
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotomechanochromic vs. mechanochromic fluorescence of a unichromophoric bimodal molecular solid: multicolour fluorescence patterning†
Li-Yun
Hsu
,
Subhendu
Maity
 ,
Yuki
Matsunaga
,
Ying-Feng
Hsu
,
Yi-Hung
Liu
,
Shie-Ming
Peng
,
Teruo
Shinmyozu
and
Jye-Shane
Yang
*
,
Yuki
Matsunaga
,
Ying-Feng
Hsu
,
Yi-Hung
Liu
,
Shie-Ming
Peng
,
Teruo
Shinmyozu
and
Jye-Shane
Yang
*
Department of Chemistry, National Taiwan University, Taipei, Taiwan 10617. E-mail: jsyang@ntu.edu.tw
First published on 2nd October 2018
Abstract
Mechanofluorochromism (MFC) of molecular solids generally results from the variation of intermolecular interactions induced by external mechanical forces. However, the use of internal photomechanical forces to perturb intermolecular interactions for multicolour fluorescence responses has yet to be demonstrated. Herein we report a unichromophoric anthracene–pentiptycene derivative (1) that displays both MFC and photomechanofluorochromism (PMFC), which lead to various fluorescence colours including red-green-blue (RGB) and near-pure white-light emission. Compound 1 crystallizes in two polymorphs, the yellow (Y) and green (G) emissive forms, in which the pairwise stacked anthracene groups undergo [4 + 4] photodimerization to form the UV (black) emissive photodimer 2 and meanwhile exert photomechanical stresses on the neighbouring molecules. While the photomechanical stresses cause an excimer-to-monomer switching that results in a blue fluorescent state for the Y form, a red-emissive “super dimer” is photomechanically produced for the G form. The recovery of the Y form demands heating, but the G form could be restored by selective photoexcitation of the super dimer. X-ray crystal structures of the Y and G forms and the photodimer 2 generated through single-crystal-to-single-crystal transformation provide a clue to the origins of polymorph-dependent PMFC. The corresponding MFC and mechano-activated vapofluorochromism (VFC) of 1 also shed light on the structure–property relationship. The ability to spatially and temporally control the fluorochromicity of 1 is demonstrated by a series of multicolour fluorescence patterning of “angelfishes”.
Introduction
The coupling of a reversible molecular photochemical reaction with a detectable property is attractive from the points of view of both basic and applied science. For example, the photoinduced charge-transfer reactions in organic photovoltaics lead to electrical power,1 the photochromic dyes in the lens of smart sunglasses change colour in response to ambient light,2 and the chemical photoswitches implanted in the retinal photoreceptors of blind animals restore the visual function.3 Regarding that photoluminescence has the advantages of high sensitivity (zero background signal) and multiple parameters (wavelength, intensity, lifetime, and phase) for detection, the development of photo-responsive luminescent molecules might further advance the progress of optical molecular devices.4,5 However, using reversible photoreactions to switch the colour of photoluminescence is inherently a challenging task, as photoreactions and photoluminescence compete with one another in the decay of an