
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple adaptation of constitutional dynamic networks and information storage in constitutional distributions of acylhydrazones†
Guangwen
Men
ab and
Jean-Marie
Lehn
 *a
*a
aLaboratoire de Chimie Supramoléculaire, Institut de Science et d’Ingénierie Supramoléculaires, Université de Strasbourg, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: lehn@unistra.fr; Fax: + 33 3 68 85 51 40; Tel: +33 3 68 85 51 44
bState Key Laboratory of Supramolecular Structure and Materials, Jilin University, 2699 Qianjin Avenue, Changchun, 130012, P. R. China
First published on 28th September 2018
Abstract
We report a study of the behavior of four dynamic covalent libraries (DCLs) based on acylhydrazones aAbB and of the corresponding square constitutional dynamic networks (CDNs) NA–ND under the effect of three agents, namely, metal cations, base + metal cations and light irradiation; in particular, the successful switching of the CDN NB between two orthogonal distributions results, respectively, from metallo-selection and photo-selection. The four DCLs undergo triple adaptation when subjected to the three agents with the generation of specific CDN distributions characteristic of each of the four DCLs. The ternary outputs displayed by the DCLs present three states (−1, 0 and 1) related to three different constitutional distributions expressed in response to the triple inputs applied. This latter process amounts to the storage of molecular information in dynamic distributions rather than in selective interactions between complementary entities undergoing molecular recognition.
Introduction
Along the path towards systems of increasing complexity,1 constitutional dynamic chemistry2 confers to chemistry a fifth dimension, that of constitution, in addition to the three dimensions of space (structure) and that of time (kinetics).2b It is based on the generation of constitutional dynamic libraries (CDLs), that may be represented in terms of constitutional dynamic networks (CDNs)1d,2b–2d,3,4 interconnecting the constituents through agonistic and antagonistic relationships.1d,2b–2d,3 The CDNs may be considered to express chemical information residing in a specific distribution of constituents, represented by a constitutional engram2b,2d subject to and characteristic of environmental/external perturbations. In this respect, CDNs give access to chemical systems that may be trained and present higher levels of behavior displaying processes of storage, recall, and erasing of information in response, for instance, to chemical effectors such as one or two metal ions.3g The simplest CDNs are formed by four constituents subject to the exchange of their components in a square [2 × 2] network. Higher order CDNs beyond the [2 × 2] case, namely, network of networks and [3 × 3] networks, have been shown to present novel features such as multiple, synergistic and competitive hierarchical adaptation.3k A further step towards systems5 of higher complexity consists of devising CDNs that would alternatively respond to multiple external perturbing agents with orthogonal features beyond two effectors, so that it may be driven along different pathways to express multiple distribution patterns, representing multiple processing of information and leading to different informational states.According to our previous works,6 pyridyl-acylhydrazones (PyAHs) derived from the reaction of hydrazides with 2-pyridine carboxaldehyde exhibit appealing triple dynamic processes, as they are able to undergo the following: (1) constitutional dynamics by exchange of the amine and/or carbonyl component through reversible transamination;1 (2) reversible metal cation binding and exchange as well as deprotonation dynamics, stemming from the tridentate NNO coordination site;7 (3) configurational dynamics by photo- and thermo-induced cis–trans (E/Z) isomerization at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond.8 However, compared to the mixed systems (comprising imines, hydrazones, acylhydrazones, etc.), the development of multi-adaptive CDNs based solely on acylhydrazone constituents and responding to diverse perturbing agents (metal ions, acid/base, light, for instance) is still challenging due to factors such as the kinetic inertia of component exchange, low specificity of metal ion binding, inefficient conversion of photoisomerization, etc.
N double bond.8 However, compared to the mixed systems (comprising imines, hydrazones, acylhydrazones, etc.), the development of multi-adaptive CDNs based solely on acylhydrazone constituents and responding to diverse perturbing agents (metal ions, acid/base, light, for instance) is still challenging due to factors such as the kinetic inertia of component exchange, low specificity of metal ion binding, inefficient conversion of photoisomerization, etc.
Herein, we demonstrate four [2 × 2] dynamic covalent libraries (DCLs)9 based on four acylhydrazone constituents aAbB (shown in Chart 1) capable of adapting to three orthogonal external agents (base, metal ions and light), through triple dynamic selection processes, respectively, coupled base-metalloselection, metalloselection and photoselection. These four DCLs, as well as their triple external perturbations, could be specifically identified by means of the overall distributions of constituents within their CDNs. The size of the colored “balls” in the [2 × 2] square networks, shown in the schemes below, gives a gross representation of the relative amounts of the four constituents. The present work reconsiders an earlier study involving acylhydrazone-based CDNs3d and achieves the switching between orthogonal metalloselection and photoselection processes. It provides a proof of principle of the ability of chemical systems of increasing complexity to display higher levels of information processing including ternary outputs under triple binary inputs.
 | ||
| Chart 1 Structures of the acylhydrazones aAbB obtained by condensation of the corresponding aldehydes and hydrazides. The fragments originating from the aldehyde components are designated by the letter A and those from the hydrazides by the letter B. | ||
Results and discussion
1. Design and construction of the acylhydrazone-based DCLs from suitably selected components and catalysts
Several para substituted benzhydrazides were studied in order to modulate the electronic properties of the acylhydrazone unit. The N-methylated benzhydrazide 1B was selected first since its electron donating group (p-dimethylamino, EDG) would be expected to increase the electron density on the carbonyl group thus increasing metal cation binding ability3e (Scheme 1, left bottom). The other hydrazide components investigated were the non-methylated N–H ones hB bearing four options of para R groups, NO2, CF3, H and N(CH3)2.
 | ||
| Scheme 1 Generation of four [2 × 2] CDNs with four constituents pA1B, bA1B, pAhB and bAhB (3.5 mM each) by component exchange between either pA1B and bAhB (left top) or pAhB and bA1B (left middle), or by component condensation of pA, bA, 1B and hB with different catalyst and temperature conditions, giving the same statistical distribution. pAhB exist in both E and Z isomers. The diagonals and edges of the square link agonistic (+) and antagonistic (−) constituents, respectively. | ||
The reaction between these aA and bB components was used to generate the DCLs constructing the CDNs NA–ND. The generation of the DCLs by the formation of the acylhydrazone functional group as well as the component exchange between constituents occur faster with the pyridine-2-carboxaldehyde components (pA) than with the benzaldehyde components (bA) (see also ref. 10a).
The introduction of Z and Z′ groups on pA and bA, respectively, was solely dependent on the R group on hB, in order to provide overall solubility of components and constituents in CD3CN. The sets of four acylhydrazones were formed either by randomization from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two preformed constituents (Scheme 1, left top and middle) or by reaction between four corresponding components.
:1 mixture of two preformed constituents (Scheme 1, left top and middle) or by reaction between four corresponding components.
