
Cobalt hexacyanoferrate supported on Sb-doped SnO2 as a non-noble catalyst for oxygen evolution in acidic medium†
Bárbara
Rodríguez-García
a,
Álvaro
Reyes-Carmona
 ab,
Ignacio
Jiménez-Morales
b,
Marta
Blasco-Ahicart
a,
Sara
Cavaliere
b,
Marc
Dupont
b,
Deborah
Jones
b,
Jacques
Rozière
b,
José Ramón
Galán-Mascarós
*ac and
Frédéric
Jaouen
*b
ab,
Ignacio
Jiménez-Morales
b,
Marta
Blasco-Ahicart
a,
Sara
Cavaliere
b,
Marc
Dupont
b,
Deborah
Jones
b,
Jacques
Rozière
b,
José Ramón
Galán-Mascarós
*ac and
Frédéric
Jaouen
*b
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans, 16. Tarragona, E-43007, Spain. E-mail: jgalan@iciq.es
bInstitute Charles Gerhardt Montpellier (ICGM), UMR 5253, Université de Montpellier, CNRS, ENSCM, F-34095 Montpellier Cedex 5, France. E-mail: frederic.jaouen@umontpellier.fr
cICREA, Pg. Lluís Companys, 23, E-08010, Barcelona, Spain
First published on 18th December 2017
Abstract
This study investigates the activity and stability of a Prussian blue analogue (PBA) as an inexpensive anode catalyst for Polymer Electrolyte Membrane Water Electrolysis (PEMWE). While some PBAs have recently been reported to catalyze the oxygen evolution reaction (OER) in acidic electrolytes, the present study focuses on their integration in a PEMWE device. Cobalt hexacyanoferrate nanoparticles were interfaced with an electrically conductive support that withstands the PEMWE anodic conditions, namely Sb-doped SnO2. The OER activity of the composite materials was first verified in liquid electrolytes and then in PEMWE. A promising current density of 50–100 mA cm−2 was reached at 2 V cell voltage. The PBA/Sb–SnO2 anode was stable up to 1.9 V, but showed more and more instability at higher potentials. Increasing leaching rates of Sn and Sb observed above 1.9 V suggest that the material instability above 1.9 V can mainly be assigned to Sb-doped SnO2 conductive support. These results are overall promising for the use of PBAs as catalytic sites at the anode of PEMWE. The study also identifies the need for more active PBAs in order to reach a higher current density at a cell voltage of 1.6–1.9 V, a potential range necessary for an acceptable energy efficiency of the PEMWE.
1 Introduction
Water electrolysis is a promising approach to convert electricity into hydrogen via water splitting, with energy-conversion efficiency up to 75%.1 The increase of electric power produced from solar and wind energy will require efficient and affordable energy storage solutions. Clean hydrogen produced via water electrolysis is a promising energy vector with key applications for transportation and electricity storage. Polymer Electrolyte Membrane Water Electrolysis (PEMWE) is a particularly appealing electrolysis technology with key advantages of fast response, production of ultrapure and electrochemically-pressurized H2 and low footprint. However, its current high cost could limit its widespread application in the near future. Its operating conditions (acidic medium, ca. 80 °C and high electrochemical anode potential) strongly limit the choice of materials that may be used for cell housing, bipolar plates and electrode catalysts and supports. The high electrochemical anode potential during operation sets particularly stringent requirements on materials. Titanium is currently used as a bipolar plate material in commercial PEMWE,1,2 while bulk IrO2–RuO2 supported on Ti prevails as the anode catalyst.3 Iridium is expensive and the rarest metal in Earth's crust, with a current production of less than 9 tons a year.4 The main ongoing lines of research for reducing the Ir loading while maintaining the same performance of PEMWE anodes are (i) nanostructuring of IrO2,5 (ii) dispersion on high surface area supports,6 (iii) formation of solid solutions with less expensive elements such as Ru and Sn,7 (iv) core–shell approaches,8 and (v) amorphous IrO2.9 The higher Oxygen Evolution Reaction (OER) activity of amorphous IrO2 has however recently been reported to be accompanied by poor stability.9Substitution of IrO2 and RuO2 by a low-cost alternative for OER catalysis in acidic medium would thus have a tremendous impact on the future large-scale deployment of PEMWE technology. While earth-abundant and inexpensive transition metal oxides such as Ni, Fe and Co catalyze the OER in alkaline medium, none of these materials that might be active toward OER in acid are thermodynamically stable in strongly acidic medium in the potential range of 1.0–1.7 V vs. RHE. While the literature on non-platinum group metals (PGM) catalysts for OER in acidic medium is scarce, recent studies have investigated stabilized metal oxides of earth-abundant metals.10–12 Early transition metals from the first row of 3d elements form more stable oxides than late transition metals from the same row, and MnO2 sits at the threshold of required stability at pH 0–1, with expected stability in the range of 1.0–1.6 V vs. RHE.13 The OER activity of MnO2 in acidic medium is however much lower than that in alkaline medium, and its stability is also insufficient.10 The Mn leaching rate during OER at 1.8 or 1.9 V vs. RHE could be reduced by ca. a factor two via the preparation of co-sputtered Mn and Ti oxides, following density-functional-theory (DFT) insights on possible stabilization paths.10 Co3O4 has also recently been reinvestigated for OER in acid medium. A cobalt film was deposited with electron-beam evaporation on fluoride-doped tin oxide (FTO) and annealed in air to form a non-smooth but crystalline Co3O4 surface.11 In 0.5 M H2SO4, the 300 nm thick Co3O4/FTO film showed a current density of ca. 8 mA cm−2 at 1.8 V vs. RHE, vs. ca. 1.58 V and 1.55 V for RuO2/FTO and IrO2/FTO films at the same current. With various control experiments, the authors could conclude that the key to observe a similar high OER activity was a proper interface between CoOx and the conductive substrate, including a small thickness of cobalt oxide. The leaching rate of cobalt during OER was also measured in galvanostatic conditions and reported to be 6 μgCo h−1 at 10 mA cm−2 (ca. 1.8 V vs. RHE applied). This relatively mild leaching rate at pH <1 is surprising considering the Pourbaix diagram of cobalt and a previous investigation of the pH-dependency of active cobalt species for OER.14 In another attempt to stabilize OER active cobalt sites in acidic medium, Nocera and co-workers recently investigated decoupled activity and stability concepts, including CoMnOx and CoFePbOx films.12 In that study, the CoFePbOx film was the only one resulting in a nearly constant potential (ca. 1.65 V vs. RHE) over 12 h at a fixed current density of 1 mA cm−2 in pH 2.5 solution, extended to 50 h in pH 2. These studies show that cobalt ions in mixed metal oxides may be relatively stable in acidic conditions, even at high electrochemical potential.