electronically excited state, and thus a critical balance between the radiative and nonradiative decay rates is required to obtain photoswitchable photoluminescence. This is manifested by typical photochromic systems such as azobenzenes,6 diarylethenes,5,7 and spiropyrans,8 in which photoisomerization of one or both of the interconverting isomers is dominant and essentially nonemissive. Although multichromophoric systems containing photoswitchable and photoluminescent chromophores might be a solution, a deliberate structural design is needed to produce photoswitchable two-colour luminescence,9 not to mention multicolour luminescence.10 There are additional obstacles to overcome for solid-state systems, including the potential problem of aggregation-caused quenching of photoluminescence11 and the geometrical constraints of photoreactions caused by the surrounding medium.12 Nevertheless, we report herein the first example of photoreaction-induced multiple fluorescence colour change of a unichromophoric molecule (1) in the solid state. In particular, the mechanism of the observed photofluorochromism (PFC) is not a switching of the emissive species from reactant 1 to photodimerized product 2 (Fig. 1a) but that of the emissive states among a monomer, an excimer, and a “super dimer” of 1 due to local photomechanical stresses exerted by the [4 + 4] photodimerization of the anthracene moiety (Fig. 1b). We call it “photomechanofluorochromism” (PMFC).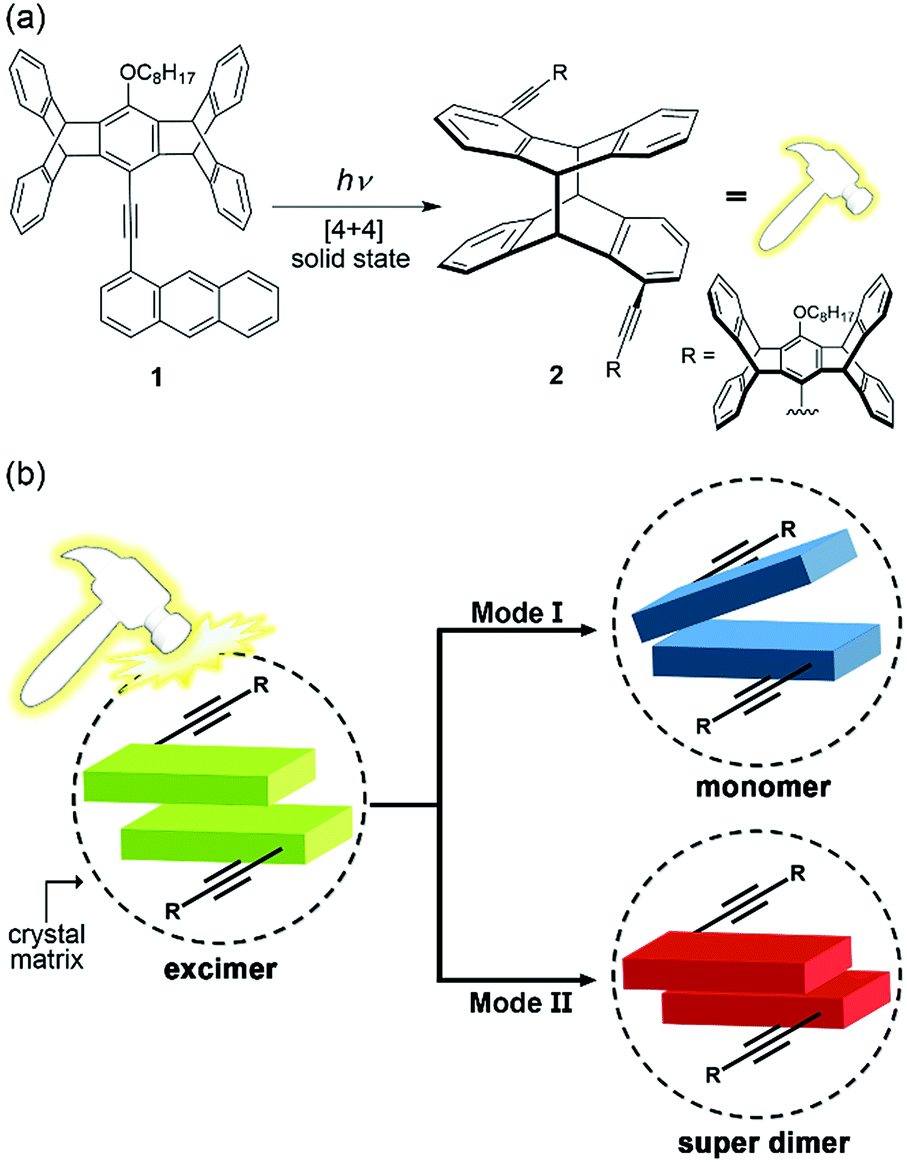 | ||
| Fig. 1 (a) Molecular structures of 1 and its photodimer 2 formed via solid-state [4 + 4] photodimerization, which exerts photomechanical stresses (symbolized by a “photohammer”) on neighbouring molecules, and (b) schematic representations of the two different modes of photomechanofluorochromism (PMFC) for 1 in crystal form. Mode (I): the blue-shifted fluorescence due to excimer-to-monomer switching. Mode (II): the red-shifted fluorescence due to excimer-to-super dimer switching. | ||
Compound 1 was originally designed to investigate the scope of multicolour mechanochromic14,15 and vapochromic16 photoluminescence in line with our recently reported examples,17,18 in which the nonplanar cogwheel-shaped pentiptycene group19 facilitates the manipulation of intermolecular π–π interactions and thus the luminescence colours via external mechanical stresses and/or vapour deposition. To our surprise, 1 displays not only intriguing mechanofluorochromism (MFC) and vapofluorochromism (VFC) that lead to multicolour fluorescence including white-light emission but also unprecedented bimodal multicolour PMFC that originated from the [4 + 4] photodimerization of the anthracene moiety (Fig. 1a). The anthracene-based [4 + 4] photodimerization is one of the typical photochemical reactions used for the design of organic photomechanical materials, which change shape or position on a macroscopic scale by the molecule-scale photoreactions.20,21 However, little attention has been paid to the photomechanical effects on the fluorescence properties of the anthracene derivatives.21 The unique PMFC behaviour of 1 is elucidated using X-ray crystal structures and illustrated with a series of multicolour fluorescence patterning.