N–N conjugation of acylhydrazones decreases the electrophilicity of the CN bond, the carbonyl group is likely to reduce this conjugation, making the CN group more reactive towards nucleophiles or water as compared to hydrazone.3e The stability of the acylhydrazone linkage is influenced by several external factors. As expected, the formation and exchange reactions of acylhydrazones can be significantly sped up by means of acid catalysis and temperature increase.7b,10b For example, one equiv. (7 mM) of each component, 2A, 3A, 1B and 8B, was mixed with 10% DCl as the acid catalyst in CD3CN. After equilibration by heating to 60 °C for 5 h, the DCL was cooled and subjected to 1H NMR measurement for the analysis of the composition of the mixture of compounds generated. The results indicated that the full set of 2A1B, 3A1B, 2A8B and 3A8B (network NA, R = NO2, 3.5 mM each) was formed from condensation, transimination, hydrolysis, and recondensation of the respective A and B components, with a statistical distribution (represented by a weighted graph of a square network, Scheme 1 right top) of 25%/25%/25% (E: 10%, Z: 15%)/21% as well as 3% of hydrolysis (Table S1,† top left), wherein 2A8B was present as both E- and Z-isomers. The same distribution was obtained starting from a pair of constituents, either 2A1B and E-2A8B or 3A1B and 3A8B under similar conditions (with a somewhat longer heating time of at least 54 h). One may note that herein the thermo-isomerization from E to Z-isomer of 2A8B occurred in the exchange case of 2A1B and E-2A8B, presumably due to the stabilization of the Z form by intramolecular N–H⋯N(pyridyl) H-bonding. Moreover, in the other cases NB, NC and ND, acid catalysis was even more efficient, leading to shorter equilibration time (Tables S2–S4, Fig. S8a–10a,† from NB-1 to ND-1), especially in the network ND (no more than 13 h) with the electron donating R group, probably because the N(Me)2 group increased the nucleophilicity of the –NH2 group in the hydrazide part.
The generation of the four DCLs constructing the four CDNs, NA–ND, could also be catalyzed in the same conditions by the addition of 20% scandium triflate instead of 10% DCl. In this case, all the CDNs retained statistical distributions of the constituents, albeit producing higher percentages of E-isomers of each pAhB presumably due to their binding with Sc3+ during the generation of libraries (Tables S1–S4,† left middle, from NA-2 to ND-2).
Organocatalysis by aniline acting as a nucleophilic catalyst has been described for hydrazone formation10,11 However, as shown in Scheme S2,† the fabrication of the aforementioned DCLs using a large amount (1% v/v) of the aniline catalyst was less than successful: either it did not work at all from two synthesized constituents or required long reaction times.
2. Zn2+ binding competition and transfer among pyridyl-acylhydrazones
Various divalent metal cations, such as Zn2+, are known to form stable octahedral complexes with PyAHs by coordination to their NNO tridentate site,12 which is a weaker and more labile binder than an NNN site formed by pyridyl-hydrazone ligands. Thus, these metal ions are expected to be selectively captured and controllably transferred by PyAHs with NNO units presenting different metal ion affinities by means of modifying the interaction strength of the carbonyl O site with metal ions through the para-substituent R on the hydrazide moiety (Chart 1). In order to design the DCLs of acylhydrazones under metalloselection, we conducted a systematic study of the effect of the substitution on Zn2+ competitions among the PyAHs.In order to investigate the relative coordination affinities towards Zn2+ between N–H PyAHs and N-methylated PyAHs (pAhB and pA1B), a 1/1 mixture of 1A1B and 1A3B (3.5 mM each) was treated with 0.5 equiv. of Zn2+. After equilibrating by heating at 60 °C for 6 h, the mixture was cooled and subjected to low-temperature (−30 °C) 1H NMR measurement for the quantitative evaluation of the composition, in particular, Zn2+ distribution between these two ligands. The analysis of the NMR spectra showed that most of the Zn2+ was trapped in the formation of the homoleptic complexes ZnII(1A1B)2 and ZnII(1A3B)2, as well as the mixed ligand complex ZnII(1A1B, 1A3B), which was confirmed by ESI mass spectrometric analysis (Fig. S12b†). After further composition analysis of this mixture, a remarkable difference in the metal ion binding ability of the aforementioned ligands was found: the N–H derived acylhydrazone 1A3B captured 89% of the total Zn2+ ions compared to only 11% of those for the N-methylated derived acylhydrazone 1A1B (Schemes 2a and S4†), indicating that methylation on the hydrazide moiety of PyAHs decreased the coordination strength with metal ions. Conversely, in a 1/1 mixture of 1A1B and 1A2B (3.5 mM each) with 0.5 equiv. of Zn2+, as shown in Schemes 2b and S3,† there is a significant preference for Zn2+ ion binding to 1A1B (85%) bearing an electron donor para-dimethylamino group, attributed to an increase in the basicity of the carbonyl and therefore the strength of its interaction with a metal ion. The results above demonstrate that the metal ion binding abilities of PyAHs are remarkably structure-dependent and can be regulated by the rational choice of the Y and R groups on the hydrazide side.
