
Revealing how molten salts promote CO2 capture on CaO via an impedance study and sorption kinetics simulation†
Liang
Huang
a,
Chunming
Xu
b,
Rongzheng
Ren
b,
Qianwen
Zheng
a,
Zhenhua
Wang
b,
Benoît
Louis
c and
Qiang
Wang
 *a
*a
aCollege of Environmental Science and Engineering, Beijing Forestry University, 35 Qinghua East Road, Haidian District, Beijing 100083, P. R. China. E-mail: qiang.wang.ox@gmail.com; qiangwang@bjfu.edu.cn; Tel: +86 13699130626
bBeijing Key Laboratory for Chemical Power Source and Green Catalysis, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, 100081, P. R. China
cLaboratoire de Synthèse, Réactivité Organiques et Catalyse, Institut de Chimie, UMR 7177, Université de Strasbourg, 1 rue Blaise Pascal, 67000 Strasbourg, France
First published on 8th November 2017
Abstract
Electrochemical impedance spectroscopy analyses were utilised to explore the oxygen ion conductivity of alkali metal salts promoted CaO, which revealed that oxysalts, such as carbonate and sulfate with good ion conductivity, promote CO2 capture.
Introduction
CaO as a high temperature CO2 sorbent is promising for some CO2-free sorption-enhanced steam reforming processes.1,2 It has been proposed to be applied in both pre- and post-combustion capture processes based on the reversible carbonation/calcination reaction,3 and could potentially be applied in the sorption-enhanced steam reforming (SESR) reaction, which is a candidate technique for producing pure H2via simultaneous reforming and adsorptive separation of CO2.4–6 By removing CO2 from the reaction products, the balance is driven to the right-hand side toward a higher conversion and yields a pure hydrogen steam. Moreover, the reaction temperature can be reduced to around 500–600 °C, which is much lower than those used in conventional steam reforming (800 °C). Hence, CaO is regarded as one of the most promising sorbents.7–9Although CaO exhibits high CO2 capture capacity under stoichiometric conditions (17.86 mmol g−1), conventional CaO sorbents are not able to achieve complete conversion at the end of the first carbonation due to a diminution of availability of CaO active sites. Along with the carbonation reaction, the blockage of internal pores prevents CO2 from reaching the remaining active surface area. This situation significantly increases the diffusion resistance of CO2 throughout the CaCO3 product layer, creating a slower diffusion-limited reaction phase.10,11 Various techniques have been proposed to improve the CO2 uptake of CaO or to reactivate the used CaO. Hu et al.,12 for instance, employed a series of organic acids to modify limestone to produce CaO sorbents and the modified sorbent maintained a higher specific surface area (SSA) that contributed to the superior performance of the sorbent. Akgsornpeak et al.13 developed synthetic CaO via a sol–gel method and investigated the effect of the Ca2+/CTAB ratio. They found that the presence of CTAB can effectively prevent the agglomeration of CaO particles, which can greatly increase the BET SSA and pore volume of the resulting CaO sorbents. Apart from the structural improvement, an alternative strategy to increase the CO2 uptake capacity of CaO is the doping of alkali metal salts. Reddy et al.14 reported that doping CaO with alkali metals can result in an improvement of CO2 uptake capacity. The addition of alkali metals improved the CO2 uptake capacity in the following order Cs > Rb > Na > K > Li. It was found that CO2 is preferably adsorbed on Cs2O rather than CaO, CsOH- and Cs2CO3-doped CaO sorbents due to the formation of a highly dispersed Cs2O on the CaO support.
Our group previously reported that mixed alkali metal carbonate molten salts promoted CaO with superior CO2 capture performance. However, there is still no clear understanding of how such alkali metal carbonates facilitate the CO2 capture by CaO. According to the reports on alkali metal nitrite coated MgO,15,16 we assume that the existence of O2− in the liquid molten salt phase might generate CO32− ions, which accelerates the CO2 capture process. To demonstrate this assumption, we compared the CO2 capture capacity of different mixed metal salts coated CaO and conducted an electrochemical impedance spectroscopy experiment to explore the ionic conductivity of each of the sorbents.