Results and discussion
Substrate preparation
The synthesis of 1 was carried out via a Sonogashira reaction between the commercially available 1-bromoanthracene and the known ethynylpentiptycene derivative22 (Scheme S1†). Detailed synthetic procedures and structural characterization data are provided as ESI.†Compound 1 crystallizes in two distinct fluorescent polymorphs: that is, yellow (Y) and green (G) fluorescent forms. The Y and G forms, either powder or crystals, are generally obtained together as a mixture (Fig. S1†). Fortunately, we were able to find optimum conditions for selective preparation of either form. For example, reprecipitation of the concentrated DCM solution (13 mM) in MeOH above 50 °C affords the powder of Y form, but the powder of G form is obtained when the temperature of MeOH is at −70 °C. The details of selective preparation of crystalline and powdered substrates are provided in the Experimental section. The G and Y forms are stable at ambient temperature, and thermal G-to-Y phase transition could not occur until the temperature is raised to 280 °C (Fig. S2†), a temperature close to the onset of melting (301 °C) of the G form as indicated by the differential scanning calorimetry (DSC) measurements (Fig. S3†).
Fig. 2 shows the optical microscopy and scanning electron microscopy (SEM) images of the crystals and powders of the Y and G forms, respectively. For the Y form, the microcrystals are in a flat parallelogram shape, but the microstructures of the powder resemble fluffy balls. While the microcrystals of the G form also show a parallelogram shape, the powder shows a granular morphology. The distinct powder morphology of the two forms denotes different kinetics of nucleation of 1 in the two types of molecular packing. In the following spectroscopic studies and fluorochromic tests, the solid-state Y and G form substrates refer to the corresponding powder unless otherwise mentioned.
 | ||
| Fig. 2 Images of the crystals (left) and powder (right) of the (a) Y and (b) G forms of 1 obtained using an optical microscope (scale bar = 400 μm; λex = 340–390 nm) and SEM (scale bar = 20 μm), respectively. | ||
MFC and VFC
The fluorescence of 1 differs dramatically in the solid state (Y or G form) vs. in dilute dichloromethane (DCM) solution. The fluorescence spectra of both Y and G forms are structureless with the fluorescence maximum (λf) at 591 and 538 nm, respectively, but the fluorescence spectrum obtained in DCM shows vibrational structures with a 0–0 transition at 415 nm (Fig. 3). The significant red shifts of 123 nm (5509 cm−1) and 176 nm (7176 cm−1) on going from the DCM solution to the G and Y forms indicate the presence of strong intermolecular excimeric interactions in the solid state. The red-shifted fluorescence excitation spectra in the solid