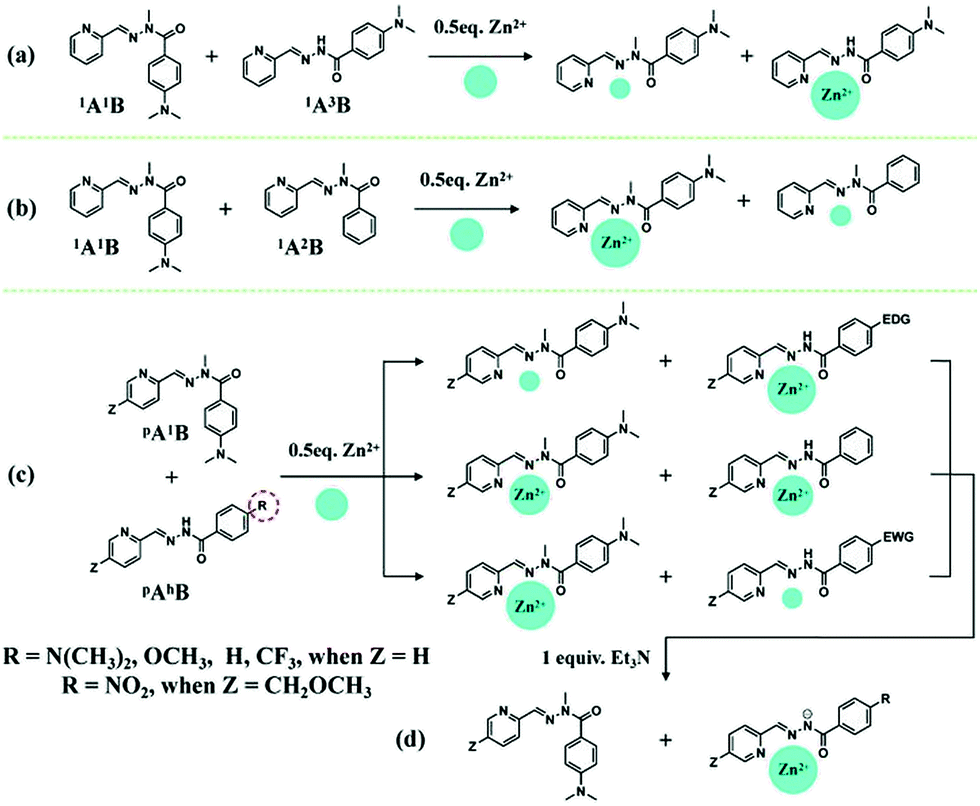 | ||
| Scheme 2 Zn2+ competition and transfer among PyAHs. (a) Zn2+ distribution in the mixture of 1 equiv. of 1A1B and 1A3B (3.5 mM each) and 0.5 equiv. of Zn2+. (b) Zn2+ distribution in the mixture of 1 equiv. of 1A1B and 1A2B (3.5 mM each) and 0.5 equiv. of Zn2+. (c) Zn2+ distribution in the mixture of 1 equiv. of N-methylated and N–H PyAHs (pA1B and bAhB) with different substituents on the R group of the hydrazide side (3.5 mM each) and 0.5 equiv. of Zn2+. (c) Zn2+ distribution in the mixture of 1 equiv. of N-methylated and N–H PyAHs with different substituents on the R group of the hydrazide side (3.5 mM each); (d) 0.5 equiv. of Zn2+ and 1 equiv. of Et3N. The CH2OCH3 group was merely used to improve the solubility of N–H PyAHs bearing a para NO2 group. In all cases, thermal equilibration was achieved by heating at 60 °C for 6 h. The size of the blue dots qualitatively indicates the relative amount of zinc complex within the pair of complexes formed in a given reaction. | ||
When a 1/1 mixture of 1A1B and 1A5B (3.5 mM each) was treated with 0.5 equiv. of Zn2+, an almost equal distribution of Zn2+ between 1A1B (45%) and 1A5B (55%) was obtained, showing no significant preference for either coordination site (Scheme S6†). Interestingly, one can readily regulate the Zn2+ distribution between N-methylated and N–H PyAHs by introducing different R groups on N–H PyAHs (Schemes 2c, S4–S8†), whereby most of the Zn2+ ions were located in complexes with N–H PyAHs bearing electron-donating groups (R = N(CH3)2 or OCH3, for instance), and conversely, the Zn2+ ions were substantially captured by N-methylated PyAHs with respect to N–H PyAHs with electron-withdrawing groups (R = CF3 or NO2, for instance).
According to our previous study,6 the deprotonation of N–H PyAHs yields a monoanionic ligand capable of coordinating with a metal ion much more efficiently. Consequently, treating a 1/1 mixture of N-methylated and N–H PyAHs (3.5 mM each) with half equiv. of Zn2+ in the presence of one equiv. of Et3N displayed the almost exclusive formation of the monoanionic ZnII(pAhB−)2 complex (close to 100%) due to the transfer of Zn2+ to the ionized N− PyAHs, regardless of the electronic properties of the R group (Scheme 2d).
3. Photoselection in CDLs of acylhydrazones
N bond and thermally activated in-plane nitrogen inversion, with their attractive features including facile preparation, high stability, efficiency of absorption, addressable and high fatigue resistance.13a
In this study, we independently investigated the spectroscopic properties under the irradiation of each constituent in the four aforementioned libraries as a function of structural variation. No clear change was found in the UV-vis in the acylhydrazones pA1B and bA1B (2A1B, 3A1B, 1A1B and 4A1B) starting from their E-isomer in CD3CN (30 μM) under constant irradiation with a 125 W Hg-lamp in the λirr = 310–400 nm domain (see Fig. S17†), a change in E to Z conversion might be induced using the appropriate wavelength. The spectral properties of E-bAhB (E-3A8B, E-4A7B, E-4A5B and E-4A3B) derived from benzaldehyde bA and non-methylated/N–H hydrazide hB were then examined. E-bAhB displayed an absorption band (λmax) in the UV region from 284 to 330 nm, showing a substantial bathochromic shift along with the electronic properties of the R group of the hydrazide moiety from the electron-withdrawing group NO2 to the electron-donating group N(Me)2 (Table S5,† the second column). Constant light irradiation (λirr = 310–400 nm) led to different decreases and hypsochromic shifts of λmax, indicating isomerization toward their Z-isomers (Fig. S18†). Clear isosbestic points suggested clean conversions from E to Z-isomer upon irradiation. The thermodynamically less stable Z-isomers of bAhB returned to their E-isomers with relatively long half-lives of the Z-isomers at 25 °C, which were also related to the electronic properties of the R group of the hydrazide part, increasing from 26 min for the NO2 group to 330 min for the N(Me)2 group. However, the half-lives of the Z isomers of these acylhydrazones were dramatically reduced by the addition of 10% HCl or 20% Sc3+, which favor the conversion of Z to E and are also the catalysts for component exchange in the DCLs (Table S5,† the last two columns). In fact, the Z-bAhB isomers were not observed by 1H NMR measurements even over a time usually larger than 10 min. According to our previous studies,6,13a the non-methylated PyAHs pAhB (2A8B, 1A7B, 1A6B, 1A5B, 1A4B and 1A3B), whose Z-isomers are stabilized by intramolecular H-bonding, are expected to be promising driving force candidates for amplifications in CDNs. In contrast to those of the E-bAhB, the λmax values of the E-pAhB were substantially independent of the R groups, except that of E-1A3B (335 nm, N(CH3)2) displaying a significant red shift relative to other E-pAhB compounds (Table S6,† the third column). Constant light irradiation resulted in decreases in absorption for λmax and bathochromic shifts with clear isosbestic points (Fig. S19a–S24a†). As expected, Z-pAhB showed high thermal stability regardless of the presence or absence of 10% HCl or 20% Sc3+ (Table S6,† the last three columns).