Results and discussion
Previously, a negative correlation between the CO2 uptake and the melting point temperature was demonstrated.17 All of the selected molar ratios of M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M2, at which the melting point of (M1–M2)X was minimal, are listed in Table 1. Fig. 1 shows the CO2 uptake of neat CaO, hydrated CaO, and 10 mol% binary alkali metal salts (M1–M2)X, (M = Li, Na, K; X = SO42−, CO32−, Cl−) coated CaO when exposed at 600 °C to 100% dry CO2 at ambient pressure (1 bar) for 1 h. It is apparent that all samples show typical two differentiated phases, a fast reaction controlled stage followed by a much slower solid state diffusion stage, which was generally accepted for CaO carbonation. However, different trends could still be observed for neat CaO, hydrated CaO, and alkali metal molten salt coated CaO. For neat CaO, both the CO2 sorption capacity and sorption kinetics were relatively poor. After 1 h of sorption, the CO2 uptake was still less than 3.26 mmol g−1, and in the first minute, only 31.81% of its maximum CO2 uptake (60 min) was achieved. For hydrated CaO, it showed a slightly higher CO2 uptake (6.44 mmol g−1) than neat CaO. It was suggested that more cracks could be formed during the hydration process, creating more channels that extend to the interior of the particles, with a consequent improvement in the CO2 capture capacity of CaO.18,19 However, its sorption kinetics were still very low; only 29.82% of its maximum CO2 uptake (60 min) was achieved within the first minute. In this process, a gas–solid reaction between CO2 and CaO on the surface of the grains took place, leading to the formation of a thin layer of CaCO3, which normally occurred in short periods of time and ended with saturation of the surface active sites.
:M2, at which the melting point of (M1–M2)X was minimal, are listed in Table 1. Fig. 1 shows the CO2 uptake of neat CaO, hydrated CaO, and 10 mol% binary alkali metal salts (M1–M2)X, (M = Li, Na, K; X = SO42−, CO32−, Cl−) coated CaO when exposed at 600 °C to 100% dry CO2 at ambient pressure (1 bar) for 1 h. It is apparent that all samples show typical two differentiated phases, a fast reaction controlled stage followed by a much slower solid state diffusion stage, which was generally accepted for CaO carbonation. However, different trends could still be observed for neat CaO, hydrated CaO, and alkali metal molten salt coated CaO. For neat CaO, both the CO2 sorption capacity and sorption kinetics were relatively poor. After 1 h of sorption, the CO2 uptake was still less than 3.26 mmol g−1, and in the first minute, only 31.81% of its maximum CO2 uptake (60 min) was achieved. For hydrated CaO, it showed a slightly higher CO2 uptake (6.44 mmol g−1) than neat CaO. It was suggested that more cracks could be formed during the hydration process, creating more channels that extend to the interior of the particles, with a consequent improvement in the CO2 capture capacity of CaO.18,19 However, its sorption kinetics were still very low; only 29.82% of its maximum CO2 uptake (60 min) was achieved within the first minute. In this process, a gas–solid reaction between CO2 and CaO on the surface of the grains took place, leading to the formation of a thin layer of CaCO3, which normally occurred in short periods of time and ended with saturation of the surface active sites.