Cobalt ions coordinated by nitrogen and/or carbon atoms have also been predicted by DFT to be OER active,15 and pyrolyzed Co–N–C catalysts have shown promising high initial OER activity, at least in alkaline medium.16 The underlying major issue with Co(Metal)–N–C catalysts for OER is the recognized thermodynamic and kinetic instability of amorphous as well as graphitic carbon at OER potentials, further exacerbated at a temperature higher than ca. 50 °C.17,18 In this work, we investigate the OER activity and stability of a Prussian-blue analogue (PBA) based on Co, Fe and cyano-ligands, free of weak C–C bonds. Despite the fact that PBAs are a well-known class of materials with interesting photocatalytic, electrochemical, and electrochromic properties, their catalytic activity towards OER was not reported until some years ago, when we reported it.19–21 Among such materials, cobalt hexacyanoferrate (CoHFe) has hitherto shown the highest OER activity in neutral and acidic electrolyte22 and, remarkable for precious-metal-free materials, has displayed strong stability even after prolonged immersion in pH 1 solution, or after OER operation in pH 2 electrolyte.20 While CoHFe was recently proposed as a potential anode catalyst for PEMWE, it has hitherto not yet been integrated in a Membrane Electrode Assembly (MEA) and characterized in a PEMWE device, in relevant conditions (80 °C, proton-conducting polymer electrolyte). For MEA integration, a major drawback of CoHFe, and PBA in general, is their low electronic conductivity. Hence, such catalysts need to be properly interfaced with an electron-conductive support that also tolerates the anode operating conditions of PEMWE. While the preparation of composites of CoHFe or PBA deposited on carbon allotropes have previously been investigated,23 carbon allotropes are unsuitable for PEMWE anodes, as mentioned earlier. Composites of CoHFe and TiO2 were recently prepared and characterized,24 but not interrogated for their OER activity.
Here, we report the first interfacing of CoHFe nanoparticles25 with a conductive oxide that withstands the PEMWE anode conditions, namely antimony-doped tin oxide (ATO). The latter has recently been investigated as a carbon-free and therefore corrosion-resistant support for platinum particles at the cathode of PEM fuel cells26 and also as a stable support for IrO2 at the anode of PEM electrolyzers.27,28 The ATO@CoHFe composite catalyst shows promising activity and short term stability for at least 20 h in PEMWE operating conditions (80 °C, Nafion® polymer electrolyte), while the upper limit of ATO stability itself is shown to be ca. 1.9 V vs. RHE.
2 Experimental
2.1 Synthesis of CoHFe and ATO
CoHFe nanoparticles were prepared by a modified literature protocol.29 Vo et al. reported a correlation between decreasing CoHFe particle size and increasing formamide content in formamide–water solutions used for PBA crystal growth. To prepare nanosized CoHFe, we used undiluted formamide as to achieve the minimum size accessible by this technique. A formamide solution of K3Fe(CN)6 (0.03 M, 50 mL, Sigma-Aldrich) was instantly poured into a formamide solution of Co(NO3)2 (0.02 M, 50 mL, Sigma-Aldrich) immediately forming a dark purple dispersion. The solution was kept under stirring for 2 h and collected by filtration. The solid material was washed by redispersion in deionized water and centrifuged at 6000 rpm and decanted. This washing/purification process was repeated three times to collect the product as a fine red powder. Finally, the CoHFe nanoparticles were dried at 60 °C. Antimony tin oxide (ATO) loose tubes were prepared by electrospinning, as previously reported by us.30 In summary, 0.1 g SbCl3 (99%, Sigma-Aldrich) was added to a solution containing 0.78 g SnCl2 (98%, Sigma-Aldrich) and 0.8 g polyvinylpyrrolidone (PVP, Mw ∼ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000, Sigma-Aldrich) dissolved in a 1.8:1 ratio mixture of ethanol and N,N-dimethylformamide. The obtained mixture was stirred overnight and electrospun at room temperature on a rotating drum (Linari Biomedical) at 15 kV, a needle-collector distance of 10 cm and a flow rate of 0.3 mL h−1. The electrospun SbCl3–SnCl2/PVP fibers were calcined at 600 °C for 4 h in air with a heating rate of 5 °C min−1 in order to remove the carrier polymer and to form crystalline ATO loose tubes. The synthesis described above results in 10 at% Sb in SnO2, a composition that was previously shown to result in high electronic conductivity.30
300000, Sigma-Aldrich) dissolved in a 1.8:1 ratio mixture of ethanol and N,N-dimethylformamide. The obtained mixture was stirred overnight and electrospun at room temperature on a rotating drum (Linari Biomedical) at 15 kV, a needle-collector distance of 10 cm and a flow rate of 0.3 mL h−1. The electrospun SbCl3–SnCl2/PVP fibers were calcined at 600 °C for 4 h in air with a heating rate of 5 °C min−1 in order to remove the carrier polymer and to form crystalline ATO loose tubes. The synthesis described above results in 10 at% Sb in SnO2, a composition that was previously shown to result in high electronic conductivity.30
2.2 CoHFe and ATO mixing and ink preparation for liquid-electrolyte electrochemistry
CoHFe nanoparticles and ATO were mixed in deionized water by a hydrothermal method. ATO (20 mg) and the desired mass of CoHFe (from 2 to 16 mg, corresponding to 9–44 wt% CoHFe on ATO) were dispersed in 10 mL of deionized water (pH 6.2). Then, the solution was heated under autogenous pressure at 100 °C for 20 h. After this, the solid material was separated from the solvent by centrifugation, washed with deionized water, and dried via successive ethanol/ether washings.The catalyst ink for the anode preparation in RDE experiments was prepared by adding the ATO@CoHFe sample (5 mg) to a mixture of ethanol (440 μL), H2O (100 μL) and Nafion solution (10 μL, 5 wt% Nafion dispersed in lower alcohols, Sigma-Aldrich). The suspension was sonicated for 30 min. An ink aliquot (4.7 μL) was deposited on a titanium support electrode of 0.07 cm2 geometric area, for a total ATO@CoHFe loading of 610 μg cm−2. Blank inks were prepared with pure ATO or pure CoHFe at the same loadings.