 | ||
| Scheme 3 Weighted graph representation of modifications of the four [2 × 2] CDNs of the four constituents pA1B, bA1B, pAhB and bAhB (3.5 mM each) under light irradiation (λirr = 310–400 nm) starting from the statistical distribution in the presence of a different catalyst and temperature conditions. (Top left) No photoselection takes place with catalysts (1)–(3). (Top right) the system undergoes photoselection in the case of NB and NC in the presence of the combined catalysts (4). pAhB exists in both E and Z isomers. The diagonal and edges of the square link agonistic (+) and antagonistic (−) constituents, respectively. See Tables S1–S4† for numerical values. | ||
In order to unravel the reason for such Sc3+-induced enhancements of the photo-conversion from E to Z isomers, the Z-isomer ratios following the photo-isomerization from E to Z isomers of N–H PyAHs pAhB (2A8B, 1A7B, 1A6B, 1A5B, 1A4B and 1A3B) under constant light irradiation (λirr = 310–400 nm) were then studied as a function of time, whereby quantitative evaluation of their photostationary states were determined by 1H NMR measurements (Fig. S25†). We found that except for 1A3B with R = N(CH3)2, all the N–H PyAHs displayed relatively low conversion (≤51%) from E to Z under sufficient duration (>150 min) of irradiation (Scheme S9,† top). The reason may be that under light irradiation with a given range of wavelengths from 310 to 400 nm, 1A3B with a maximum absorption band at 335 nm may have the strongest absorption compared to the other pAhB (λmax around 300 nm). Surprisingly, in the presence of a catalytic amount of 2% Sc3+ (Fig. S26 and Scheme S9,† bottom), long time (>300 min) light irradiation led to larger conversion (≥79%) for almost all cases except that for 2A8B with R = NO2 (one possible explanation is due to the weak interaction between Sc3+ and 2A8B). By comparing the ratio of Z-isomers in the absence and presence of Sc3+, enhancements of the Z-isomer ratios with different R groups in the presence of ScIII(OTf)3 under sufficient duration of constant light irradiation (λirr = 310–400 nm) are demonstrated in Fig. S27.† Large enhancements were found in the photoisomerization of PyAHs with R = CF3, Cl, H and OCH3, with unsuccessful catalysis in the case when R = NO2 or R = N(CH3)2. The reason for these enhancements may be attributed to the increase in light absorption caused by the Sc3+-induced red shift of the absorption bands λmax, extending into the irradiation range of 310–400 nm (See Table S6 and Fig. S19b–S24b†).
N bond;14 (ii) increasing the E to Z conversion of PyAHs under irradiation by increasing light absorption. Indeed, by using the catalyst pair of 2% Sc3+ and 1% v/v aniline and inducing E to Z photoisomerization, photoselection was successfully achieved for the CDNs NB and NC in response to constant light irradiation leading to the amplification of a single diagonal of the network (Scheme 3, top right). The results for the photoselection of these CDNs are described in more detail below.
4. Triple adaptation in CDNs based on four acylhydrazone CDLs
On the basis of the results above, four DCLs, each with four constituents, were subjected to the application of triple external perturbations: metalloselection, base-metalloselection and photoselection. The following results (A)–(D) were obtained.(A) First, we investigated the multiple adaptations of the DCL formed by constituents 2A1B, 3A1B, 2A8B and 3A8B (R = NO2) starting from its statistical distribution within the CDN NA.
(A-1) Metalloselection: this initial mixture of four constituents (2A1B, 3A1B, E/Z-2A8B and 3A8B, 3.5 mM each) was treated with 0.5 eq. of Zn2+ (with respect to the amount of components, 3.5 mM) under equilibration by heating at 60 °C for 10 h either with or without the addition of catalysts (10% DCl or 20% Sc3+). The zinc ions were distributed between 2A1B and 2A8B, with 67% (of total Zn2+) binding to the former constituent and 33% binding to the latter (NA-Zn in Table S7,† middle center), which is in accordance with the discussion above (Scheme S8†). Such a marked bias in the Zn2+ capture between the two PyAHs led to the reorganization of the DCL to amplify the constituent 2A1B, presenting the strongest Zn2+ binding, in the form of its octahedral complex ZnII(2A1B)2, the mixed ligand complex ZnII(2A1B, 2A8B) together with E-2A1B, as well as simultaneously its diagonally linked agonist 3A8B (agonist amplification). The resulting modified network NA-Zn presented the distribution 32%/16%/18%/32% for the constituents 2A1B/3A1B/2A8B/3A8B, respectively, with 2% hydrolysis (Scheme 4, middle center).
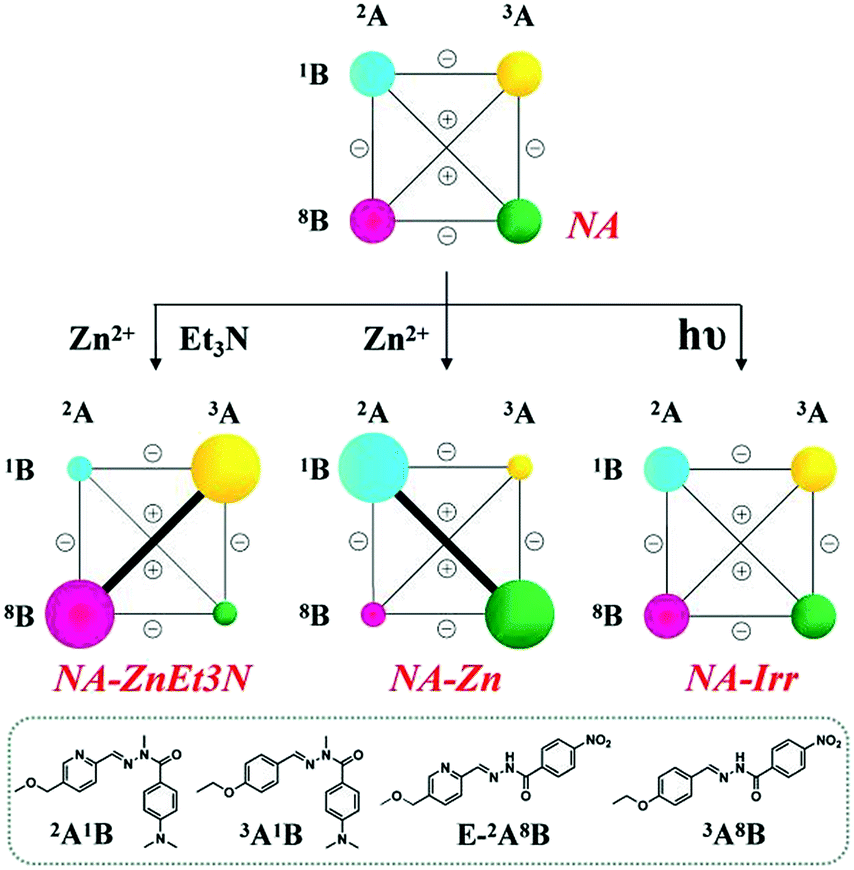 | ||
| Scheme 4 Weighted graph representation of triple adaptation within the [2 × 2] CDN NA from the statistical distribution (top) of the four constituents 2A1B, 3A1B, 2A8B and 3A8B (3.5 mM each) in response to 0.5 eq. Zn2+ & 1 eq. Et3N (middle left), 0.5 eq. Zn2+ (middle center) and light irradiation (λirr = 310–400 nm, middle right)a. aSee text and ESI for conditions, catalyst and temperature. See Table S7† for numerical values. | ||
(A-2) Base-metalloselection: the base-induced deprotonation of constituent 2A8B is expected to dramatically enhance its affinity toward zinc ions; therefore, treating the same DCL with 0.5 eq. Zn2+ and 1eq. Et3N caused a rearrangement of the CDN NA to the almost exclusive formation of the ZnII(2A8B−)2 complex (NA-ZnEt3N in Table S7,† middle left). This acted as a driving force for the modification of the network from the initial statistical uniformity to the diagonal amplification of 2A8B and its agonist 3A1B, a distribution orthogonal to the metalloselection distribution (NA-Zn). The resulting distribution was 16%/34%/33%/11% for the 2A1B/3A1B/2A8B/3A8B, constituents respectively, with 5% hydrolysis. These results showed that NA can undergo dual orthogonal adaptation.