| Component | Ratio (M1:M2)a |
Melting pointa (°C) | Uptake capacity (mmol g−1) |
|---|---|---|---|
| a The molar ratio and melting point were obtained from FactSage database.21 | |||
| (Na–K)2SO4/CaO | 0.742:0.258 |
831 | 7.52 |
| (Na–K)2CO3/CaO | 0.555:0.445 |
710 | 8.55 |
| (Na–K)Cl/CaO | 0.506:0.494 |
657 | 8.02 |
| (Li–Na)2SO4/CaO | 0.617:0.383 |
591 | 9.73 |
| (Li–K)2SO4/CaO | 0.81:0.19 |
530 | 10.93 |
| (Li–Na)2CO3/CaO | 0.52:0.48 |
500 | 10.27 |
| (Li–K)2CO3/CaO | 0.428:0.578 |
498 | 10.38 |
| (Li–Na)Cl/CaO | 0.72:0.28 |
554 | 1.40 |
| (Li–K)Cl/CaO | 0.592:0.418 |
353 | 2.41 |
| CaO | — | — | 3.26 |
| Hydrated CaO | — | — | 6.44 |
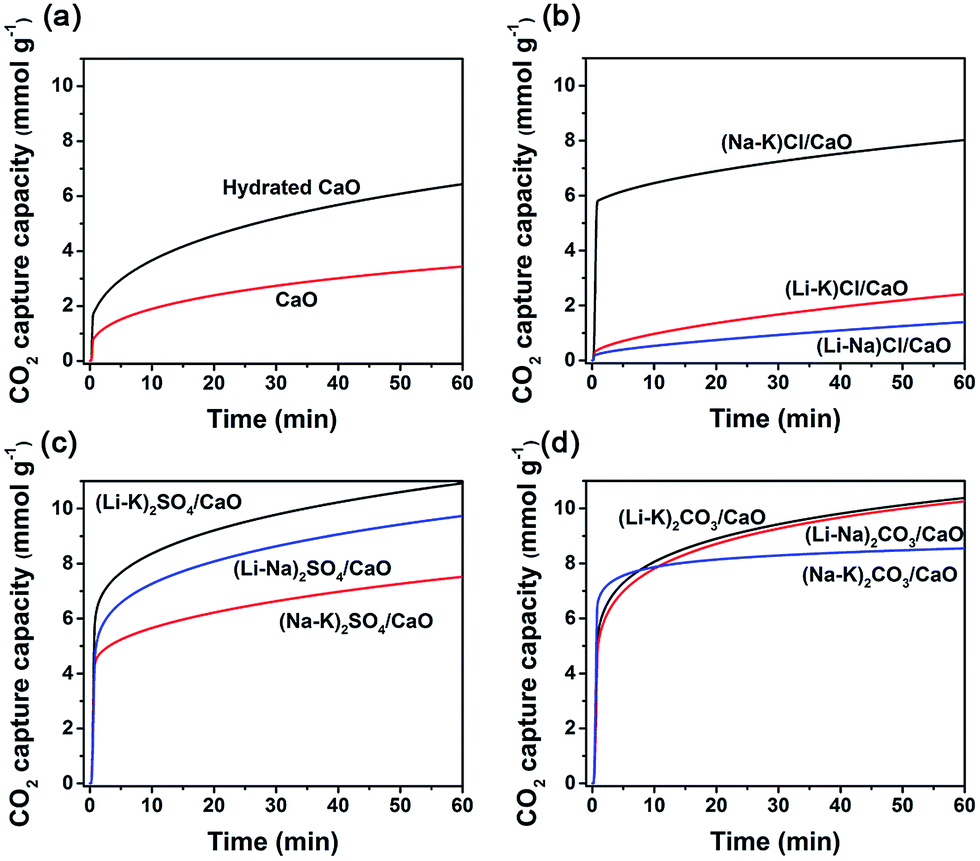 | ||
| Fig. 1 CO2 uptake by (a) CaO; (b) alkali metal chloride coated CaO (MCl/CaO); (c) alkali metal carbonate coated CaO (MSO4/CaO); and (d) alkali metal sulfate coated CaO (MCO3/CaO). | ||
While for (Li–K)Cl/CaO and (Na–K)Cl/CaO, the CO2 uptakes for the fast reaction stage were even lower than neat CaO, demonstrating a decrease in the active sites on the surface. The melting points of (Li–K)Cl and (Na–K)Cl were below the sorption temperature (600 °C), leading the phase transformation from solid to liquid, which covered the surface of the active sorbents and hindered the reaction. This data suggested that the alkali chloride molten salts could not provide any promotion effect on the CO2 capture on CaO, but, on the contrary, resulted in an inhibiting effect.