2.3 Ink preparation and catalyst layer preparation for PEMWE anodes
ATO@17% CoHFe (e.g. obtained from 39.7 mg ATO and 8 mg CoHFe) was selected on the basis of its OER activity measured in liquid electrolyte. The catalyst ink for the PEMWE anode was obtained by mixing ATO@17% CoHFe (37.5 mg), Nafion solution (215 μL of 5 wt% Nafion solution, Sigma-Aldrich), 3 mL isopropanol and 1 mL deionized water in a 5 mL vial. This results in 20 wt% dry Nafion to the total mass of solids. The ink was sonicated for 45 min and entirely sprayed with an aerograph (Badger Air-brush, universal®) on a Teflon-glass fiber fabric masked with a second, thicker, Teflon-glass fiber fabric (Plastiques Élastomères). The targeted catalyst load was 3 mg cm−2. Both masks were placed on a heating pad at 80 °C. The exact catalyst loading deposited on the active area was deduced from the weight change of the Teflon sheet before and after the spraying and drying.2.4 MEA preparation
The Nafion® 115 membrane (Ion Power inc.) was first cleaned with 3% H2O2 solution at 80 °C for 1 h, then with 1 M H2SO4 at 80 °C for 1 h and finally with deionized water at 80 °C for 1 h. The membrane was dried at 80 °C in an oven and stored. The anode catalyst layer deposited on Teflon glass-fiber was transferred onto the membrane by hot pressing. The membrane and anode/Teflon sheet were sandwiched between two metallic plates and pressed at 8.1 MPa and 80 °C. Then the temperature was increased to 135 °C and once reached, the pressure was increased to 15.7 MPa for 5 min. In a second step, the catalyst coated cathode gas diffusion layer (Sigracet 10BC, 0.5 mgPt cm−2, Baltic Fuel Cells) was hot pressed onto the anode-membrane assembly in the same conditions as described above, to complete the MEA fabrication.2.5 Characterization methods
Powder X-ray diffraction (XRD) data were collected in Bragg–Brentano configuration using a PANAlytical X'pert diffractometer, equipped with a hybrid monochromator, operating with Cu Kα radiation (λ = 1.514 Å), and using a step size of 0.1° within the 2θ domain starting from 10° angle. Fourier Transform Infra-Red (FT-IR) measurements were carried out on a Thermo Nicolet iS50 spectrometer equipped with a DTGS detector, KBr beam splitter at 4 cm−1 resolution and were collected in absorbance mode on pellets of KBr and of the material of interest. Scanning Electron Microscopy (SEM) images were obtained on CoHFe nanoparticles with an environmental microscope JEOL-JMS6400. ATO loose tubes were observed by field emission-scanning electron microscopy (FE-SEM) using a Hitachi S-4800 microscope and by transmission electron microscopy (TEM) using a JEOL 1200 EXII microscope operating at 120 kV equipped with a CCD camera SIS Olympus Quemesa. For TEM analysis, the samples were suspended in ethanol and sonicated before deposition onto carbon-coated copper grids. Nitrogen sorption measurements on ATO tubes were conducted at 77 K with a Micromeritics ASAP 2020 apparatus after outgassing overnight under vacuum (10−5 Torr) at 200 °C. The specific surface area was calculated using the Brunauer Emmett Teller (BET) equation. The elemental composition of ATO was analyzed with X-ray fluorescence, previously calibrated for an appropriate range of Sb and Sn contents.2.6 Electrochemical characterization in liquid electrolyte
For OER activity measurements on ATO@CoHFe composites, cyclic voltammetry experiments were performed at room temperature with a RDE setup, cycling the potential from 1 V vs. RHE to 2.2 V vs. RHE at 50 mV s−1 with a Bio-Logic SP-150 Potentiostat. The reference electrode was an Ag/AgCl (3.5 M KCl) electrode, the counter electrode a Pt mesh (2 cm2 geometric area) and the working electrode support was a titanium rotating disk electrode (RDE) with 0.07 cm2 geometric area and with a rotating speed of 1600 rpm. All potentials were converted to the reversible hydrogen electrode (RHE) scale using the equation E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.264 V. The electrolyte was an aqueous 0.1 M H2SO4 solution. The faradaic current density was obtained from the total current density by subtracting the plateau of current density observed at 1.2 V vs. RHE on the positive-going scan.To investigate the electrochemical corrosion of ATO at anodic potentials, chronoamperometry was carried out in a three-electrode cell comprising a titanium foil coated with an ATO ink as a working electrode (200 μm thick, geometric area 4 cm2), a RHE reference electrode and a platinum wire counter electrode. An ATO loading of 2.8 mg cm−2 was deposited from an ink prepared by mixing 11.2 mg of ATO support with 55 μL of 5% Nafion solution, 1800 μL of ethanol and 600 μL of deionized water. After 10 min of sonication, the ink was sprayed using an aerograph onto the Ti foil, the foil itself being placed on a heating plate at 80 °C. Chronoamperometry measurements were performed with a potentiostat (Pine Instruments AFCBP1) by holding the working electrode for 4 h at a constant potential (OCP, 1.9, 2.0 or 2.2 V vs. RHE) in 0.5 M H2SO4 at 80 °C. In order to quantify the amount of metal leached from ATO, 10 mL of electrolyte were withdrawn at the end of the potentiostatic control and analyzed using an Inductively-Coupled Plasma Mass Spectrometer (Agilent ICP-MS 7900).
