
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissolution and homogeneous photocatalysis of polymeric carbon nitride†
Chaofeng
Huang‡
a,
Jing
Wen‡
b,
Yanfei
Shen
 a,
Fei
He
a,
Li
Mi
a,
Ziyu
Gan
a,
Jin
Ma
a,
Songqin
Liu
a,
Haibo
Ma
*b and
Yuanjian
Zhang
*a
a,
Fei
He
a,
Li
Mi
a,
Ziyu
Gan
a,
Jin
Ma
a,
Songqin
Liu
a,
Haibo
Ma
*b and
Yuanjian
Zhang
*a
aJiangsu Engineering Laboratory of Smart Carbon-Rich Materials and Device, Jiangsu Province Hi-Tech Key Laboratory for Bio-Medical Research, School of Chemistry and Chemical Engineering, Medical School, Southeast University, Nanjing 211189, China. E-mail: Yuanjian.Zhang@seu.edu.cn
bSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: haibo@nju.edu.cn
First published on 2nd October 2018
Abstract
As a metal-free conjugated polymer, carbon nitride (CN) has attracted tremendous attention as a heterogeneous (photo)catalyst. By following the example of enzymes, making all of the catalytic sites accessible via homogeneous reactions is a promising approach toward maximizing CN activity, but hindered due to the poor solubility of CN. Herein, we report the dissolution of CN in environmentally friendly methanesulfonic acid, and homogeneous photocatalysis (two biomimetic/pharmaceutical photocatalytic oxidation reactions) driven by CN for the first time with the activity boosted up to 10-times compared to the heterogeneous counterparts. Moreover, facile recycling and reusability, the hallmarks of heterogeneous catalysts, were kept for the homogeneous CN photocatalyst via reversible precipitation using poor solvents. This study opens a new vista for CN in homogeneous catalysis and offers a successful example of a polymeric catalyst that bridges the gap between homo/heterogeneous catalysis.
Introduction
Awareness of a sustainable society calls for the development of highly efficient catalysts: heterogeneous and homogeneous. Each of them demonstrate characteristic sets of advantages and limitations with regards to accessible catalytic sites, separation, and recycling; thus, the combination of all merits in a single catalyst is highly anticipated.1 As a promising metal-free heterogeneous catalyst, polymeric carbon nitride (CN) has stimulated substantial interest in the conversion of organic molecules and solar-to-chemical energy conversion and beyond, e.g. in optoelectronic biosensing, due to its unique electronic structure and surface properties.2 By following the example of enzymes, making all of the catalytic sites accessible via homogeneous reactions is a promising approach toward maximizing CN activity. However, the poor solubility of CN, ascribed to the interlayer van der Waals forces,3 makes the use of CN for homogeneous catalysis difficult. Concentrated H2SO4 was