EC/DMC (a) and 1 M NaPF6 EC/DMC (b). | ||
The galvanostatic curves of Lig/PEDOT 20/80 with and without C65 (Fig. S6†) showed the same general trend. Nevertheless, when C65 was added to the biopolymer and the composite tested in LiPF6 (Fig. S6a†), the peak intensity in the dQ/dV plots was increased, showing that C65 improved the Li+ diffusion inside the Lig/PEDOT biopolymer.
Moreover, when the composite was tested in NaPF6 (Fig. S6b†), the insertion/de-insertion occurred at higher/lower voltages than when Lig/PEDOT 20/80 was tested and the intensity of certain peaks was enhanced. The combination of these two phenomena resulted in the increase of the capacity values when C65 was added to the biopolymer.
The difference in the rate capabilities of the biopolymers in LiPF6 and NaPF6 might be caused by the difference in reduction potential between the two anodes. For example, in the case of NaPF6, the highest oxidation peak observed during the second cycle was at 4.25 V vs. Na/Na+, while in LiPF6 the peak was not completely resolved (onset at 3.9 V vs. Li/Li+). This might lead to an initial enhanced capacity for NaPF6, but also a decreased capacity retention. However, the complete mechanism should be further investigated in future studies.
The performance of these biopolymer Lig/PEDOT hybrid electrodes is comparable with that of other electrode materials and surpasses that of other lignin based bulk electrode materials. Hydrolysis lignin has been studied as a cathode for Li-ion primary batteries at low rates. A hydrolysis lignin cathode was constructed with 76% active component and the final electrode was heated at 280 °C for 2 hours and tested in 1 M LiBF4 in γ-butyrolactone and 1 M LiClO4 in propylene carbonate, delivering up to 185 mA h g−1 at 75 μA cm−2.31 Lignin/PEDOT hybrids have recently been studied for sodium battery applications in BMPyrTFSI:NaTFSI (20 mol%) and EMImFSI:NaFSI (20 mol%) electrolytes, achieving up to 46 and 70 mA h g−1 at 25 mA g−1, respectively.32 By deriving our electrodes from black liquor, rather than commercial lignin, we are actually using a low cost by-product from the paper industry. Moreover, by employing more conventional electrolytes with smaller anions, the ion exchange was improved and therefore the capacity values were enhanced. Furthermore, in the present work the percentage of active material within the electrode was increased from 65 to 80%, therefore enhancing the gravimetric energy density. Further capacity improvements could be expected from electrode optimization in the form of thin films.
Conclusions
To sum up, in this work we synthesized and investigated lignin, PEDOT and lignin/PEDOT hybrid biopolymers as bulk electrode materials for electrochemical energy storage in lithium and sodium ion batteries. The interesting synergistic behaviour of lignin/PEDOT biopolymer hybrids leads to high storage capacity, i.e. ∼50 mA h g−1 at C/20 in LiPF6 and NaPF6. To enhance to the maximum the delivered capacity, a composite made from C65 and a lignin/PEDOT biopolymer was tested as the cathode against lithium and sodium, and peak values up to 159 mA h g−1 and a stable value of 74 mA h g−1 were attained. This work demonstrates that biopolymers based on lignin, a renewable and inexpensive material, can be used as sustainable organic electrodes with highly efficient electrochemical energy storage for lithium- and sodium-ion batteries. Surface and interface engineering is essential to improve the electrochemical performance of lignin/PEDOT materials for lithium- and sodium-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council through Starting Grant Innovative Polymers for Energy Storage (iPes) 306250 (DM), the Etortek Program (ENERGIGUNE12) of the Basque Government and the Ministerio de Economía y Competitividad (MINECO) of the Spanish Government through the Proyectos I+D Retos 2013 program (LINABATT project, reference number: ENE2013-44330-R). AMNS (PRE_2014_1_62) and NC were supported by the Basque Government Scholarship for pre-doctoral formation. JCG would like to thank the Spanish Ministry of Economy, Industry and Competitiveness, for funding through a Ramón y Cajal fellowship (RYC-2015-01627). Finally, the kind donation of black liquor samples from Smurfit Kappa Nervion (Basque Country, Spain) and Papelera Guipuzcoana de Zicuñaga, S. A. (Basque Country, Spain) is gratefully acknowledged.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- G. L. Soloveichik, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 503–527 CrossRef CAS PubMed.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312 CAS.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS.