2.7 PEMWE measurements
MEAs were assembled in a square 6.25 cm2 active-area single cell, having Ti and Au-coated stainless steel anode and cathode end plates, respectively. A Ti-sintered mesh (Bekaert, ST/Ti/20/450/70, 0.3 mm) gas diffusion layer was used on the anode. Deionized water (18 MOhm) was pumped through the anode with a flow rate of 200 mL h−1. The cathode was previously flooded with deionized water. The MEA was assembled in the cell with fluorinated ethylene propylene gaskets to give a compression of 43 ± 2% and assembled with a torque of 10 Nm. To characterize the cell, a Bio-Logic SP-150 Potentiostat with a 20 A booster was used. The cell and MEA were conditioned at 80 °C for 24 hours at open circuit voltage (OCP) to ensure full hydration of the membrane. Polarization curves with the ATO@CoHFe anode were recorded by stepping the current density from 0 to 100 mA cm−2 keeping the current density constant until the variation of the steady-state potential was less than 1 mV min−1. Electrochemical impedance spectroscopy (EIS) was performed in galvanostatic mode, using scanning frequencies from 20 KHz to 50 mHz with a wave amplitude of 10% of the steady-state current.3 Results and discussion
3.1 Structure and morphology of CoHFe nanoparticles, ATO tubes and ATO@17% CoHFe
The CoHFe nanoparticles were first characterized by TEM. Fig. 1A shows a few particles with the typical anisotropic shape with characteristic length and width of ca. 100 and 25–50 nm, respectively. This is a smaller size than the CoHFe cubic particles of ca. 250 nm edge previously investigated by us that were electrochemically prepared.19 The smallest particle size of ca. 25 nm is in agreement with the original report of the preparation of CoHFe nanoparticles in formamide.29 The XRD pattern in the 2θ range 10–40° (Fig. 1B) shows diffraction lines assigned to the (111), (200), (220), (400) and (420) crystal planes of the face-centered cubic PBA crystal structure.31 The ATO morphology was evaluated by electron microscopies. The FE-SEM micrograph shown in Fig. 1C indicates fiber-in-tube structures with outer diameter of ca. 170 nm. The outer surface of the loose tubes is homogeneously covered with grains having an average diameter of 15 nm (Fig. 1C and ESI Fig. S1†). The XRD pattern of the ATO tubes (Fig. 1D) displays diffraction peaks corresponding to the rutile SnO2 structure. The SnO2 crystallite size estimated using the Scherrer equation was 16.5 nm, in agreement with the grain size observed in the SEM micrograph (ESI Fig. S1†). The nitrogen sorption isotherm of ATO was typical of a mesoporous material and its specific surface area obtained by the BET equation was 35 m2 g−1. The enhanced surface area compared to SnO2 materials with lower Sb content is attributed to a smaller grain size.32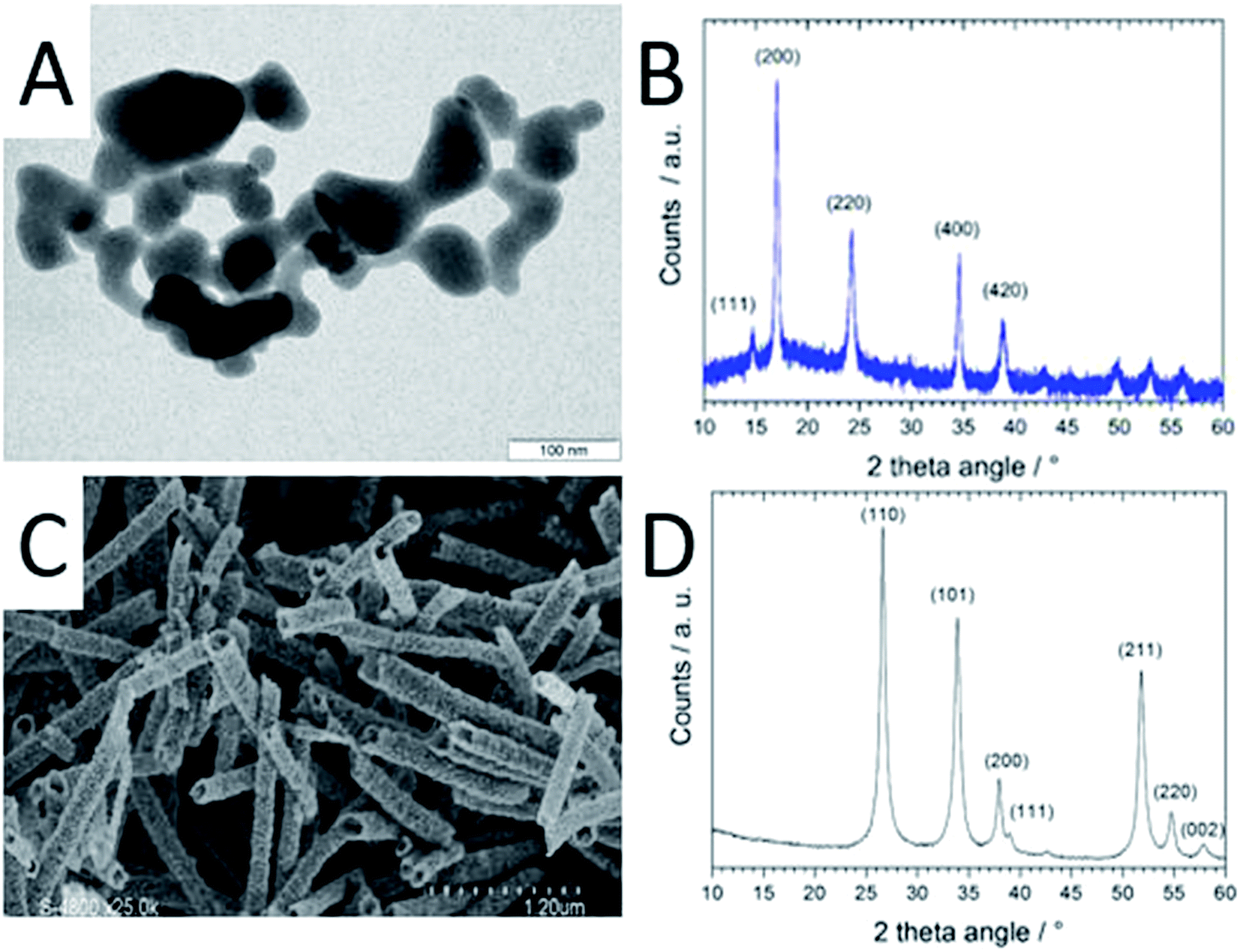 | ||
| Fig. 1 Characterisation of pristine CoHFe and ATO. (A) TEM micrograph of CoHFe nanoparticles, (B) XRD of CoHFe, (C) SEM micrograph of ATO tubes (some fibre-in-tube structures are seen for tubes that are oriented nearly normal to the image plane, as for example on the lower right handside corner), (D) XRD pattern of ATO tubes. Indexes for the standard rutile structure of SnO2 are indicated. | ||
As reported later, ATO@17% CoHFe leads to the highest OER activity. Its morphology and structure were thus investigated in detail. Fig. 2A shows a high magnification TEM image, revealing the partial coverage of the outer surface of ATO tubes by CoHFe particles (light grey). Due to the transparency of CoHFe in TEM, CoHFe particles located on top of the ATO tube may not be all visible. Also, any presence of CoHFe inside the ATO tubes cannot be assessed. While some CoHFe particles seem to be loosely connected to the ATO tubes (ESI Fig. S2†), FT-IR analyses presented later suggest a modified electronic state for all CoHFe particles in ATO@17% CoHFe relative to pure CoHFe, implying that most of the CoHFe particles are electrically connected to the ATO support.
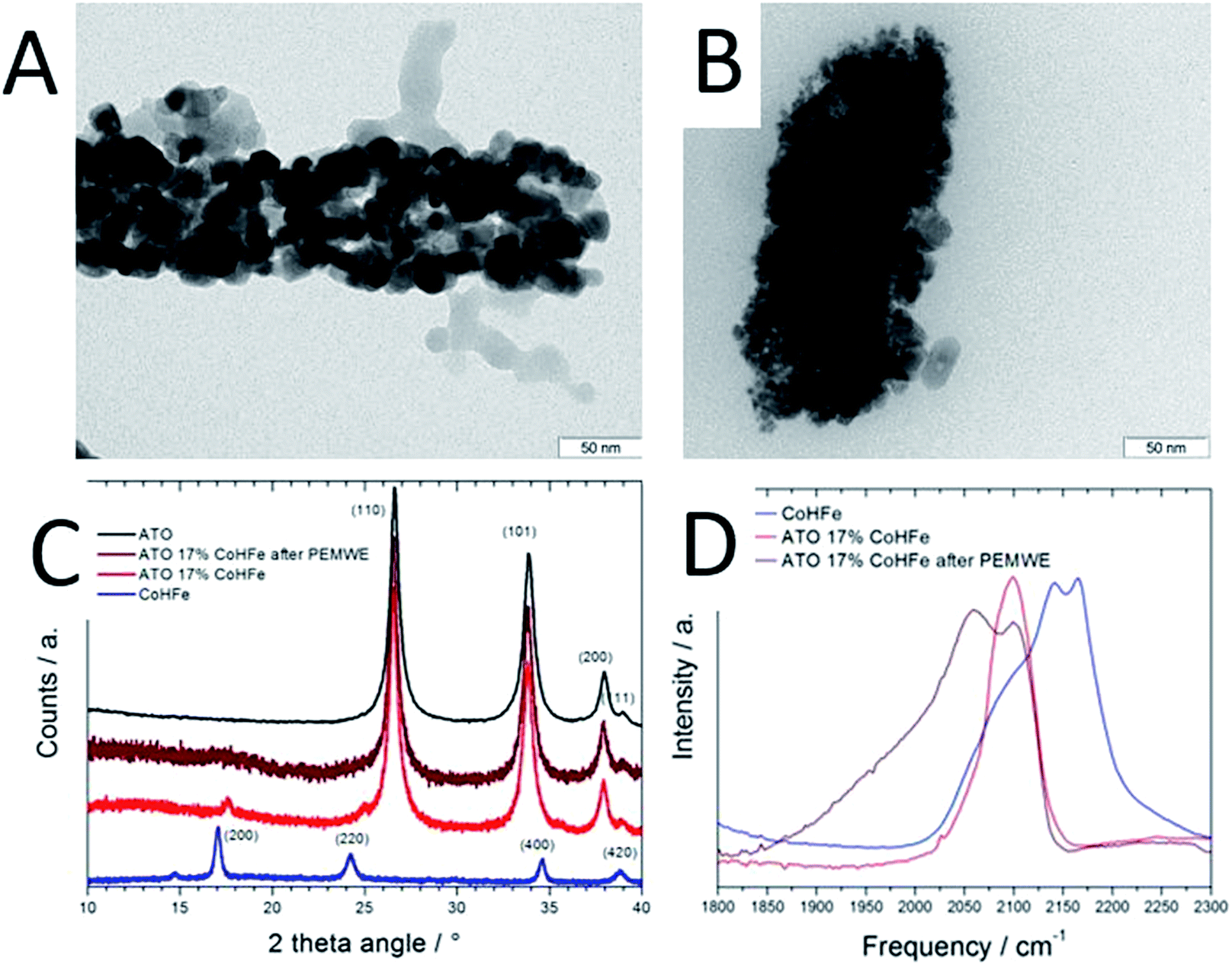 | ||
| Fig. 2 Characterisation of ATO@17% CoHFe before and after PEMWE operation. (A) TEM micrograph of ATO@17% CoHFe, (B) TEM micrograph of ATO@17% CoHFe after 2 h at 2 V in PEMWE, (C) XRD of ATO@17% CoHFe anode before and after PEMWE for 2 h at 2 V, (D) FT-IR spectra of CoHFe, ATO@17% CoHFe, and an anode comprising ATO@17% CoHFe after PEMWE for 2 h at 2 V. | ||