reported as the first solvent for CN, but the harsh conditions hindered the further application of this approach. Polar aprotic solvents, such as dimethyl sulfoxide (DMSO), have been disclosed to dissolve bulk poly(triazine imide) (PTI·LiBr)-based CN with controllable luminescent properties.4 As an innovative alternative, exfoliating bulk CN into nanosheets greatly boosted its photocatalytic activity.5 Similarly, in the early days of graphene and carbon nanotube research, their insolubility in all solvents remained as one of the biggest impediments to the realization of their potential applications.6 Therefore, seeking more general solvents to solubilize CN and perform homogeneous catalysis would be fascinating for the subsequent research of CN.Results and discussion
Herein, we report the dissolution of CN in CH3SO2OH (MSA), an environmentally friendly solvent. As controls, other sulfonic acid group-containing solvents, including ClSO2OH (ClSA), HOSO2OH (H2SO4), and C2H5OSO2CH3 (EMS), were also investigated. Taking two photocatalytic oxidations as examples, up to 10-fold faster catalytic kinetics of homogeneous CN were achieved relative to those of the heterogeneous one. Moreover, due to its capability for reversible dissolution and recovery using good and poor solvents, CN could be facilely separated from the homogeneous system and reused, thus combining the advantages of both homogeneous and heterogeneous catalysts as shown in Fig. 1a.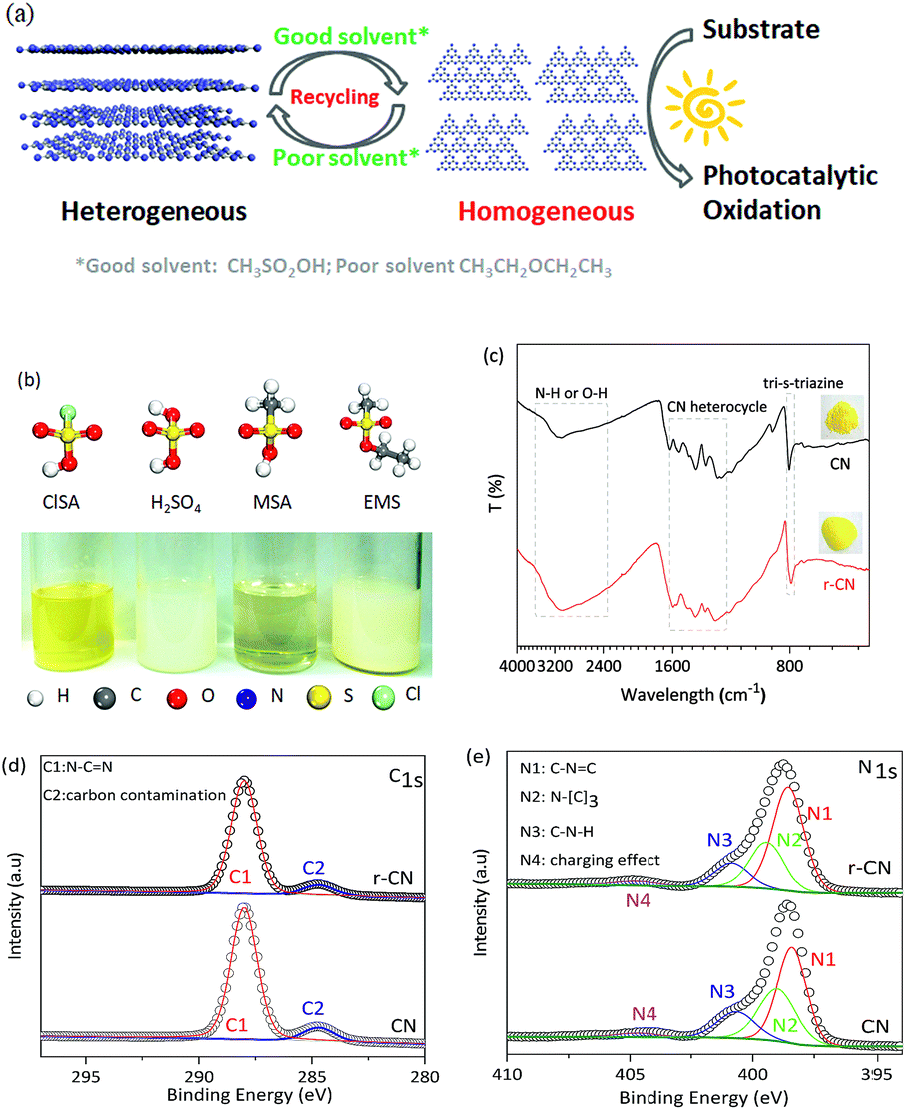 | ||
| Fig. 1 (a) Bridging homo/heterogeneous photocatalysis. (b) A photo of CN in sulfonic acid group-containing solvents (50 mg mL−1) at room temperature, and a ball-and-stick model of the solvents. Bulk CN was only completely dissolved in MSA. (c) FT-IR, (d) XPS C 1s and (e) N 1s spectra of bulk CN and that recovered from the MSA solution (r-CN). Inset: photos of CN and r-CN. | ||
The starting CN with interlinked heptazine-based units (Fig. S1–S3†) and a stacked lamellar texture (Fig. S4†) was prepared by a conventional thermal polymerization of dicyandiamide at 550 °C in air. The dissolution ability of CN in MSA and control solvents at room temperature is demonstrated in Fig. 1b. CN was completely dissolved in MSA, exhibiting a clear and yellow solution, and stirring would accelerate the dissolution speed (Fig. S5†). Different to CN nanosheets prepared by liquid-exfoliation,5c no evident Tyndall effect was observed for CN in MSA (Fig. S6a†), indicating CN macromolecules of minimal lateral dimensions rather than nanosheets in MSA. As controls, CN only partially dissolved in concentrated H2SO4, ClSA, and EMS under the same conditions. Therefore, the dissolution ability of CN in the solvents followed the order MSA, H2SO4, ClSA and EMS.