All other sorbents coated with alkali carbonates and sulphates showed much higher CO2 uptake capacity during the carbonation. With 10 mol% (Li–K)2SO4, (Li–Na)2SO4, and (Na–K)2SO4, the CO2 uptake of CaO was promoted to be 10.93, 9.73, and 7.52 mmol g−1, respectively. With 10 mol% (Li–K)2CO3, (Li–Na)2CO3, and (Na–K)2CO3, the CO2 uptake of CaO was promoted to be 10.38, 10.27, and 8.55 mmol g−1, respectively. The sorption kinetics were also significantly improved by doping with alkali carbonates and sulphates. For instance, with 10 mol% (Li–K)2SO4, (Li–Na)2SO4, (Li–K)2CO3, and (Li–Na)2CO3 coating, 56%, 51%, 64%, and 54% of their maximum CO2 uptakes (60 min) were achieved within the first minute, which were much higher than that for neat CaO. After the relatively fast initial reaction, a slower reaction stage controlled by diffusion in the pores or in the product layer takes place, resulting in a slower reaction rate. The increased carbonation rate resulted in fast cessation of carbonation, and the quickly formed CaCO3 product layer may hinder further carbonation.20
The components, molar ratio of molten salts, melting point and CO2 uptake capacity of promoted CaO are listed in Table 1. As the melting point of the eutectics can be tuned by changing the molar ratio to influence the CO2 uptake of the promoted CaO sorbents, the minimum melting point eutectics were optimized for loading. When the sorption temperature was kept at 600 °C, different phases of salt mixtures were formed; for example, (Na–K)2SO4 which has a melting point of 831 °C will be in solid phase, which showed limited promotion on CO2 uptake, maybe due to the formation of a chemically bound Ca–O–K surface species, the same as the K2CO3-promoting mechanism in Layered Double Hydroxide (LDH).22,23 In contrast, for (Li–Na)2SO4/CaO or those listed downward, which were used as a sorbent, the coating mixture salts melted under identical conditions and led to a significant promoting effect on CO2 uptake. To explore the intrinsic reason for this promotion effect, (Li–K)2CO3/CaO, (Li–K)2SO4/CaO, and (Li–K)Cl/CaO were chosen as the representative molten salts.
To explore the impact of structure and morphology, XRD, SEM, and BET analyses were measured (Fig. S2–S4, ESI†). The results showed that all samples consisted of alkali metal salts and Ca(OH)2. Upon calcination, Ca(OH)2 transformed into crystalline CaO and no other impurities were detected. The grain diameters of these three modified CaO samples were similar in shape, which led to an analogous microstructure and surface area (Table S1, ESI†).
Ion-diffusion efficiencies can be measured through a determination of mobility or more directly through an electrical measurement of ion conductivity.24 Impedance is the response of an electrochemical system to an applied alternating voltage. The frequency dependence of impedance can reveal the underlying processes in electrochemical systems. Fig. 2 shows the impedance spectra in Nyquist form obtained from CaO, (Li–K)Cl/CaO, (Li–K)2SO4/CaO, and (Li–K)2CO3/CaO. For CaO and (Li–K)Cl/CaO, the impedance spectra were totally irregular, which showed similar trends with an open circuit (Fig. 2(a) and (b)) corresponding to the CV test (Fig. S5a and b†). The real part (Z′) in the Nyquist plot coordinated values was around 105 Ω, corresponding to an impedance of about 105 Ω in magnitude. Fig. 2(c) and (d) show typical impedance spectra for (Li–K)2SO4/CaO and (Li–K)2CO3/CaO; the resistance was calculated from the intersection with the real axis at high frequency by using ZSimDemo software. (Li–K)2SO4/CaO and (Li–K)2CO3/CaO showed much lower resistance (72.36 Ω and 27.09 Ω), which corresponds to a high ionic conductivity according to the formula σ = L/RA, where σ is the ionic conductivity, R is the bulk resistance, and L and A are the thickness and sectional area of the pellets.24L and A are constants when a sample is prepared, thus σ was numerical inversely proportional to A. It can be seen that the oxygen ion conductivity for (Li–K)2SO4/CaO and (Li–K)2CO3/CaO was several orders of magnitude higher than that for CaO and (Li–K)Cl/CaO. These results are in concordance with CO2 sorption tests that (Li–K)2SO4 and (Li–K)2CO3 coated CaO resulted in excellent CO2 uptakes due to good O2− conductivity, which offered a continuous stream of CO32− for the carbonation reaction.