vs. solution phase (422 vs. 400 nm) further reveal that the intermolecular interactions are already present in the ground state (Fig. S4†). The ground-state interactions (i.e., a physical dimer) might account for the shoulder band (∼460 nm) of the fluorescence spectra, and the yellow and green fluorescence should be mainly from the excimers because of the large Stokes shifts and the structureless emission profiles. The fluorescence quantum yield (Φf) is 0.17 for the Y form and 0.19 for the G form, which are significant for a solid-state excimer but lower than that (0.66) for a monomer emission in DCM solution (Table S1†).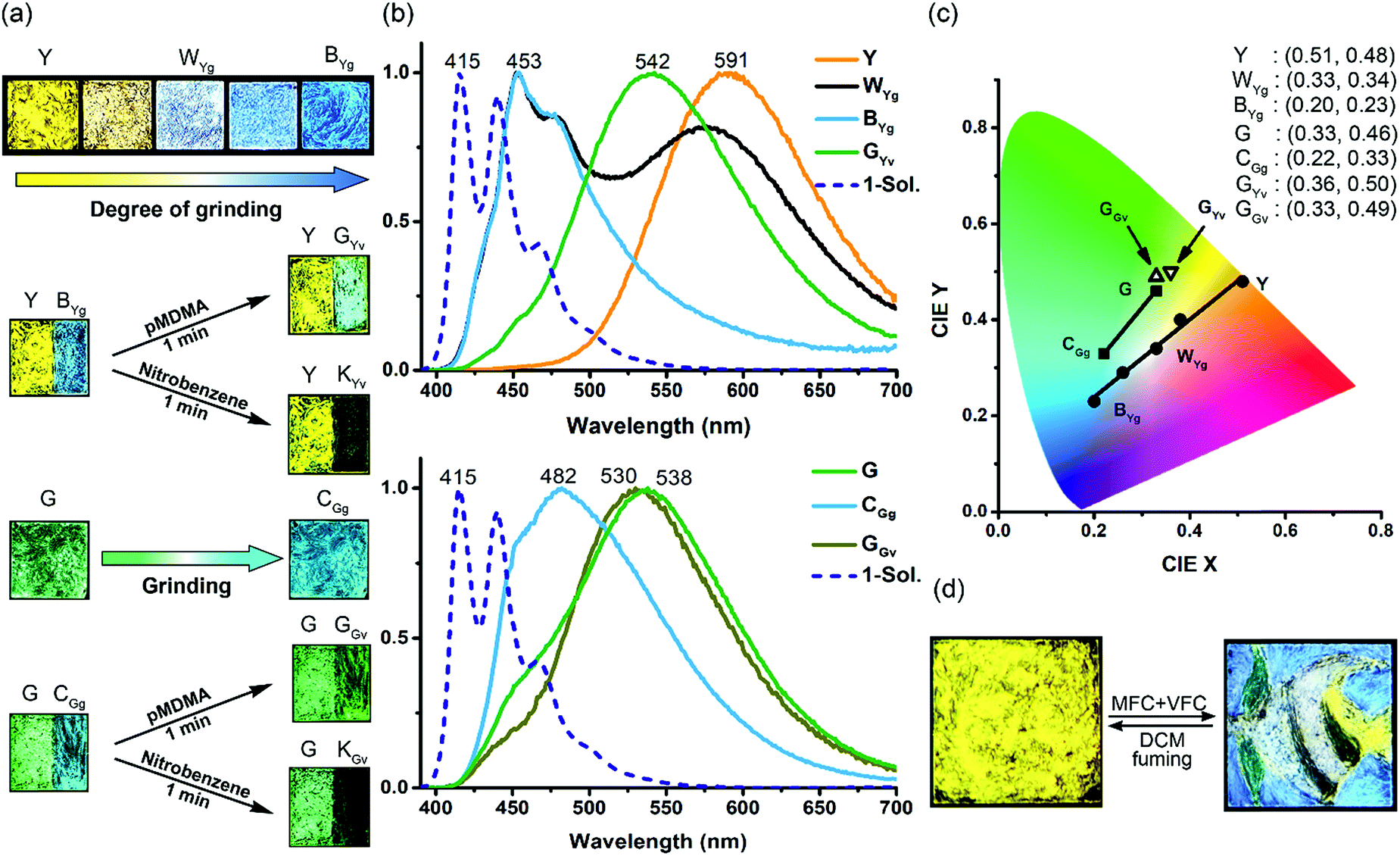 | ||
| Fig. 3 MFC and VFC of the Y and G powder films of 1: (a) photographs of the fluorescence colour (λex = 365 nm); (b) the corresponding normalized fluorescence spectra (λex = 380 nm); (c) the corresponding coordinates and positions on the CIE chromaticity diagram; (d) multicolour fluorescence patterning of “an angelfish and an aquatic plant” on a powder film (1.5 cm × 1.5 cm) of the Y form. See Fig. S8† for detailed drawing and patterning procedures and intermediate stages. The fluorescence spectrum of 1 in DCM (blue dashed line) is included in (b) for comparison. | ||
Interestingly, the solid-state excimeric interactions in the Y and G forms could be readily disrupted by gentle manual grinding with a spatula, as indicated by the grinding-induced blue (BYg form, λf = 453 nm, Φf = 0.11) and cyan (CGg form, λf = 482 nm, Φf = 0.13) emission from the Y and G form, respectively (Fig. 3a and b). Note that we use a three-letter notation XYz to describe each form obtained from the treatment of the Y or G form with a stimulus in this work: the capital letter X represents the colour of fluorescence, and the two subscript letters Y (in uppercase) and z (in lowercase) represent the original sample polymorph (Y or G) and the nature of stimulus (g for grinding, v for vapour fuming, and i for irradiation), respectively. Therefore, the notation BYg refers to the blue form obtained from the yellow polymorph via grinding. Powder X-ray diffraction (PXRD) analysis shows that the Y and G forms are crystalline but the BYg and CGg forms are amorphous (Fig. S5†). The longer fluorescence lifetimes for the Y (47.0 ns) and G (64.4 ns) vs. the BYg (2.8 ns) and CGg (8.5 ns) forms are consistent with the excimeric vs. monomeric nature of the emission (Table S1†). To our knowledge, the Y-to-BYg switching represents the largest monomer–excimer-based MFC shift of 138 nm (5155 cm−1) to date for metal-free organic compounds.23 More interestingly, the colours of Y and BYg forms are complementary such that the grinding processes result in a linear colour transformation on the Commission Internationale d'Eclairage (CIE) chromaticity diagram (Fig. 