(A-3) Photoselection attempt: the same four-membered network underwent light irradiation (310–400 nm) in the presence of the catalysts, 2% Sc3+ and 1% v/v aniline in order to increase the isomerization conversion from the E to Z isomer and activate the component exchange reaction of the system. The latter did not take place and the stated statistical distribution remained unchanged (NA-Irr in Scheme 4, middle right), albeit limited enhancement of the Z-isomer from 11% to 12% was observed after light irradiation (NA-Irr in Table S7, middle right; Table S1c†).
It is worth noting that each perturbation (base-metalloselection, metalloselection and photoselection) induces a specific distribution of the four dynamic constituents, allowing, in principle, for the identification of the perturbing agent that gives rise to it.
(B) Under the action of the same perturbation agents, base, metal ion and light, the DCL consisting of the four constituents 1A1B, 4A1B, 1A7B and 4A7B exhibited triple adaptation, expressing different biased distributions within network NB (R = CF3) compared to network NA.
(B-1) Metalloselection: on addition of 0.5 eq. Zn2+ (3.5 mM) to this DCL with the initial statistical distribution, the difference in the zinc ion affinities between 1A1B and 1A7B resulted in a marked metalloselection by 61% of Zn capture for 1A1B with respect to 39% for 1A7B; the decreased ability for zinc ion binding is attributed to its electron-withdrawing CF3 group (NB-Zn in Table S8,† middle center). As a result, DCL recombination resulted in the pronounced amplification of the former ligand 1A1B at the expense of its antagonist constituents containing either 1A or 1B and consequently a corresponding up-regulation of its agonist 4A7B (NB-Zn in Scheme 5, middle center).
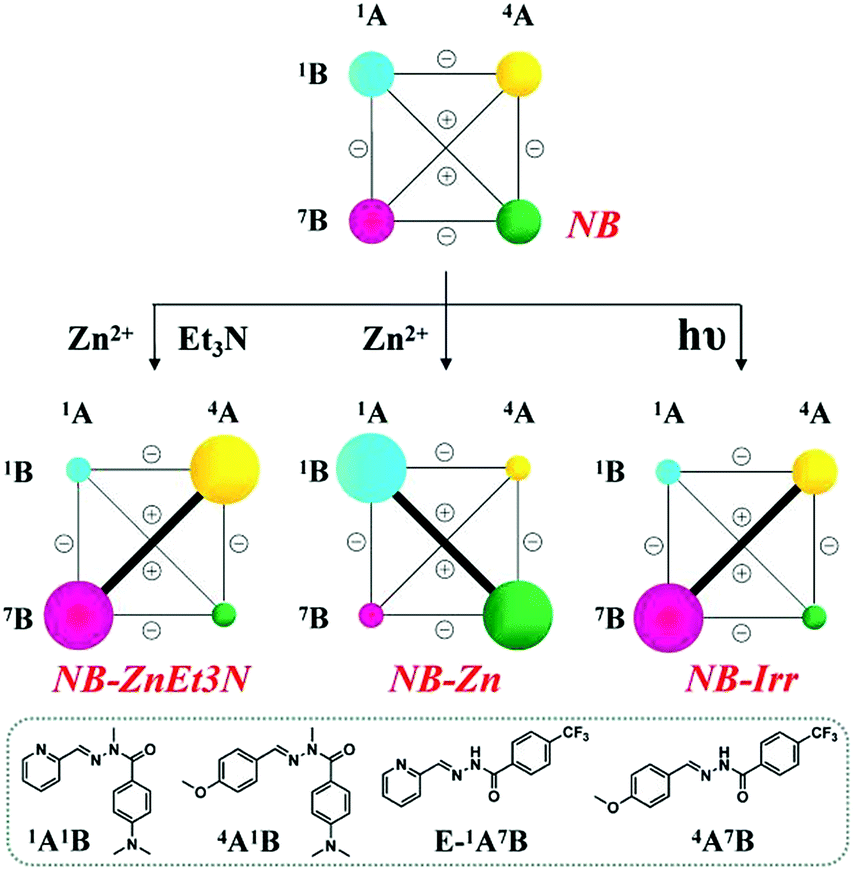 | ||
| Scheme 5 Weighted graph representation of the triple adaptation of the [2 × 2] CDN NB from the statistical distribution (top) of the four constituents 1A1B, 4A1B, 1A7B and 4A7B (3.5 mM each) in response to 0.5 eq. Zn2+ & 1 eq. Et3N (middle left), 0.5 eq. Zn2+ (middle center) and light irradiation (λirr = 310–400 nm, middle right)a. aSee text and ESI for conditions, catalyst and temperature. See Table S8† for numerical values. | ||
(B-2) Base-metalloselection: as expected, in the presence of 3.5 mM Zn2+ (0.5 eq. with respect to the concentration of components) and 7 mM Et3N (1 eq.), constituent 1A7B underwent strong up-regulation on deprotonation of its N–H site with concomitant amplification of its agonist 4A1B (NB-ZnEt3N in Scheme 5, middle left). This reorganization process of the DCL was kinetically slow and required acceleration using catalysts (1% v/v aniline, for instance).
(B-3) Photoselection: light irradiation (310–400 nm) promoted the E to Z photoisomerization of PyAHs 1A7B with the assistance of N–H⋯N hydrogen bonding and in the presence of 2% Sc3+ ions from an initial Z-isomer ratio of 7% to 25% (see Tables S2c and S8†). When the irradiation of the DCL was conducted in the presence of both 2% Sc3+ and 1% v/v aniline to catalyze component exchange, the composition of the library was modified giving a distribution of 19%/20%/30%/18% for constituents 1A1B/4A1B/1A7B/4A7B, respectively, together with 13% hydrolysis (NB-Irr in Table S8,† middle right). The corresponding rearranged CDN thus showed a simultaneous diagonal amplification of constituent 1A7B and of its agonist 4A1B (NB-Irr in Scheme 5, middle right).
Remarkably, one may note that this CDN undergoes orthogonal dual adaptation in response to two orthogonal external agents: a chemical effector, Zn2+, and a physical stimulus, light irradiation.
(C) We then investigated the multiple adaptations of the DCL containing the four interconvertible constituents 1A1B, 4A1B, 1A5B and 4A5B (R = H) within the network NC.
(C-1) Metalloselection attempt: in view of the lack of preference for zinc ions between 1A1B and 1A5B as discussed above, applying Zn2+ to the evenly distributed CDN NC did not change the distribution pattern of the network (NC-Zn in Scheme 6, middle center).