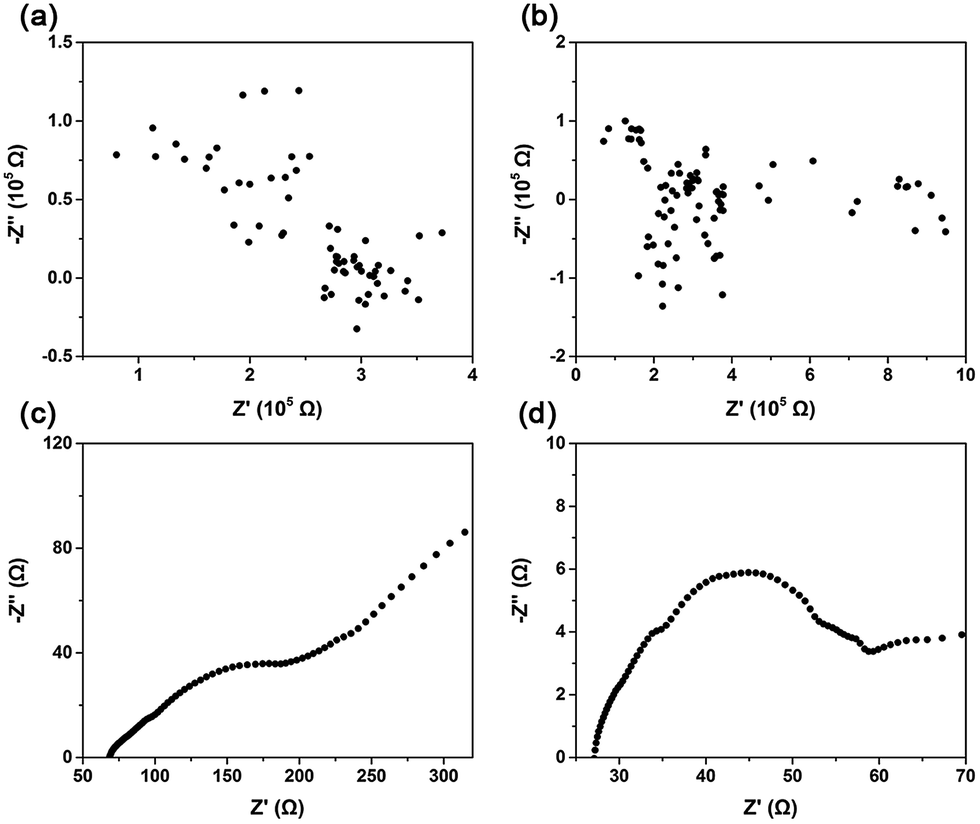 | ||
| Fig. 2 Impedance spectroscopy of (a) CaO; (b) (Li–K)Cl/CaO; (c) (Li–K)2SO4/CaO; and (d) (Li–K)2CO3/CaO. | ||
To understand the detailed reaction process for CO2 capture on neat and promoted CaO samples, the dynamic variations during the reaction of the particles with CO2 were further examined by fitting to the well accepted double exponential model (Fig. S6, ESI†) as reported for other sorbents.25,26 The double exponential model is:
| y = Aexp(−k1t) + Bexp(−k2t) + C | (1) |
In eqn (1), y represents the CO2 uptake of sorbents, t is the sorption time in minute, and k1 and k2 are the kinetic parameters in the surface chemical reaction and bulk diffusion process, respectively. Constants A and B are the coefficient of each process that controls the whole CO2 sorption process, and C was the y value when t tending to infinity indicates the maximum sorption capacity. For (Li–K)Cl/CaO and hydrated CaO which did not show O2− transmittance capacity, A is larger than B (see Table S2, ESI†), suggesting that the diffusion stage has a great influence on the overall CO2 sorption process. By changing the coating mixtures to (Li–K)2CO3 and (Li–K)2SO4, B became larger than A, demonstrating that the diffusion stage was no longer a limiting factor, the chemisorption process with a fast reaction rate contributed more to the total uptake capacity.