To evaluate the possible changes of the electronic structure of CN after dissolution, CN was recovered from the MSA solution by using poor solvents (e.g. acetonitrile). As shown in Fig. 1c inset, the recovered CN (r-CN) was still yellow, suggesting that CN did not disintegrate after dissolution. The UV-Vis spectrum of r-CN (Fig. S7†) showed a minor blue-shift relative to that of CN. Such a blue-shift was also observed in the photoluminescence spectrum of r-CN (Fig. S8†), presumably due to the protonation of CN by MSA.3a,7
The molecular structure of r-CN was further studied by Fourier transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1c, for both CN and r-CN, the typical stretching mode of the CN heterocycles (1200–1600 cm−1) and the breathing mode (810 cm−1) were mostly retained. Broad and intense bands at 2800–3500 cm−1 and some peak-shifts were also observed, which might result from protonation by MSA.3a,7 The C 1s XPS spectra of r-CN and CN showed two main peaks at 288.3 eV and 284.6 eV (Fig. 1d), which are attributed to N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and random adventitious carbon contamination, respectively.5a,b,8 Meanwhile, four N 1s XPS peaks ascribed to CN–C (398.2 eV, tri-s-triazine rings), N–(C)3 (399.4 eV, bridging N atoms), C–N–H (400.9 eV, bonded with H atoms) and the charge effect were observed for both CN and r-CN (Fig. 1e), further verifying that these two compounds had similar C–N bonding characteristics.3a Elemental analysis (Table S1†) quantified the very similar molar C/N ratios of CN (0.68) and r-CN (0.67). The BET surface areas and pore volume of CN and r-CN (Fig. S9†) were also not evidently changed. In addition, despite the fact that the precipitation recovery led to a lower crystallinity, the X-ray diffraction (XRD) pattern of r-CN still exclusively demonstrated a similar predominant (002) diffraction peak to CN, rather than peaks related to its intermediate precursors (Fig. S2†). All of these facts jointly demonstrated that the typical structure of CN was maintained after dissolution, rather than it being decomposed into smaller fragments.
N and random adventitious carbon contamination, respectively.5a,b,8 Meanwhile, four N 1s XPS peaks ascribed to CN–C (398.2 eV, tri-s-triazine rings), N–(C)3 (399.4 eV, bridging N atoms), C–N–H (400.9 eV, bonded with H atoms) and the charge effect were observed for both CN and r-CN (Fig. 1e), further verifying that these two compounds had similar C–N bonding characteristics.3a Elemental analysis (Table S1†) quantified the very similar molar C/N ratios of CN (0.68) and r-CN (0.67). The BET surface areas and pore volume of CN and r-CN (Fig. S9†) were also not evidently changed. In addition, despite the fact that the precipitation recovery led to a lower crystallinity, the X-ray diffraction (XRD) pattern of r-CN still exclusively demonstrated a similar predominant (002) diffraction peak to CN, rather than peaks related to its intermediate precursors (Fig. S2†). All of these facts jointly demonstrated that the typical structure of CN was maintained after dissolution, rather than it being decomposed into smaller fragments.