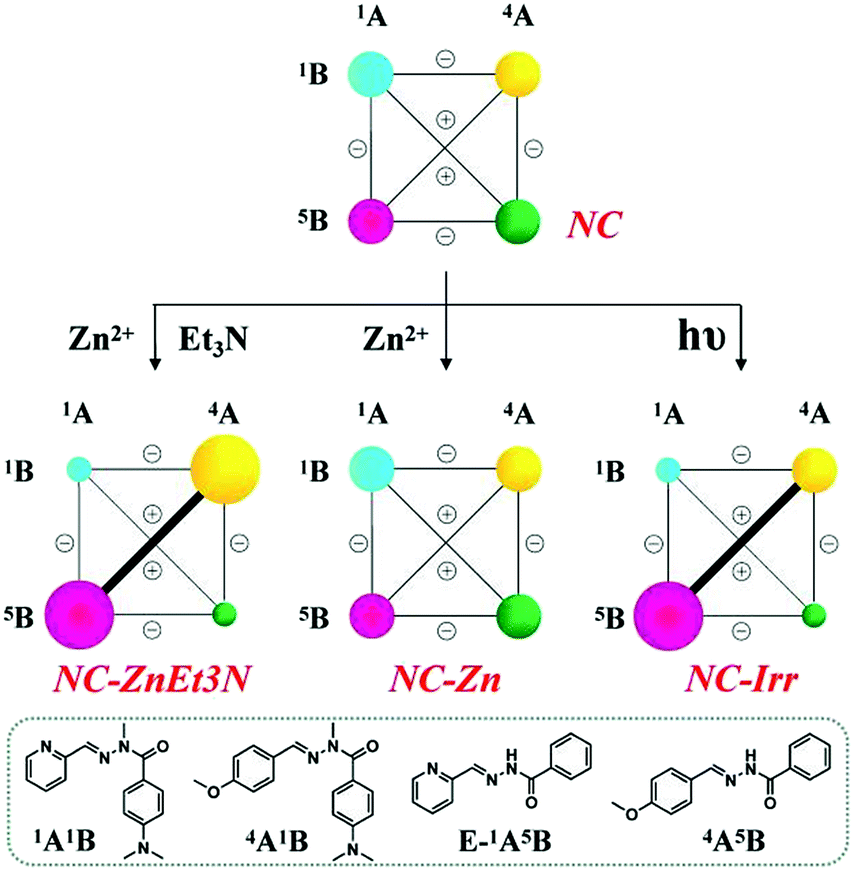 | ||
| Scheme 6 Weighted graph representation of triple adaptation within the [2 × 2] CDN NC from the statistical distribution (top) of the four constituents 1A1B, 4A1B, 1A5B and 4A5B (3.5 mM each) in response to 0.5 eq. Zn2+ and 1 eq. Et3N (middle left), 0.5 eq. Zn2+ (middle center) and light irradiation (λirr = 310–400 nm, middle right)a aSee text and ESI for conditions, catalyst and temperature. See Table S9† for numerical values. | ||
(C-2) Base-metalloselection: as expected, the constitutional distribution of this DCL underwent a similar diagonal amplification of the agonist constituents 1A5B and 4A1B (NC-ZnEt3N in Scheme 6, middle left; see Table S9† for numerical values) as observed above for the CDNs NA and NB.
(C-3) Photoselection: according to the results shown in Fig. S27 and Table S9,† irradiation of the above library in the presence of 2% Sc3+ enhanced the E to Z conversion of 1A5B from 11% to 30% (percentages of the Z-isomers). As in the case of network NB after 1 h of constant irradiation (310–400 nm), the percentage of the constituents of the diagonal 1A5B – 4A1B underwent up-regulation relative to the other diagonal 1A1B – 4A5B, although there was no amplification of 4A1B with respect to the starting network (from 25% to 24%, see Table S9†) due to the light-induced hydrolysis. The result is a markedly biased distribution of 16%/24%/34%/18% for the constituents 1A1B/4A1B/1A5B/4A5B, respectively, together with around 7% hydrolysis (NC-Irr in Table S9,† middle right).
(D) Finally, the fourth DCL containing equimolar amounts of constituents 1A1B, 4A1B, 1A3B and 4A3B was similarly subjected to the application of the three agents, metal ions, base and light within the CDN ND.
(D-1) Metalloselection: according to the data above, the added Zn2+ ions are expected to be selectively captured by PyAH 1A3B in preference to other constituents in this DCL, as it presents both a favorable N–H site and a strongly electron-donating N(Me)2 group that increases electron density on the carbonyl group. Such metalloselection yields a distribution of 17%/32%/33%/15% for the constituents 1A1B/4A1B/1A3B/4A3B, respectively, together with 3% hydrolysis (ND-Zn in Table S10,† middle center), displaying a diagonal amplification in the network (ND-Zn in Scheme 7, middle center), unlike the behavior of the former cases NA-Zn, NB-Zn and NC-Zn.
 | ||
| Scheme 7 Weighted graph representation of triple adaptation within the [2 × 2] CDN ND from the statistical distribution (top) of the four constituents 1A1B, 4A1B, 1A3B and 4A3B (3.5 mM each) in response to 0.5 eq. Zn2+ and 1 eq. Et3N (middle left), 0.5 eq. Zn2+ (middle center) and light irradiation (λirr = 310–400 nm, middle right)a. aSee text and ESI for conditions, catalyst and temperature. See Table S10† for numerical values. | ||
(D-2) Base-metalloselection: the synergistic perturbation of the same DCL by Zn2+ and Et3N together gave the same distribution bias as observed on application of these agents to the previous networks, NA-ZnEt3N, NB-ZnEt3N and NC-ZnEt3N (ND-ZnEt3N in Scheme 7, middle left).
(D-3) Photoselection attempt: on exposure to constant light irradiation (λirr = 310–400 nm, for 1 h), the photoresponsive constituent 1A3B underwent only a small increase in isomerization with enhancement of the Z-isomer ratio from 12% to 17%, whereas the statistical distribution of the constituents of the DCL remained unchanged.