3c) and produce near-pure white light (WYg form, Φf = 0.12) with CIE coordinates of (0.33, 0.34). The use of MFC to create white-light emission from single small organic molecules has recently been achieved with inherently dual-emissive compounds24 but not with common single-emissive systems. Therefore, the Y form of 1 represents the first example of MFC-based white-light emission from a single chromophoric and single emissive molecular solid, which requires not only the formation of complementary colours, namely, blue-yellow, cyan-red, or green-magenta, for the MFC but also a fraction-controllable phase transformation for colour mixing.
Furthermore, the grinding-generated BYg and CGg forms are responsive to the vapours of p-methyl-N,N-dimethylaniline (pMDMA) and nitrobenzene (NB) and form green emissive (GYv and GGv forms with λf = 542 and 530 nm and Φf = 0.17 and 0.15, respectively) and dark (KYv and KGv forms due to fluorescence quenching) states, respectively (Fig. 3a–c). The GYv and GGv forms could be attributed to the formation of exciplexes between the electron-donating pMDMA and the electron-accepting 1, and the different degrees of VFC shifts for the BYgvs. CGg form (3625 vs. 1879 cm−1) reveal the dependence of the solid–vapour host–guest interactions on the molecular packing mode (vide infra). Note that the exciplex emission also occurs in toluene solutions, and the presence of solvation and feasible structural relaxation for the exciplexes lead to a longer emission wavelength (λf = 594 nm, Fig. S6†). For the KYv and KGv forms, the occurrence of electron transfer from the excited monomers of 1 to the electron-deficient NB could account for the fluorescence quenching.25 In contrast, the pristine Y and G forms are inert to the vapours of pMDMA and NB, attributable to the lower-lying excimeric states of these two forms. Such a mechano-activated VFC for 1 is opposite to the mechano-deactivated VFC previously observed for a centrosymmetric pentiptycene–anthracene–pentiptycene system.17 Evidently, the VFC properties of pentiptycene derivatives are very sensitive to the crystal packing (e.g., Y vs. G form) and excited-state energetics (e.g., Y vs. BYg form) of the systems, which in turn depends on the number and position of the pentiptycene groups.
Both the MFC and VFC processes are reversible (Fig. S7†), and the original Y and G forms could be restored by fuming the ground and/or pMDMA-/NB-fumed samples with DCM. Since DCM fuming is inherently a process of “recrystallization” of the mechanically perturbed molecules on the film, the recovery of the pristine fluorescence colours of the films without messing up the Y and G forms reveals a memory effect. This could be rationalized by the presence of seed crystals of the pristine form on the film even after extensive grinding.