The possible mechanism for the effect of different alkali metal coatings was illustrated schematically in Fig. 3, in regard to (Li–K)2CO3, (Li–K)2SO4 coated CaO particles (Fig. 3(a)), the alkali metal mixtures show a lower melting point indicating that it will be in a molten phase at 600 °C with good oxygen ion conductivity, the adsorbed CO2 combined with O2− to generate CO32−, which formed CaCO3 by reaction with CaO at the CaO–CaCO3 interface as shown in eqn (2)–(4).11
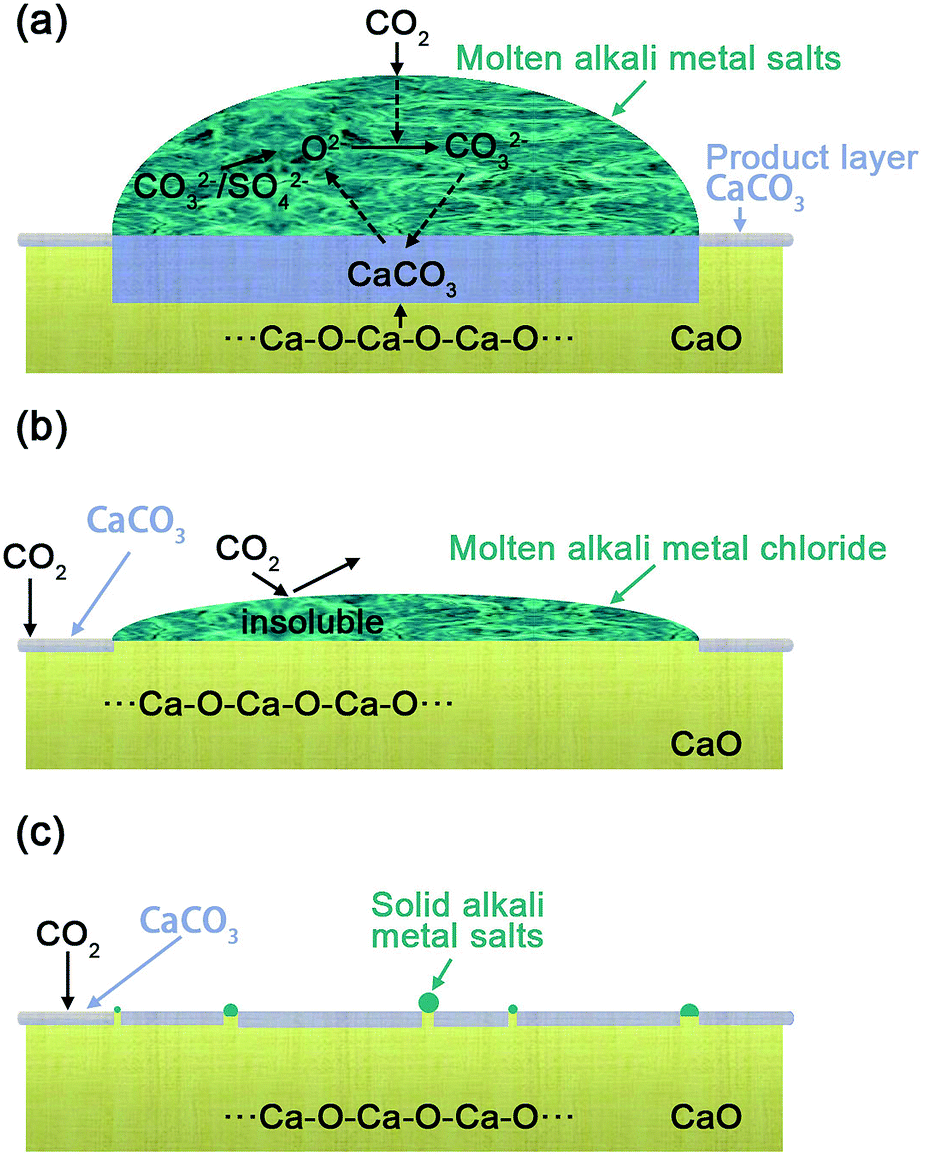 | ||
| Fig. 3 Schematic diagrams of the reaction for CO2 with coated CaO particle at 600 °C, (a) CaO coated with low melting point oxysalt eutectic: (b) CaO coated with anaerobic salts and (c) CaO coated with high melting point mixtures. | ||
At the CaO–CaCO3 interface:
| CO32− + CaO → CaCO3 + O2− | (2) |
At the pore surface:
| CO2(g) ⇄ (CO2)ads | (3) |
| (CO2)ads + O2− → CO32− | (4) |
The generated O2− interacted with adsorbed CO2 in the carbonates molten salts to generate the carbonate ions (CO32−).27 (Li–K)2CO3 and (Li–K)2SO4 possessed a good O2− transmission ability (as discussed in Fig. 2), which ensured a continuous counter diffusion of CO32− and O2− and made a great contribution to the diffusion stage, leading to a complete carbonation. While (Li–K)Cl does not possess any O2− transmission capacity at 600 °C, even when it was in the liquid phase. On the contrary, the (Li–K)Cl molten mixtures lay over the surface of the CaO sorbents and cover the basicity, leading to a low rate of carbonation in the first fast reaction stage. In (Li–K)Cl molten mixtures, the generated O2− and CO32− cannot diffuse from the gas–liquid surface to the liquid–solid and solid–solid interface, and the interrupted counter-diffusion of CO32− and O2− hindered the continuous reaction of CaO and CO2 as shown in Fig. 3(b). For (Na–K)X (X= SO42−, CO32−, Cl−) that have a higher melting point, the mixtures will be in the solid state at the CO2 adsorption temperatures. This solid mixture could not make a contribution to the