To gain insight into MSA dissolving CN, we carried out first-principles density functional theory (DFT) calculations to analyze the adsorption sites, and adsorption abilities using a polarizable continuum model. The hollow (H) position was found to be the most stable adsorption site relative to the top (T) and bridge (B) positions (Fig. 2a). Generally, a solvent molecule with a stronger adsorption ability could more competitively interact with CN and dissolve it.9 The calculated Eads values for MSA (−0.84 eV), H2SO4 (−0.73 eV), and ClSA (−0.67 eV) were all evidently higher than that for H2O (−0.27 eV), supporting the fact that the sulfonic acid group-containing solvents were more effective than H2O in dissolving CN, and MSA was the best one in this study (Fig. 2b–d). Besides, because of the lack of protonation, the adsorption energy (−0.14 eV) and distance (2.38 Å) between the nearest H atom of EMS and the CN plane was apparently larger than that of the other three solvents, making EMS the poorest among them. Considering the contribution of entropy to the dissolution, the Gibbs energy was also calculated (see Computational methods in the ESI†), in agreement with the above results. This was also consistent with the recent computations for sulfonic acid group-containing surfactants in liquid-exfoliation of other layered materials.10
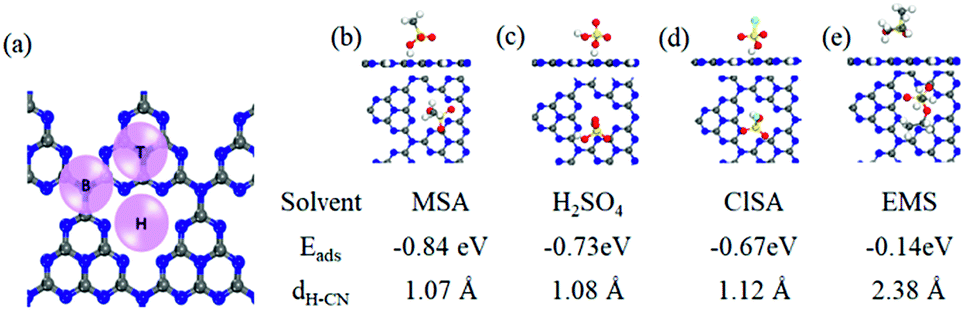 | ||
| Fig. 2 (a) Structural model of a single CN layer for calculating the adsorption energies in different solvents. The insets show the typical adsorption sites: top (T), hollow (H), and bridge (B). The most stable adsorption structures of MSA (b), H2SO4 (c), ClSA (d) and EMS (e) molecules on CN. Eads and dH–CN represent the adsorption energy and the distance between the nearest H atom and the CN plane, respectively. | ||
CN catalysts have been widely explored in heterogeneous conversion of organic molecules and harnessing solar energy to form chemical fuels; however, the activity was limited by low exposure of potential active sites. As a result, many attempts have been made, e.g. to prepare nanostructured CN with a higher density of exposed active sites.5,11 Driving a homogeneous catalytic reaction by CN at the molecular level is the ultimate goal. Taking advantage of the excellent dissolution of CN in MSA,12 the photocatalytic dehydrogenation of 3,3′,5,5′-tetramethylbenzidine (TMB), a typical substrate for studies on enzymatic or biomimetic dehydrogenation oxidation (Fig. S10†),13 was firstly investigated. TMB was originally colorless in MSA and turned reddish brown after photocatalytic reaction; thus, the reaction rate could be quantitatively determined by using UV-Vis spectroscopy and the Beer–Lambert law when the concentration of oxidized TMB (TMBox) was low.