The XRD pattern of ATO@17% CoHFe (red curve in Fig. 2C) reveals the most intense peaks of the SnO2 rutile structure (2θ = 27, 34, 38°, see Fig. 1D), with very weak peaks assigned to CoHFe (see Fig. 1B). These peaks appear at higher angles than for pure CoHFe, indicating a decreased interplanar distance. This is possibly due to a different charge distribution in the metal centers. A charge transfer mechanism between CoHFe and TiO2 anatase was recently reported by Berrettoni and co-workers.33,34 The study concluded that TiO2 stabilizes the low spin (LS) electronic configuration CoIII(LS)–N–C–FeII(LS) vs. the high spin (HS) configuration CoII(HS)–N–C–FeIII(LS), typically observed in the bulk. This results in a shortened Co–N bond. This is in good agreement with the decreased unit cell parameter from 10.34 Å (CoHFe particles) to 10.07 Å (TiO2@CoHFe composite) observed by Berrettoni.34 Decrease of the unit cell dimension as a result of a change in the Co(II)/Co(III) oxidation state is well known for pure CoHFe compounds, with the most stable configuration being controlled by stoichiometry, counter-cations, solvent or temperature.35
In order to further investigate whether such charge transfer also occurs for ATO@17% CoHFe, the electronic state and coordination of Fe and Co ions were investigated by FT-IR spectroscopy (Fig. 2D). The spectrum in the 2000–2300 cm−1 range is characteristic for vibration bands of the cyanide triple bond, that are highly sensitive to the nature, spin and oxidation states of linked metal cations.36 In the infrared spectrum of bulk CoHFe (blue curve in Fig. 2C), two bands centered at 2170 and 2140 cm−1 are observed, as well as a shoulder around 2080–2110 cm−1. The vibration mode at 2170 cm−1 can be assigned to Co(II)–N–C–Fe(III) bridges (2156–2166 cm−1 reported in ref. 36 and 37) and Co(III)–N–C–Fe(III) bridges (2190 cm−1, ref. 37). The band at 2140 cm−1 can be assigned to Co(III)–N–C–Fe(II) bridges (2120–2130 cm−1, ref. 36 and 37). The shoulder around 2080–2110 cm−1 is attributed to Co(II)–N–C–Fe(II) bridges or to non-bridging cyano groups on the surface or at defect sites.35,37,38 The most intense infrared peaks of ATO are observed at 1613, 1396, 1130 and 701 cm−1 (assigned to SnO2), while three weak peaks are observed for ATO at 1950, 2120 and 2270 cm−1 (ESI Fig. S3†).39,40 For ATO@CoHFe (red curve in Fig. 2D), a single band is observed at 2100 cm−1, overlapping with the shoulder seen in the spectrum of CoHFe alone. This clear spectral change unambiguously demonstrates a long-range electronic effect of ATO on CoHFe, since they were separately synthesized before being mixed. The same type of spectral modification was previously observed for TiO2@CoHFe composites. While two vibration bands at 2157 and 2119 cm−1 were visible for pure CoHFe, a single band at 2119–2133 cm−1 (depending on the ratio CoHFe:TiO2) was visible for TiO2@CoHFe composites.24 From FT-IR but also X-ray photoelectron and X-ray absorption spectroscopies, the authors concluded that the composites with 1:1 and 10:1 molar ratios of TiO2:CoHFe do not contain ferric species. In the FT-IR spectrum of ATO@17% CoHFe, we observe an even stronger shift to lower frequencies relative to CoHFe. The single band observed for ATO@17% CoHFe located at the lower wave number of 2100 cm−1 possibly corresponds to the vibration mode of Co(II)–N–C–Fe(II). This may be related to the higher energy of formation of TiO2vs. SnO2 (higher stability of TiO2). This red shift from ca. 2150 to 2100 cm−1 of the FT-IR spectra from CoHFe to ATO@17% CoHFe perfectly matches the spectro-electrochemical red shift observed when electrochemically cycling CoHFe films from 0.9 to 0.0 V vs. SCE (converting CoII3[FeIII(CN)6]2 into K2CoII3[FeII(CN)6]2).37 In summary, XRD and FT-IR show that CoHFe nanoparticles were not denatured by the hydrothermal treatment used in preparation of the supported catalyst (XRD pattern typical for CoHFe), that the unit cell parameter is slightly decreased and the FT-IR is red-shifted, both changes being explained by different Co–N–C–Fe electronic states existing in CoHFe, with respect to a single dominant Co(II)–N–C–Fe(II) state in ATO@17% CoHFe.
3.2 Electrocatalytic OER activity in sulfuric acid electrolyte
We investigated the OER activity of ATO@CoHFe materials by RDE experiments. First we optimized the ATO/CoHFe ratio. As seen in Fig. 3A, ATO@17% CoHFe results in the highest OER activity, for a 610 μg cm−2 loading.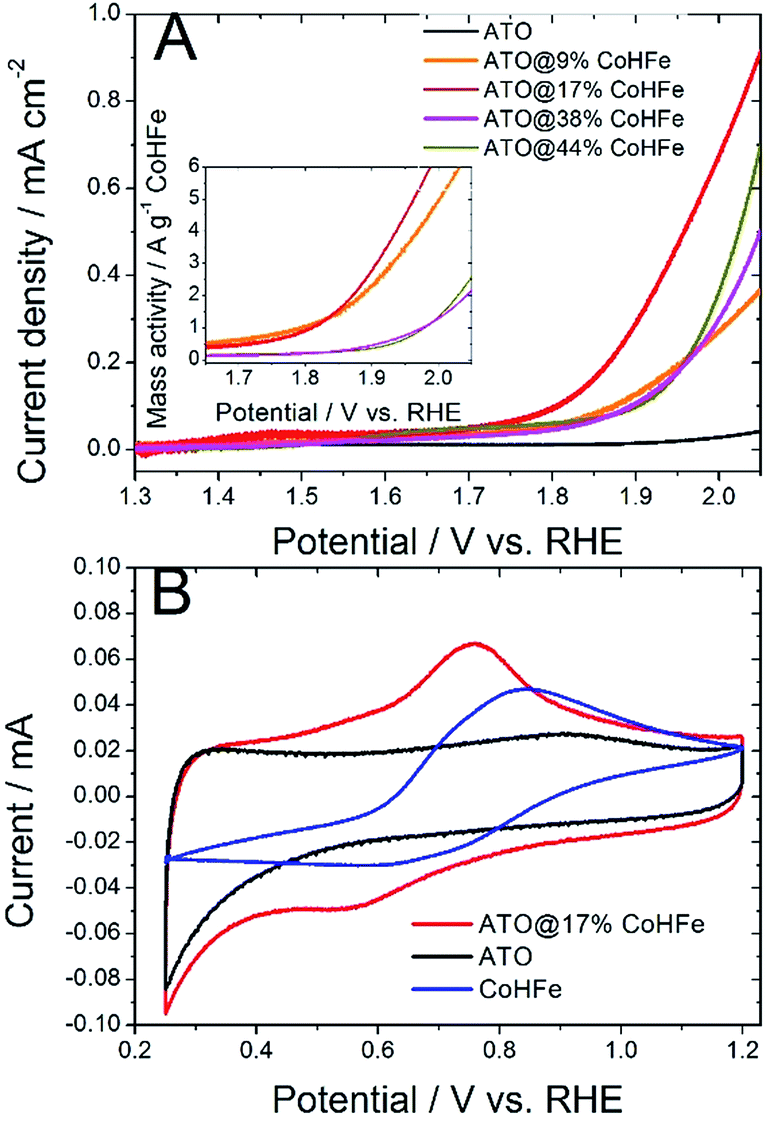 | ||
| Fig. 3 (A) OER activity in acidic electrolyte of ATO@x% CoHFe and pure ATO. Linear scan voltammetry with a scan rate 50 mV s−1, 1600 rpm, 0.1 M H2SO4. The inset shows the faradaic current density after normalization by the mass of CoHFe only. Total catalyst loading 610 μg cm−2. (B) Cyclic voltammetry for CoHFe, ATO and ATO@17% CoHFe. Scan rate 20 mV s−1, 0.1 M H2SO4. A titanium RDE tip was used for all experiments. | ||
Also after normalization by the mass of CoHFe, the catalyst ATO@17% CoHFe results in the highest mass activity, at par with that of ATO@9% CoHFe (inset of Fig. 3A). Higher CoHFe contents are detrimental, probably due to poor electronic conductivity, because of the insulating character of CoHFe. This results in loss of electron percolation through the active layer. Fig. 3A also shows the negligible OER activity for both ATO (black curve) and CoHFe alone (not shown, because it almost perfectly superimposed on the ATO curve), highlighting the synergy between OER catalyst and ATO support.