With a phase-selective vapochromic response of the ground vs. unground substrates, one could readily perform multicolour fluorescence patterning via the combined MFC and VFC behaviour of 1 without the need for using masks or templates. This is illustrated with the five-colour patterning of “an angelfish and an aquatic plant” on a single film of the Y form (Fig. 3d). The drawing/patterning started with the creation of the black stripes (KYv form) and ended with the formation of the blue water background (BYg form). The intact Y form corresponds to the tail of the fish, the WYg form to the white stripes, and the GYv form to the aquatic plant. Details of the drawing/patterning procedures and photographs of the intermediate stages are shown in Fig. S8.† Although several multistimuli, multiresponsive, and multicolour luminescent molecular solids have been reported,11,15,17,26,27 those that allow one to perform mask-/template-free fluorescence patterning with three or more colours are rare,17,27 because it requires selective or orthogonal responses to external stimuli such that the later stimuli would not overwrite the colour of the earlier created patterns. The five-colour pattern shown in Fig. 3d extends the scope of our previous illustration17 of collaborative MFC and VFC in terms of the colours (i.e., green-yellow-blue-white-black vs. green-yellow-orange-red-black) and the phase-selectivity of VFC (i.e., mechanical activation vs. deactivation). These features could be applied for sequential and orthogonal sensing of stresses and chemical vapours.
[4 + 4] photodimerization
The X-ray single-crystal structures of the Y and G forms have been determined, which not only account for the polymorph-dependent fluorescence colour but also guide us toward the discovery of PMFC. As shown in Fig. 4a and b, the anthracene groups in both the Y and G forms adopt antiparallel pairwise stacking. However, the interplanar distance is shorter (3.40 vs. 3.69 Å) and the overlap ratio is larger (66 vs. 52%) for the Y vs. G form. This is consistent with the expectation of a stronger electronic coupling and thus a lower energy of the excimer state for the Y form. We also notice that a centre-to-centre distance of 3.63 Å for the anthracene pairs in the Y form conforms to an ideal topochemical geometry for the [4 + 4] anthracene photodimerization.28,29 Indeed, the [4 + 4] photodimerization occurred when a single crystal of the Y form was subjected to UV irradiation (340–390 nm). The formation of photodimer 2 does not change the monoclinic crystal system and the P21/c space group, and the dimensions of the unit cell (cif in ESI†) and the PXRD patterns (Fig. S9†) resemble those of the