The UV-Vis absorbance of the TMBox in MSA increased dramatically with time when CN was used as a homogeneous catalyst (CN-homo, CN dissolved in MSA) under irradiation (Fig. 3a circles and Fig. S11†). In contrast, only negligible color changes were observed without CN or without light irradiation (Fig. 3a, triangles). Control experiments also showed that MSA itself did not promote the catalytic activity (Fig. S12†). These facts jointly indicated that the oxidation of TMB was exclusively driven by CN-homo via a photocatalytic pathway. As a control, the same amount of CN was dispersed in an aqueous solution as a heterogeneous catalyst (CN-hetero) to drive the same reaction, but only very moderate activity was observed (Fig. 3a, squares). Although the blue-shift of CN-homo was not in favor of improving the photocatalytic activity due to the eliminated absorbance of light, Fig. 3b and S13† still show that the turnover frequency (TOF)14 ratio of the CN-homo system to the CN-hetero system increased up to 10-fold when the concentration of CN increased up to 0.5 mg mL−1, demonstrating the inherent advantage of the CN-homo in exposing active sites, especially at a high concentration, in driving a practical catalytic reaction. Notably, no activity was observed in the first several minutes for the CN-homo system, which may be attributed as an induction stage.15
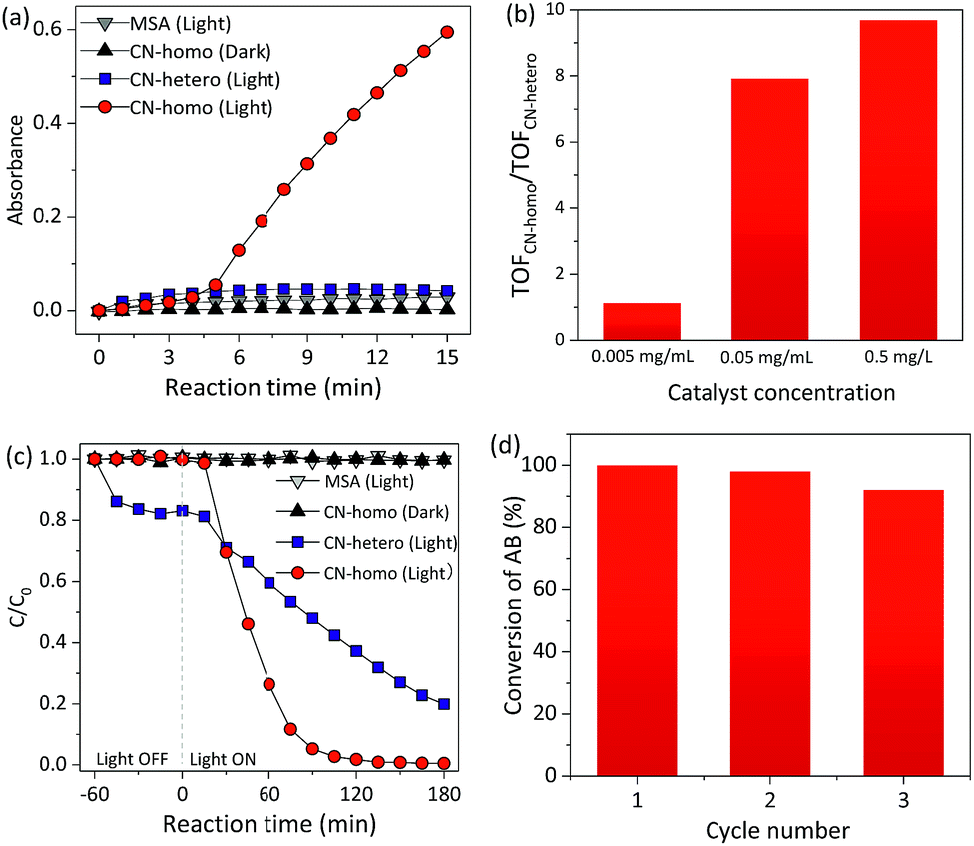 | ||
| Fig. 3 (a) Absorbance of TMBox at 456 nm as a function of time during photocatalytic dehydrogenation using a homogeneous CN catalyst (CN-homo, CN dissolved in MSA) and heterogeneous CN catalyst (CN-hetero, CN dispersed in H2O). Catalyst loading: 0.5 mg mL−1 (see optimization in Fig. S12†). (b) Ratio of the TOFs for different concentrations of CN-homo and CN-hetero catalysts. (c) C/C0 of AB concentration as a function of time during photocatalytic N-demethylation using CN-homo and CN-hetero. (d) Re-use catalytic efficiency of the CN-homo catalyst in the photocatalytic N-demethylation of AB. | ||
Recycling and reusability are challenging for homogeneous catalysts.16 Interestingly, this limitation did not happen for CN-homo, because of the unique dissolution/recovery ability of CN in MSA. The collection yield of CN in MSA using diethyl ether as a poor solvent was up to ca. 95%. To verify this assumption, in the second set of experiments, the CN-homo photocatalyst was used in the N-demethylation of Azure B (AB), which is an important chemical transformation in the pharmaceutical industry (Fig. S14†).17 In contrast to the darkening of TMB, the color of AB lightened after oxidation; thus, the Beer–Lambert law was valid for kinetic studies, especially when a high concentration of CN photocatalyst was used to eliminate the systematic loss of the catalyst during recycling.