We performed cyclic voltammetry on ATO@17% CoHFe to investigate the presence of redox peaks and the capacitive current. In Fig. 3B, we observe a single apparent pair of redox process, with a reduction peak at ca. 0.55 V vs. RHE and an oxidation peak at ca. 0.75 V vs. RHE. This redox pair is also observed on unsupported CoHFe, and can be assigned to the Fe(III)/Fe(II) redox switch.41 The position of the peaks, though, is shifted to lower potentials for ATO@17% CoHFe, indicating the electronic interaction between catalyst and support. Possibly, the shoulders at 0.4–0.6 V vs. RHE (oxidation peak) and 0.7–0.9 V vs. RHE (reduction peak) belong to a second pair of redox peaks, as observed for CoHFe in ref. 41 (and references therein). Both pairs of redox peaks where assigned to Fe(III)/Fe(II) redox switch, with a redox position slightly depending on the counter-cation and exact CoHFe stoichiometry. Fig. 3B also shows that the electrochemical signal for ATO@17% CoHFe, to a first approximation, results from the superimposition of the capacitive current of ATO and of the characteristic CoHFe redox peaks. The fact that the double layer current of ATO@17% CoHFe is as high as that of ATO alone (compare the regions 0.25–0.40 V and 0.95–1.15 V vs. RHE, where CoHFe redox peaks' contribution is negligible) indicates that the ATO surface is mostly unblocked by CoHFe nanoparticles. FT-IR analysis suggest that all CoHFe particles are in electric contact with ATO, implying that the CoHFe content in ATO@17% CoHFe is sufficiently low to avoid the formation of a thick layer of CoHFe particles on ATO tubes.
3.3 Investigation of ATO@17% CoHFe in PEMWE
Nafion content lower than 20 wt% was insufficient to allow proper decal transfer of the sprayed catalyst layer onto the Nafion membrane, while Nafion contents higher than 30 wt% lead to overly high cell ohmic resistance during break-in of the PEMWE, as determined by EIS. Following this initial screening, ATO@17% CoHFe was mixed with 20 wt% Nafion (see Experimental section), sprayed and transferred onto a Nafion® 115 membrane at a catalyst loading of ca. 3 mg cm−2 (ESI Fig. S4†). Following the break-in procedure, the initial polarization curve was recorded (Fig. 4A). The onset potential is ca. 1.75 V, in line with the RDE results (Fig. 3A). The low ohmic resistance (high frequency intercept in the Nyquist plot, Fig. 4B) is comparable to the ohmic-resistance value expected for the Nafion membrane only. This confirms that the ATO@17% CoHFe composite with 20 wt% Nafion leads to an electrode with percolation for electrons.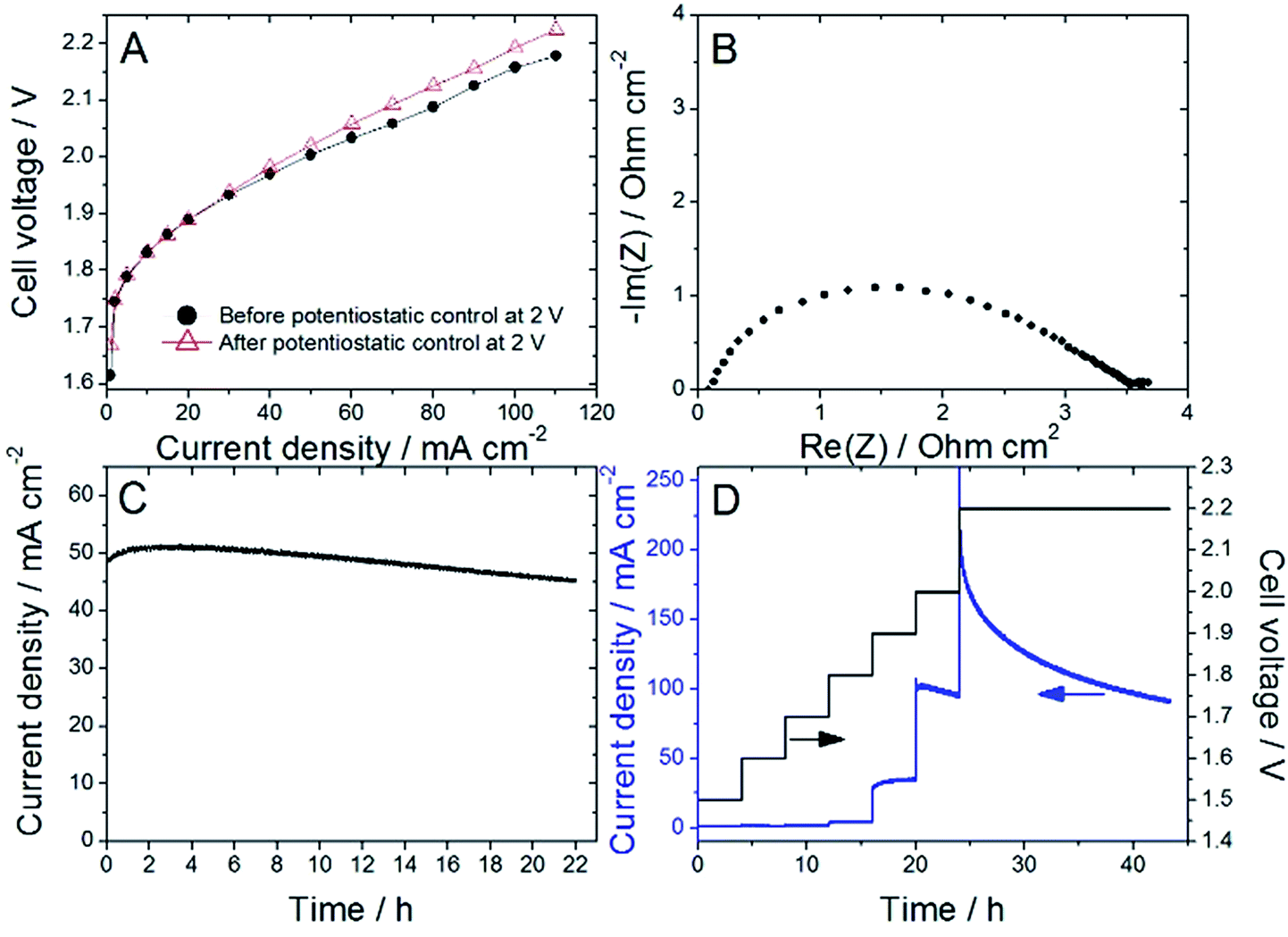 | ||
| Fig. 4 (A) PEMWE polarization curves before (filled circles) and after the 22 h control at 2 V (empty triangles), (B) EIS Nyquist plot recorded at 50 mA cm−2 before the 22 h potentiostatic control at 2 V, (C) current vs. time during the potentiostatic control at 2 V, (D) series of potentiostatic controls for 4 h from 1.5 to 2.0 V and ended with 20 h at 2.2 V. (A–C) recorded with a first MEA and (D) recorded on a second MEA. All experiments were performed at 80 °C, anode ATO@17% CoHFe (3 mg cm−2), cathode Pt/C (0.5 mgPt cm−2) and Nafion® 115 membrane. | ||
To investigate the stability, the cell voltage was fixed at 2 V for 22 h, and the current density recorded (Fig. 4C). A decrease of only 10% of the current density was observed during this time, attesting to the relative stability of the anode catalyst in acid medium even at high electrochemical potential. To further explore the potential-dependence of this apparent instability, we gradually increased the cell potential from 1.5 to 2 V, maintaining the new potential after each step for 4 h, and completing the experiment with a 2.2 V potentiostatic control for 20 h (Fig. 4D). The current density was found constant (or even slowly increasing) at each step up to 1.9 V. A small decrease was recorded during the 4 h step at 2 V, and a strong decrease was observed during the final step at 2.2 V.