transmission of CO32− and O2−. However, the K ions dispersed into the CaO sorbents and created more Ca–O–K bonds to enhance the promotion effect, the same as the LDH reveals.22 This explains the reason that (Na–K)X (X= SO42−, CO32−, Cl−) only showed a limited promotion on CO2 uptake as shown in Fig. 3(c).
Conclusions
This study demonstrates that the uptake of CaO can be influenced by different mechanisms. By loading the eutectic mixtures with a low melting point, but no O2− conduction capability, such as (Li–K)Cl, (Li–Na)Cl, the CO2 uptake decreased dramatically due to the reduction of the basic active sites. By loading the eutectic mixtures with a high melting point, the CO2 uptake was only slightly improved. By loading the eutectic mixtures with a low melting point (lower than the sorption temperature) and a good O2− transmission capacity, such as (Li–K)2CO3 or (Li–K)2SO4, the CO2 uptake can be improved significantly. The highest capacity of over 10.9 mmol g−1 at 600 °C can be attained. The peculiar effect of alkali metal salts was attributed to the presence of a high concentration of oxygen ions in the molten alkali metal salts that facilitated the generation of carbonate ions (CO32−), resulting in the rapid formation of CaCO3 and ease of regeneration of the particles at high temperatures. This combined effect of high O2− concentration and O2− conductivity was seen to have the most benefit for the whole carbonation process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (2016ZCQ03), the National Natural Science Foundation of China (51622801, 51572029, and 51308045), and the Beijing Excellent Young Scholar (2015000026833ZK11).Notes and references
- A. Perejón, L. M. Romeo, Y. Lara, P. Lisbona, A. Martínez and J. M. Valverde, Appl. Energy, 2016, 162, 787–807 CrossRef.
- Z. H. Lee, S. Ichikawa, K. T. Lee and A. R. Mohamed, J. Energy Chem., 2015, 24, 225–231 CrossRef.
- J. Blamey, M. Zhao, V. Manovic, E. J. Anthony, D. R. Dugwell and P. S. Fennell, Chem. Eng. J., 2016, 291, 298–305 CrossRef CAS.