Fig. 3c and S15† show the variation in the AB concentration with reaction time. When CN-homo was used, AB was almost completely oxidized (remnant < 5%, see Fig. S16 and more discussion in the ESI†) upon irradiation for 90 min, while for CN-hetero, more than 48% of unreacted AB remained, suggesting that the CN-homo photocatalyst had a much faster oxidation rate. Control experiments without CN and without light irradiation (Fig. 3c, triangles) resulted in negligible activity within the prescribed time, confirming that the oxidation of AB was exclusively driven by CN via a photocatalytic pathway. The first-order rate constant (Fig. S17†) of the reaction by CN-homo (0.041 min−1) was nearly six times that of the reaction driven by CN-hetero (0.007 min−1). CN-homo was further explored in recycling experiments, in which the catalyst was separated by using diethyl ether as a poor solvent after the reaction and recycled in the next experiment. As shown in Fig. 3d, CN-homo maintained most of the original high photocatalytic activity for at least three cycles. The stability of the catalyst was also reflected in the similar XRD patterns of the CN after the photocatalytic reaction (Fig. S18†). A minor deterioration in the catalytic activity was observed, presumably due to mechanical loss of the catalyst during the recycling process. Therefore, the recycling experiment demonstrated the unique features of the proposed CN photocatalytic system, which combines the advantages of both homogeneous catalysts that often exhibit superior activity, and heterogeneous catalysts that can be easily recycled and reused without evident activity loss. The catalytic activities of the dehydrogenation of TMB and the N-demethylation of AB were also compared using other (photo)catalysts (Tables S2 and S3†) and showed very competitive activities. Besides, the dissolved CN in MSA was also explored as a photocatalyst for water splitting, but no significant H2 evolution activity was observed. This may be caused by the fact that the current photocatalytic reaction conditions, e.g. the special nature of the MSA solvent and the protonated surface of CN, were not favorable for H2 evolution. To overcome this issue, more studies are needed in the future.