This apparent performance loss at high potentials was surprising. CoHFe has been reported as stable in acidic media, and at applied potential differences well above 2 V.20 On the other hand, ATO has been reported to be stable in anodic conditions in PEMWE up to 1.8 V. However, its long term stability in the range of 1.9–2.2 V has not been studied yet, to the best of our knowledge. While instability of ATO has been reported after galvanostatic control leading to potentials as high as 3.5 V,42,43 such high potentials would mean very low energy efficiency of water electrolysis. In order to better assign the origin of this instability at potentials above 1.9 V but up to 2.2 V maximum, we investigated the fate of ATO and of ATO@CoHFe anodes after electrolysis.
To further investigate the stability of ATO to high potential in acid medium, an ATO-coated Ti electrode was potentiostatically controlled at OCP, 1.9, 2.0 or 2.2 V vs. RHE for 4 h. Significant leaching of Sn and Sb was detected by ICP-MS at 1.9 and 2.0 V vs. RHE (Table 1) and massive leaching at 2.2 V vs. RHE. These results suggest that the main degradation mechanism leading to the small decrease in current density observed at 2 V in PEMWE with the ATO@17% CoHFe anode and the much faster decrease observed at 2.2 V is the electrochemical corrosion of the ATO support.
| Potential/V vs. RHE | Sn loss/% | Sb loss/% | Remaining Sb in corroded material/at% |
|---|---|---|---|
| OCP | <10−5 | <10−4 | 10.0 |
| 1.9 | 0.14 | 7.7 | 9.5 |
| 2.0 | 0.16 | 8.5 | 9.4 |
| 2.2 | 22.6 | 52.7 | 6.0 |
The leaching from ATO is clearly associated with the applied electrochemical potential (see Table 1, no leaching at OCP). The preferential loss of Sb (Table 1) must have as a consequence a reduction in the electronic conductivity of ATO, especially at the surface. Thus, disrupting the conductive network of the anode layer is detrimental to its performance, even if CoHFe itself is stable in such conditions. Sn is already fully oxidized in ATO, and therefore cannot lead to an oxidation current (ESI Fig. S5†). It has been recently reported that for OER-active metal oxides such as RuO2 in acidic medium and MnOx in alkaline electrolyte, electrochemically-induced degradation cannot, in practice, be detected by recording the anodic dissolution current that is orders of magnitude lower than the OER current.44
In contrast, ICP-MS can quantitatively determine very small amounts of dissolved elements, and allows off-line and online monitor of the dissolution of metals as a function of the electrochemical potential.45,46
The end of test ATO@17% CoHFe anode was also characterized (after a potentiostatic control for 2 h at 2 V) by TEM, XRD and FT-IR spectroscopy. The TEM image after PEMWE shows smaller ATO grain size on the surface (Fig. 2Bvs.Fig. 2A). However, similar width of the SnO2 peaks in XRD (Fig. 2C, brown and red curves) suggests that either the grain size in the bulk of ATO fibres did not change, or that the small grains in Fig. 2B do not correspond to the domain sizes that coherently diffract. The low intensity diffraction lines given by the CoHFe component in the XRD pattern is less marked after PEMWE (Fig. 2C, brown curve) and no peak related to CoHFe is distinct. It is therefore difficult to discuss from this result possible electronic or structural changes. FT-IR (Fig. 2D) of the used ATO reveals two bands. The original one located at 2100 cm−1, assigned to Co(II)–N–C–Fe(II), that confirms the robustness of the OER catalysts, preserving its initial state. There is also a new band at ca. 2060 cm−1, which has been associated with the same Co(II)–N–C–Fe(II) pairs, with higher occupancy of cations in tetrahedral sites.47 This suggests that protons may be integrated in these sites during OER, promoting the appearance of the new IR band. Still, these bands can only be assigned to bridging CN moieties, and are experimental evidence that the PBA structure is conserved, subject to amorphization and partial restructuring. Operando characterization is needed to further investigate evolution in Fe and Co coordination during OER.
Conclusions
The OER activity of the optimized ATO@CoHFe composite was ca. 6 mA cm−2 at 1.8 V in PEMWE conditions for a CoHFe loading of 0.5 mg cm−2. This compares favorably with another non-PGM OER catalyst based on Ti-stabilized MnO2 that showed ca. 3 mA cm−2 at 1.8 V in 0.05 M H2SO4 (ref. 10) and with an electrodeposited film of CoFePbOx that showed ca. 1 mA cm−2 at 1.8 V vs. RHE (corresponding to 1.65 V vs. NHE at pH 2.5).12 Recently reported CoOx films and CoOx nanoparticles physically deposited as metallic cobalt on FTO and calcined in air reached 6–20 mA cm−2 at 1.8 V vs. RHE in 0.5 M H2SO4.11 Improved coverage of CoHFe on ATO or other conducting nanostructured oxides could lead to further improvement in OER activity. The direct growth of CoHFe on the support could be a promising approach, while detailed end of test and operando characterization is required to better understand the coordination of cobalt in CoHFe when it catalyzes the OER. While the OER activity of such non-PGM catalysts is so far too low to rival IrO2 for utilization in PEMWE operating at high current density, their application in photo-electrochemical water splitting devices with maximum photocurrent of ca. 10–14 mA cm−2 can be foreseen.48 A PEM-based electrolyzer with a PGM-free anode can thus be designed, the acidic polymer electrolyte being more favorable than alkaline electrolytes for the hydrogen evolution reaction at the cathode, on both PGM and non-PGM catalysts.49,50Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement No. 306682 SPINAM and under the H2020 Programme/ERC StG Grant Agreement No. 279313 CHEMCOMP is gratefully acknowledged. This work was also partially supported by the Spanish Ministerio de Economía y Competitividad (MINECO) through project CTQ2015-71287-R, and the Severo Ochoa Excellence Accreditation 2014-2018 SEV-2013-0319; and the Generalitat de Catalunya (2014-SGR-797, and the CERCA program). ARC thanks the Marie Curie COFUND Action from the European Commission for co-financing his postdoctoral fellowship. MBA thanks the Generalitat Catalana (AGAUR) for a predoctoral fellowship.References
- C. Lamy, Int. J. Hydrogen Energy, 2016, 41, 15415–15425 CrossRef CAS.