5. Information processing in CDNs with triple inputs and ternary outputs
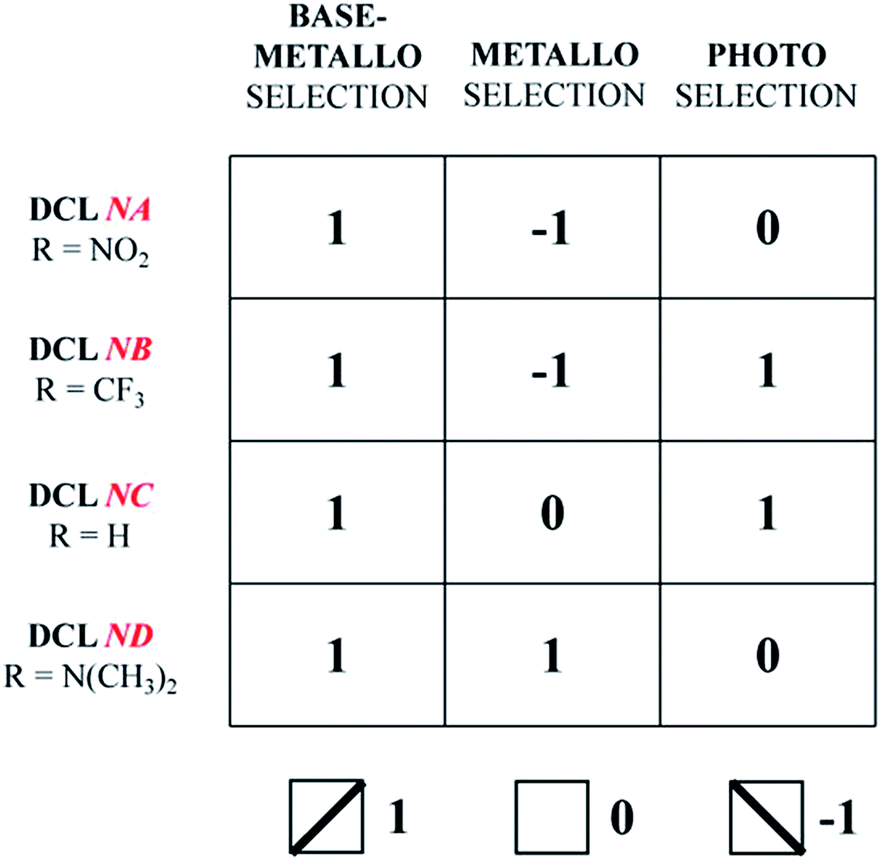
The four DCLs above adapt/respond to the application of the three agents, base-metalloselection, metalloselection and photoselection, by generating/expressing a constitutional state, characteristic of a given DCL/agent pair, represented by specific distributions of the constituents within the corresponding square CDN through the agonistic/antagonistic relationships between the constituents. The triple input perturbations (metal ion, base and light) enforce different constitutional states that define the response to a given agent and provide a readout that is characteristic of a given DCL. The present set of four acylhydrazone-based CDNs may thus be developed as information devices based on the storage of information in a distributional pattern. As shown in Table 1, a given input agent yields a specific output represented by −1, 0 and 1 for statistical distribution (0) and expression of a network diagonal (−1 and 1). The ternary outputs generated by the three perturbations encode a specific horizontal readout defining each of the DCLs/CDNs investigated. As indicated in Table 1, the four DCLs NA–ND are specifically encoded into the horizontal readouts (1, −1, 0), (1, −1, 1), (1, 0, 1) and (1, 1, 0), respectively, under the action of the three inputs. On the other hand, the vertical readouts (1, 1, 1, 1), (−1, −1, 0, 1) and (0, 1, 1, 0) for the four DCLs, respectively, are characteristic of the applied input agent, base-metalloselection, metalloselection and photoselection, respectively. Thus, Table 1 characterizes both a given DCL (horizontal; line) and a specific perturbing agent (vertical; column) in an informational double entry CDN matrix.
|
Experimental
For experimental details, see the ESI.†Conclusions
The present study of four acylhydrazones-based DCLs, represented by the corresponding CDNs, has achieved three goals: (1) the successful switching of the CDN NB between two orthogonal distributions under metallo- and photo-selection, respectively; (2) the expression of specific CDN distributions in DCLs subjected to three different agents, metal cation, base + metal cation and light irradiation, acting on four different CDLs; (3) the storage of molecular information in dynamic distributions that link triple inputs with ternary outputs (Table 1).The latter deserves some further elaboration. It confirms the interest of such constitutional dynamic systems for exploring the storage and processing of molecular information not through specific molecular recognition processes between complementary components but through distributions of constituents in dynamic interconnection in a network. The network explored here is the simplest one involving four constituents located at the vertexes of a square and connected by agonistic (diagonals) and antagonistic (edges) relationships. This may in principle be extended to networks of higher complexity, such as the reported [3 × 3]3k and 3D networks.15
In general terms, the perturbation of a DCL by a given chemical or physical agent induces a redistribution of all linked constituents in the CDN that will result in a specific distribution corresponding to a given output code (such as those displayed in Table 1 for the present study), which may be considered as a sort of fingerprint, a constitutional engram2b,2d of dynamic nature, characteristic of that perturbation. Such storage of chemical information in dynamic constitutional distributions has been described for the effect of different alkali metal cations on a library of dynamic covalent polymers.16 On the other hand, the response of a given CDN to the application of multiple chemical/physical agents generates a specific combination of distributional outputs. Such processes establish a two-way link between a driving agent and a set of outputs in an informational double entry CDN matrix. Of course, the features of DCLs hold as well for non-covalent constituents, i.e. altogether for constitutional dynamic libraries operating on the covalent molecular, and the non-covalent supramolecular levels.2,3
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the ERC (Advanced Research Grant SUPRADAPT 290585), the ANR Grant DYNAFUN and the University of Strasbourg Institute for Advanced Study (USIAS) for financial support. GWM gratefully acknowledges Professor Shimei Jiang (Jilin University, Changchun), Dr Ghislaine Vantomme and Dr Martin Herder for extensive discussions, as well as the National Natural Science Foundation of China (51673082) and the above ERC grant for post-doctoral fellowship support.Notes and references
- For complexity and complex systems, see, for instance: (a) K. Mainzer, Thinking in Complexity, Springer, Berlin, 5th edn, 2007 Search PubMed; (b) R. Cohen and S. Havlin, Complex Networks, Cambridge University Press, Cambridge, 2010 CrossRef; (c) G. Nicolis and C. Nicolis, Foundations of Complex Systems, World Scientific Publishing Co., Singapore, 2nd edn, 2012 CrossRef; (d) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS; (e) I. Alfonso, Chem. Commun., 2016, 52, 239–250 RSC.
- For a selection of papers on constitutional dynamic chemistry, see, for instance: (a) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS; (b) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (c) Constitutional Dynamic Chemistry: Topics in Current Chemistry, ed. M. Barboiu, Springer, 2012, vol. 322 Search PubMed; (d) J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- For recent examples of constitutional dynamic networks, see, for instance: (a) N. Giuseppone and J.-M. Lehn, Chem.–Eur. J., 2006, 12, 1715–1722 CrossRef CAS; (b) S. Ulrich and J.-M. Lehn, Chem.–Eur. J., 2009, 15, 5640–5645 CrossRef CAS; (c) L. Lao, J.-L. Schmitt and J.-M. Lehn, Chem.–Eur. J., 2010, 16, 4903–4910 CrossRef CAS; (d) N. Hafezi and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 12861–12868 CrossRef CAS; (e) G. Vantomme, S. M. Jiang and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS ; corrigendum: J. Am. Chem. Soc., 2015, 137, 3138–3138 and J. Am. Chem. Soc., 2018, 140, 1179–1180; (f) G. Vantomme, N. Hafezi and J.-M. Lehn, Chem. Sci., 2014, 5, 1475–1483 RSC; (g) J. Holub, G. Vantomme and J.-M. Lehn, J. Am. Chem. Soc., 2016, 138, 11783–11791 CrossRef CAS; (h) J. J. Armao IV and J.-M. Lehn, Angew. Chem., Int. Ed., 2016, 55, 13450–13454 CrossRef; (i) S. Dhers, J. Holub and J.-M. Lehn, Chem. Sci., 2017, 8, 2125–2130 RSC; (j) L. Yue, S. Wang, A. Cecconello, J.-M. Lehn and I. Willner, ACS Nano, 2017, 11, 12027–12036 CrossRef CAS; (k) G. W. Men and J.-M. Lehn, J. Am. Chem. Soc., 2017, 139, 2474–2483 CrossRef CAS; (l) S. Wang, L. Yue, Z. Shpilt, A. Cecconello, J. S. Kahn, J.-M. Lehn and I. Willner, J. Am. Chem. Soc., 2017, 139, 9662–9671 CrossRef CAS PubMed; (m) M. Herder and J.-M. Lehn, J. Am. Chem. Soc., 2018, 140, 7647–7657 CrossRef CAS PubMed.