- L. Barelli, G. Bidini and F. Gallorini, Appl. Energy, 2015, 143, 110–118 CrossRef CAS.
- A. Di Giuliano, J. Girr, R. Massacesi, K. Gallucci and C. Courson, Int. J. Hydrogen Energy, 2017, 42, 13661–13680 CrossRef CAS.
- M. Z. Memon, X. Zhao, V. S. Sikarwar, A. K. Vuppaladadiyam, S. J. Milne, A. P. Brown, J. Li and M. Zhao, Environ. Sci. Technol., 2017, 51, 12–27 CrossRef CAS PubMed.
- A. Antzara, E. Heracleous, D. B. Bukur and A. A. Lemonidou, Int. J. Greenhouse Gas Control, 2015, 32, 115–128 CrossRef CAS.
- I. Zamboni, C. Courson and A. Kiennemann, Appl. Catal., B, 2017, 203, 154–165 CrossRef CAS.
- G. Ji, X. Xu, H. Yang, X. Zhao, X. He and M. Zhao, Environ. Sci. Technol., 2017, 51, 11484–11492 CrossRef CAS PubMed.
- A. Ebrahimi, M. Arab, M. Saffari, A. I. Minett and T. Langrish, Chem. Eng. J., 2016, 291, 1–11 CrossRef CAS.
- S. K. Bhatia and D. D. Perlmutter, AIChE J., 1983, 29, 79–86 CrossRef CAS.
- Y. Hu, W. Liu, J. Sun, M. Li, X. Yang, Y. Zhang, X. Liu and M. Xu, Fuel, 2016, 167, 17–24 CrossRef CAS.
- A. Akgsornpeak, T. Witoon, T. Mungcharoen and J. Limtrakul, Chem. Eng. J., 2014, 237, 189–198 CrossRef CAS.
- E. P. Reddy and P. G. Smirniotis, J. Phys. Chem. B, 2004, 108, 7794–7800 CrossRef CAS.
- W. Gao, T. Zhou, Y. Gao, B. Louis, D. O'Hare and Q. Wang, J. Energy Chem., 2007, 26, 830–838 CrossRef.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- L. Huang, Y. Zhang, W. Gao, T. Harada, Q. Qin, Q. Zheng, T. A. Hatton and Q. Wang, Energy Technol., 2017, 5, 1328–1336 CrossRef CAS.
- Y. Wu, J. Blamey, E. J. Anthony and P. S. Fennell, Energy Fuels, 2010, 24, 2768–2776 CrossRef CAS.
- N. Phalak, N. Deshpande and L. S. Fan, Energy Fuels, 2012, 26, 3903–3909 CrossRef CAS.
- H. Chen, P. Zhang, Y. Duan and C. Zhao, Appl. Energy, 2016, 162, 390–400 CrossRef CAS.
- FactSage FTsalt Salt Database List of Systems and Phases, http://www.factsage.cn/fact/documentation/FTsalt/FTsalt_list.htm Search PubMed.
- S. Li, Y. Shi, Y. Yang, Y. Zheng and N. Cai, Energy Fuels, 2013, 27, 5352–5358 CAS.
- S. Walspurger, L. Boels, P. D. Cobden, G. D. Elzinga, W. G. Haije and R. W. van den Brink, ChemSusChem, 2008, 1, 643–650 CrossRef CAS PubMed.
- X. Wang, W. Zhou, J. B. DeLisio, G. C. Egan and M. R. Zachariah, Phys. Chem. Chem. Phys., 2017, 19, 12749–12758 RSC.
- H. P. R. Rodríguez-Mosqueda, J. Phys. Chem. A, 2010, 114, 4535–4541 CrossRef PubMed.
- H. Kim, H. D. Jang and M. Choi, Chem. Eng. J., 2015, 280, 132–137 CrossRef CAS.
- R. I. Olivares, Sol. Energy, 2012, 86, 2576–2583 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00502d |
| This journal is © The Royal Society of Chemistry 2018 |