- A. S. Gago, A. S. Ansar, P. Gazdzicki, N. Wagner, J. Arnold and K. A. Friedrich, J. Electrochem. Soc., 2014, 64, 1039–1048 CAS.
- P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukaloo and C. Etiévant, Int. J. Hydrogen Energy, 2011, 36, 4134–4142 CrossRef CAS.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- D. F. Abbott, D. Lebedev, K. Waltar, M. Povia, M. Nachtegaal, E. Fabbri, C. Copéret and T. J. Schmidt, Chem. Mater., 2016, 28, 6591–6604 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- C. Felix, T. Maiyalagan, S. Pasupathi, B. Bladergroen and V. Linkov, Int. J. Electrochem. Sci., 2012, 7, 12064–12077 CAS.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- S. Geiger, O. Kasian, B. R. Shrestha, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2016, 163, F3132–F3138 CrossRef CAS.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- J. S. Mondschein, J. F. Callejas, C. G. Read, J. Y. C. Chen, C. F. Holder, C. K. Badding and R. E. Schaak, Chem. Mater., 2017, 29, 950–957 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- Y. Su, Y. Zhu, H. Jiang, J. Shen, X. Yang, W. Zou, J. Chen and C. Li, Nanoscale, 2014, 6, 15080–15089 RSC.
- V. Goellner, C. Baldizzone, A. Schuppert, M. T. Sougrati, K. J. J. Mayrhofer and F. Jaouen, Phys. Chem. Chem. Phys., 2014, 16, 18454–18462 RSC.
- L. Dubau and F. Maillard, Electrochem. Commun., 2016, 63, 65–69 CrossRef CAS.
- S. Pintado, S. Goberna-Ferron, E. C. Escudero-Adan and J. R. Galan-Mascaros, J. Am. Chem. Soc., 2013, 135, 13270–13273 CrossRef CAS PubMed.
- L. Han, P. Tang, A. Reyes-Carmona, B. Rodriguez-Garcia, M. Torrens, J. R. Morante, J. Arbiol and J. R. Galan-Mascaros, J. Am. Chem. Soc., 2016, 138, 16037–16045 CrossRef CAS PubMed.
- M. Aksoy, S. V. K. Nune and F. Karadas, Inorg. Chem., 2016, 55, 4301–4307 CrossRef CAS PubMed.
- S. Goberna-Ferron, W. Y. Hernandez, B. Rodriguez-Garcia and J. R. Galan-Mascaros, ACS Catal., 2014, 4, 1637–1641 CrossRef CAS.
- L. Qian and X. Yang, Talanta, 2006, 69, 957–962 CrossRef CAS PubMed.
- M. Berrettoni, M. Ciabocco, M. Fantauzzi, M. Giorgetti, A. Rossi and E. Caponetti, RSC Adv., 2015, 5, 35435–35447 RSC.
- L. Catala and T. Mallah, Coord. Chem. Rev., 2017, 346, 32–61 CrossRef CAS.
- I. Savych, S. Subianto, Y. Nabil, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2015, 17, 16970–16976 RSC.
- X. Wu and K. Scott, Int. J. Hydrogen Energy, 2011, 36, 5806–5810 CrossRef CAS.
- A. Marshall, B. Borresen, G. Hagen, M. Tsypkin and R. Tunold, Electrochim. Acta, 2006, 51, 3161–3167 CrossRef CAS.
- V. Vo, M. N. Van, H. I. Lee, J. M. Kim, Y. Kim and S. J. Kim, Mater. Chem. Phys., 2008, 107, 6–8 CrossRef CAS.
- S. Cavaliere, I. Jimenez-Morales, G. Ercolano, I. Savych, D. Jones and J. Rozière, ChemElectroChem, 2015, 2, 1966–1973 CrossRef CAS.
- A. Bleuzen, C. Lomenech, V. Escax, F. Villain, F. Varret, C. Cartier dit Moulin and M. Verdaguer, J. Am. Chem. Soc., 2000, 122, 6648–6652 CrossRef CAS.
- S. Cavaliere, S. Subianto, I. Savych, M. Tillard, D. Jones and J. Rozière, J. Phys. Chem. C, 2013, 117, 18298–18307 CAS.
- M. Ciabocco, M. Berrettoni, M. Giorgetti, M.-T. Sougrati, N. Louvain and L. Stievano, New J. Chem., 2016, 40, 10406–10411 RSC.
- M. Giorgetti, G. Aquilanti, M. Ciabocco and M. Berrettoni, Phys. Chem. Chem. Phys., 2015, 17, 22519–22522 RSC.
- N. R. de Tacconi, K. Rajeshwar and R. O. Lezna, Chem. Mater., 2003, 15, 3046–3062 CrossRef CAS.
- N. Shimamoto, S.-I. Ohkoshi, O. Sato and K. Hashimoto, Inorg. Chem., 2002, 41, 678–684 CrossRef CAS PubMed.
- R. O. Lezna, R. Romagnoli, N. R. de Tacconi and K. Rajeshwar, J. Phys. Chem. B, 2002, 106, 3612–3621 CrossRef CAS.
- O. Sato, Y. Einaga, A. Fujishima and K. Hashimoto, Inorg. Chem., 1999, 38, 4405–4412 CrossRef CAS PubMed.
- T. Krishnakumar, R. Jayaprakash, N. Pinna, A. R. Phani and S. Santucci, J. Phys. Chem. Solids, 2009, 70, 993–999 CrossRef CAS.
- J. Zhang and L. Gao, Inorg. Chem. Commun., 2004, 7, 91–93 CrossRef CAS.
- C.-X. Cai, K.-H. Xue and S.-M. Xu, J. Electroanal. Chem., 2000, 486, 111–118 CrossRef CAS.
- B. Correa-Lozano, C. Comninellis and A. D. Battisti, J. Appl. Electrochem., 1997, 27, 970–974 CrossRef CAS.
- F. Montilla, E. Morallón, A. De Battisti and J. L. Vázquez, J. Phys. Chem. B, 2004, 108, 5036–5043 CrossRef CAS.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- E. Pizzutilo, S. Geiger, J. P. Grote, A. M. Mingers, K. J. J. Mayrhofer, M. Arenz and S. Cherevko, J. Electrochem. Soc., 2016, 163, F1510–F1514 CrossRef CAS.
- J. P. Grote, A. R. Zeradjanin, S. Cherevko and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2014, 85, 104101 CrossRef PubMed.
- R. O. Lezna, R. Romagnoli, N. R. De Tacconi and K. Rajeshwar, J. Phys. Chem. B, 2002, 106, 3612–3621 CrossRef CAS.
- J. Jia, L. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. Wei, D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 CrossRef CAS.
- A. Morozan, V. Goellner, Y. Nedellec, J. Hannauer and F. Jaouen, J. Electrochem. Soc., 2015, 162, H719–H726 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images of Sb–SnO2 tubes and of the composite catalyst, photograph of the membrane electrode assembly and graph showing the anodic current of Sb–SnO2 during the electrochemical corrosion test. See DOI: 10.1039/c7se00512a |
| This journal is © The Royal Society of Chemistry 2018 |