Conclusions
In summary, CN was dissolved in environmentally friendly MSA at room temperature. The DFT results verified that the sulfonic acid group significantly reduced the adsorption energies. Taking the dehydrogenation of TMB and the N-demethylation of AB as examples, the homogeneous CN photocatalyst exhibited a dramatic enhancement in activity by a factor of up to 10 relative to the heterogeneous one due to the increase of potential accessible catalytic sites. Moreover, the reversible dissolution of CN using good/poor solvents allowed the homogeneous CN to be effectively recycled and reused, which united the advantages of both homo/heterogeneous catalysts. This work may extend the application of CN as a homogeneous (photo)catalyst and highlights the utility of polymeric catalysts with intrinsic catalytic properties in bridging the gap between homo/heterogeneous catalysis. Besides, the detailed structure information of dissolved CN in MSA is very interesting, and requires further experiments and molecular dynamics simulations in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21775018, 21675022), the Natural Science Foundation of Jiangsu Province (BK20160028, BK20170084), the Open Funds of the State Key Laboratory of Electroanalytical Chemistry (SKLEAC201703) and the Fundamental Research Funds for the Central Universities. We thank Prof. Shuxin Ouyang (Tianjin University, China) for helpful discussion.Notes and references
- (a) D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS PubMed; (b) A. Corma and H. Garcia, Top. Catal., 2008, 48, 8 CrossRef CAS.
- (a) X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed; (b) Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed; (c) S. W. Cao, J. X. Low, J. G. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed; (d) R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216 CrossRef CAS PubMed; (e) Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298 RSC.
- (a) Z. X. Zhou, J. H. Wang, J. C. Yu, Y. F. Shen, Y. Li, A. R. Liu, S. Q. Liu and Y. J. Zhang, J. Am. Chem. Soc., 2015, 137, 2179 CrossRef CAS PubMed; (b) J. S. Xu and M. Antonietti, J. Am. Chem. Soc., 2017, 139, 6026 CrossRef CAS PubMed.
- T. S. Miller, T. M. Suter, A. M. Telford, L. Picco, O. D. Payton, F. Russell-Pavier, P. L. Cullen, A. Sella, M. S. P. Shaffer, J. Nelson, V. Tileli, P. F. McMillan and C. A. Howard, Nano Lett., 2017, 17, 5891 CrossRef CAS PubMed.
- (a) P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS; (b) S. B. Yang, Y. J. Gong, J. S. Zhang, L. Zhan, L. L. Ma, Z. Y. Fang, R. Vajtai, X. C. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed; (c) X. D. Zhang, X. Xie, H. Wang, J. J. Zhang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS PubMed; (d) K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730 CrossRef CAS PubMed.
- N. Behabtu, J. R. Lomeda, M. J. Green, A. L. Higginbotham, A. Sinitskii, D. V. Kosynkin, D. Tsentalovich, A. N. Parra-Vasquez, J. Schmidt, E. Kesselman, Y. Cohen, Y. Talmon, J. M. Tour and M. Pasquali, Nat. Nanotechnol., 2010, 5, 406 CrossRef CAS PubMed.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50 CrossRef CAS PubMed.
- (a) Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135 CrossRef CAS; (b) W. Wu, J. Zhang, W. Fan, Z. Li, L. Wang, X. Li, Y. Wang, R. Wang, J. Zheng, M. Wu and H. Zeng, ACS Catal., 2016, 6, 3365 CrossRef CAS.
- B. Zhu, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2018, 224, 983 CrossRef CAS.
- M. Monajjemi, J. Mol. Liq., 2017, 230, 461 CrossRef CAS.
- (a) Q. Han, B. Wang, J. Gao and L. Qu, Angew. Chem., Int. Ed., 2016, 55, 10849 CrossRef CAS PubMed; (b) K. S. Lakhi, D. H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J. H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72 RSC.
- M. D. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127 RSC.
- X. J. Cui, Y. H. Li, S. Bachmann, M. Scalone, A. E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467 CrossRef CAS PubMed.
- B. An, L. Zeng, M. Jia, Z. Li, Z. Lin, Y. Song, Y. Zhou, J. Cheng, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 17747 CrossRef CAS PubMed.
- D. E. Bergbreiter, J. H. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530 CrossRef CAS PubMed.
- G. B. Kok, C. C. Pye, R. D. Singer and P. J. Scammells, J. Org. Chem., 2010, 75, 4806 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03855d |
| ‡ C. Huang and J. Wen contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2018 |