- G. Crabtree, Nature, 2015, 526, S92 CrossRef CAS PubMed.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- E. Gonzalo, M. H. Han, J. M. López del Amo, B. Acebedo, M. Casas-Cabanas and T. Rojo, J. Mater. Chem. A, 2014, 2, 18523–18530 CAS.
- G. Sandu, B. Ernould, J. Rolland, N. Cheminet, J. Brassinne, P. R. Das, Y. Filinchuk, L. Cheng, L. Komsiyska, P. Dubois, S. Melinte, J.-F. Gohy, R. Lazzaroni and A. Vlad, ACS Appl. Mater. Interfaces, 2017, 9, 34865–34874 CAS.
- M. Arbabzadeh, J. X. Johnson, G. A. Keoleian, P. G. Rasmussen and L. T. Thompson, Environ. Sci. Technol., 2016, 50, 1046–1055 CrossRef CAS PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef.
- N. Casado, G. Hernández, H. Sardon and D. Mecerreyes, Prog. Polym. Sci., 2016, 52, 107–135 CrossRef CAS.
- R. Zhao, L. Zhu, Y. Cao, X. Ai and H. X. Yang, Electrochem. Commun., 2012, 21, 36–38 CrossRef CAS.
- W. Deng, X. Liang, X. Wu, J. Qian, Y. Cao, X. Ai, J. Feng and H. Yang, Sci. Rep., 2013, 3, 2671 CrossRef PubMed.
- A. M. Navarro-Suárez, J. Carretero-González, T. Rojo and M. Armand, J. Mater. Chem. A, 2017, 5, 23292–23298 Search PubMed.
- L. Cheng, X. Du, Y. Jiang and A. Vlad, Nano Energy, 2017, 41, 193–200 CrossRef CAS.
- Y. Ma, L. Lv, Y. Guo, Y. Fu, Q. Shao, T. Wu, S. Guo, K. Sun, X. Guo, E. K. Wujcik and Z. Guo, Polymer, 2017, 128, 12–23 CrossRef CAS.
- M. Yao, H. Senoh, T. Sakai and T. Kiyobayashi, Int. J. Electrochem. Sci., 2011, 6, 2905–2911 CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 CAS.
- G. Milczarek, Electroanalysis, 2007, 19, 1411–1414 CrossRef CAS.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed.
- K. Hernández-Burgos, G. G. Rodríguez-Calero, W. Zhou, S. E. Burkhardt and H. D. Abruña, J. Am. Chem. Soc., 2013, 135, 14532–14535 CrossRef PubMed.
- B. R. Eggins and J. Q. Chambers, J. Electrochem. Soc., 1970, 117, 186 CrossRef CAS.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- S. V. Gnedenkov, D. P. Opra, S. L. Sinebryukhov, A. K. Tsvetnikov, A. Y. Ustinov and V. I. Sergienko, J. Solid State Electrochem., 2013, 17, 2611–2621 CrossRef CAS.
- N. Casado, M. Hilder, C. Pozo-Gonzalo, M. Forsyth and D. Mecerreyes, ChemSusChem, 2017, 10, 1783–1791 CrossRef CAS PubMed.
- A. M. Navarro-Suárez, N. Casado, J. Carretero-González, D. Mecerreyes and T. Rojo, J. Mater. Chem. A, 2017, 5, 7137–7143 Search PubMed.
- F. Blanchard, H. Pagès, B. Carré, F. Bonhomme, P. Biensan and D. Lemordant, J. New Mater. Electrochem. Syst., 2003, 6, 245–249 CAS.
- H. Senoh, M. Yao, H. Sakaebe, K. Yasuda and Z. Siroma, Electrochim. Acta, 2011, 56, 10145–10150 CrossRef CAS.
- W. Huang, Z. Zhu, L. Wang, S. Wang, H. Li, Z. Tao, J. Shi, L. Guan and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9162–9166 CrossRef CAS PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- M. Dubarry, V. Svoboda, R. Hwu and B. Yann Liaw, Electrochem. Solid-State Lett., 2006, 9, A454–A457 CrossRef CAS.
- T. Waldmann, A. Iturrondobeitia, M. Kasper, N. Ghanbari, F. Aguesse, E. Bekaert, L. Daniel, S. Genies, I. J. Gordon, M. W. Löble, E. De Vito and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2016, 163, A2149–A2164 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00551b |
| ‡ Current address: Department of Physics, Chalmers University of Technology, 41296 Gothenburg, Sweden. |
| § Current address: Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2018 |