- For a chemical interaction network, see: S. Ghosh, P. Mukhopadhyay and L. Isaacs, J. Syst. Chem., 2010, 1, 1–6 CrossRef.
- For recent reviews about systems chemistry, see, for instance: (a) S. A. Kauffman, ACS Symp. Ser., 2007, 981, 310–324 CrossRef CAS; (b) R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC; (c) J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS; (d) J. J. P. Peyralans and S. Otto, Curr. Opin. Chem. Biol., 2009, 13, 705–713 CrossRef CAS; (e) R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847–858 RSC; (f) O. Taran and G. von Kiedrowski, in Chemical Synthetic Biology, ed. P. L. Luisi and C. Chiarabelli, John Wiley & Sons, Chichester, 2011, pp. 289–319 Search PubMed; (g) J. Huck and D. Philp, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley & Sons, Chichester, 2012, pp. 1415–1445 Search PubMed; (h) N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS PubMed; (i) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS; (j) B. Grzybowski, S. Otto and D. Philp, Chem. Commun., 2014, 50, 14924–14925 RSC; (k) E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J., 2011, 17, 248–258 CrossRef CAS.
- (a) A. Choudhury, B. Geetha, N. R. Sangeetha, V. Kavita, V. Susila and S. J. Pal, Coord. Chem., 1999, 48, 87–95 CrossRef CAS; (b) S. Choudhary and J. R. Morrow, Angew. Chem., Int. Ed., 2002, 41, 4096–4098 CrossRef CAS; (c) P. V. Bernhardt, P. Chin, P. C. Sharpe and D. R. Richardson, Dalton Trans., 2007, 30, 3232–3244 RSC; (d) C. F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007–5010 CrossRef CAS; (e) X. Y. Cao, J. Harrowfield, J. Nitschke, J. Ramirez, A. M. Stadler, N. Kyritsakas-Gruber, A. Madalan, K. Rissanen, L. Russo, G. Vaughan and J.-M. Lehn, Eur. J. Inorg. Chem., 2007, 18, 2944–2965 CrossRef.
- (a) G. Palla, A. Mangia and G. Predieri, Ann. Chim., 1984, 74, 153–158 CAS; (b) D. G. Belov, B. G. Rogachev, L. I. Tkachenko, V. A. Smirnov and S. M. Aldoshin, Russ. Chem. Bull., 2000, 49, 666–668 CrossRef CAS; (c) V. V. Syakaev, S. N. Podyachev, B. I. Buzykin, S. K. Latypov, W. D. Habicher and A. I. Konovalov, J. Mol. Struct., 2006, 788, 55–62 CrossRef CAS.
- For a selection of reviews specifically on dynamic covalent/combinatorial chemistry, see, for instance: (a) J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (b) S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (c) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS; (d) S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 RSC; (e) Dynamic Combinatorial Chemistry, ed. B. L. Miller, Wiley, Chichester, 2010 Search PubMed; (f) J. N. H. Reek and S. Otto, Dynamic Combinatorial Chemistry, Wiley-VCH, Weinheim, 2010 CrossRef; (g) R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847–855 RSC; (h) M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC; (i) J. W. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS; (j) A. Herrman, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (k) Dynamic Covalent Chemistry: Principles, Reactions and Applications, ed. W. Zhang and Y. Jin, Wiley, 2018 Search PubMed.
- (a) E. T. Kool, D. H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS; (b) D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef.
- (a) E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS; (b) W. P. Jencks, Catalysis in Chemistry and Enzymology; McGraw-Hill, New York, 1969, pp. 493–496 Search PubMed; (c) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS; (d) V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS PubMed; (e) M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- (a) S. E. Livingstone and J. E. Oluka, Transition Met. Chem., 1978, 3, 261–267 CrossRef CAS; (b) O. Pouralimardan, A.-C. Chamayou, C. Janiak and H. Hosseini-Monfared, Inorg. Chim. Acta, 2007, 360, 1599–1608 CrossRef CAS; (c) A.-M. Stadler and J. Harrowfield, Inorg. Chim. Acta, 2009, 362, 4298–4314 CrossRef CAS; (d) N. A. Mangalam, S. Sivakumar, S. R. Sheeja, M. R. Prathapachandra Kurup and E. R. T. Tiekink, Inorg. Chim. Acta, 2009, 362, 4191–4197 CrossRef CAS; (e) A. A. Recio Despaigne, J. G. da Silva, A. C. M. do Carmo, F. Sives, O. E. Piro, E. E. Castellano and H. Beraldo, Polyhedron, 2009, 28, 3797–3803 CrossRef; (f) N. A. Mangalam, S. R. Sheeja and M. R. Prathapachandra Kurup, Polyhedron, 2010, 29, 3318–3323 CrossRef CAS; (g) M. S. Shongwe, S. H. Al-Rahbi, M. A. Al-Azani, A. A. Al-Muharbi, F. Al-Mjeni, D. Matoga, A. Gismelseed, I. A. Al-Omari, A. Yousif, H. Adams, M. J. Morris and M. Mikuriya, Dalton Trans., 2012, 41, 2500–2514 RSC.
- (a) D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed; (b) I. Cvrtila, H. Fanlo-Virgós, G. Schaeffer, G. M. Santiago and S. Otto, J. Am. Chem. Soc., 2017, 139, 12459–12465 CrossRef CAS PubMed.
- (a) N. Giuseppone, J.-L. Schmitt and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 4902–4906 CrossRef CAS PubMed; (b) N. Giuseppone, J.-L. Schmitt, E. Schwartz and J.-M. Lehn, J. Am. Chem. Soc., 2005, 127, 5528–5539 CrossRef CAS; (c) N. Giuseppone, J.-L. Schmitt and J.-M. Lehn, J. Am. Chem. Soc., 2006, 128, 16748–16763 CrossRef CAS PubMed.
- G. Vantomme, N. Hafezi and J.-M. Lehn, Chem. Sci., 2014, 5, 1475–1483 RSC.
- (a) S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2009, 48, 7635–7638 CrossRef CAS PubMed; (b) See also Fig. 11 in J.-M. Lehn, Adv. Polym. Sci., 2013, 261, 155–172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03858a |
| This journal is © The Royal Society of Chemistry 2019 |