Y form, leading to a single-crystal-to-single-crystal (SCSC) transformation (Fig. 4c). The length of the newly formed C–C bond is 1.65 Å, which is longer than a regular C(sp3)–C(sp3) bond length of 1.54 Å but comparable to those of other anthracene photodimers.29,30 For the G form, the centre-to-centre distance of 4.55 Å for the anthracene pairs is beyond the 4.20 Å threshold for effective photodimerization.28,29 Nevertheless, the photochemical [4 + 4] cycloaddition reaction that leads to photodimer 2 also took place, albeit in lower yield, for the G form (vide infra). The overlapping crystal structures of the Y and G forms of 1 and photodimer 2 shown in Fig. 4d reveal that the photodimerization requires a larger translational movement for molecules in the G vs. Y form. This might account for the lower photoreactivity of the G form and the poorer stability of the resulting photodimer 2 in the G form (vide infra).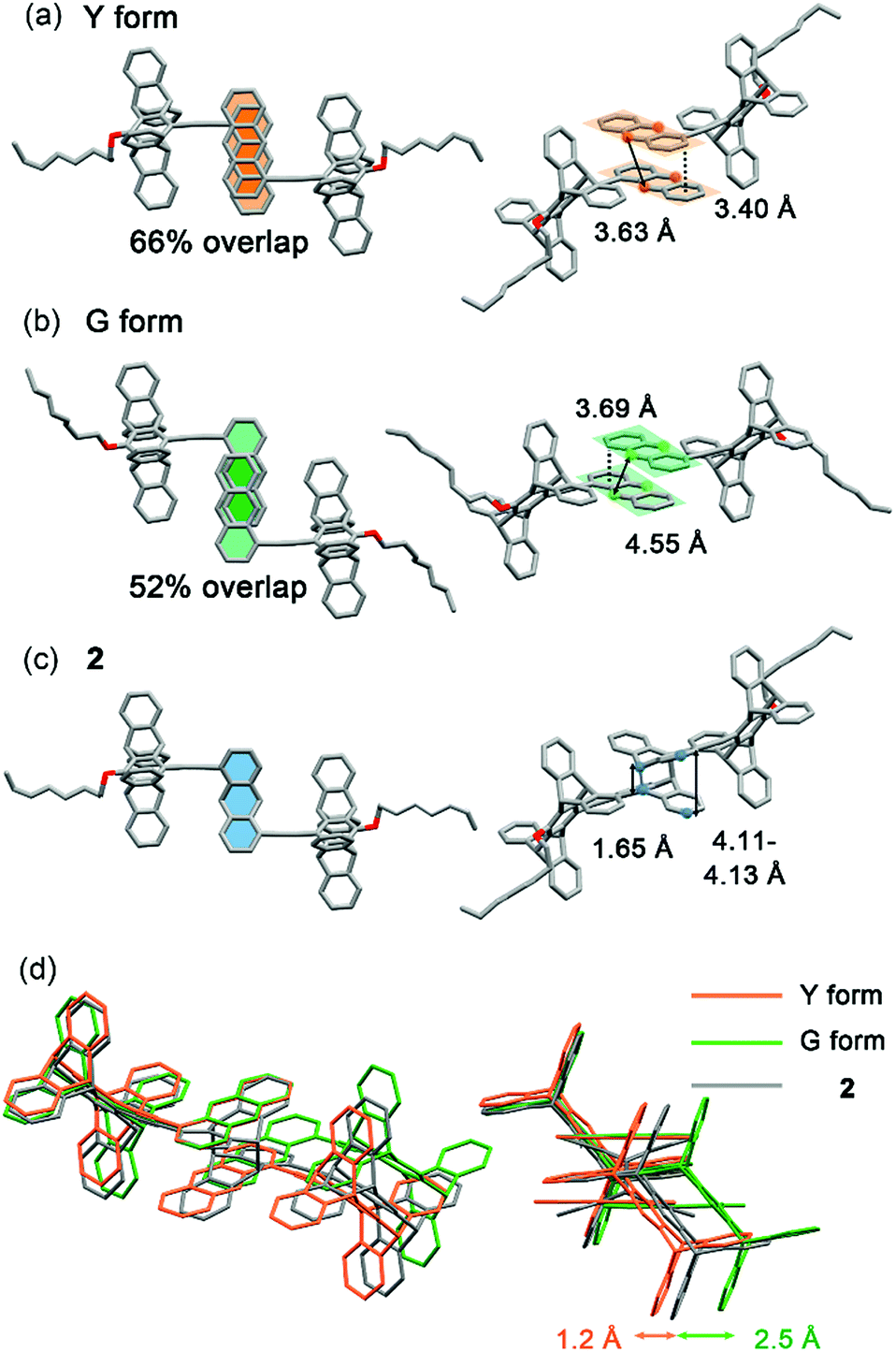 | ||
| Fig. 4 X-ray crystal structures of (a) 1 (Y form), (b) 1 (G form), and (c) 2 in two different views of a molecular pair and (d) superimposed images of the three structures (the octyloxy groups are omitted) for comparison. | ||
PMFC of the Y form
Intriguing and unexpected fluorescence colour changes took place along with the photochemical [4 + 4] cycloaddition reactions for the Y form. For a drop-cast film of the Y form, the fluorescence colour first turned white (WYi form, Φf = 0.08) and then blue (BYi form, λf = 439 nm, Φf = 0.06) upon irradiation, resembling the MFC phenomenon but with an even larger spectral shift of 152 nm (5859 cm−1) (Fig. 5a). Interestingly, the WYi form has the same CIE coordinates of (0.33, 0.34) as the WYg form (Fig. 5b). Besides the powder films, the crystals of the Y form also show the same sequential photoinduced Y-to-WYi-to-BYi fluorescence colour changes. Because the fluorescence of pure photodimer 2 is “black”, located in the UV region (λf = 329 nm and Φf = 0.93 in DCM; λf = 341 nm and Φf = 0.03 in solid state), the blue emission of BYi is not from 2; instead, it is from the monomer of 1, as the blue emission bands of BYi coincide with the monomer emission of 1 in DCM (Fig. 5c and S10†). Indeed, the photodimerization reaction was incomplete in the BYi form, and the percentage of photochemical conversion is 87% according to the analyses by UV-vis absorption spectroscopy. For comparison, the percentage of photodimerization is 31% in the WYi form. An incomplete photodimerization explains not only the blue emission of the BYi form but also the decreased crystallographic quality (only 30% of the reflections observed) of 2 obtained from SCSC transformation of the Y form.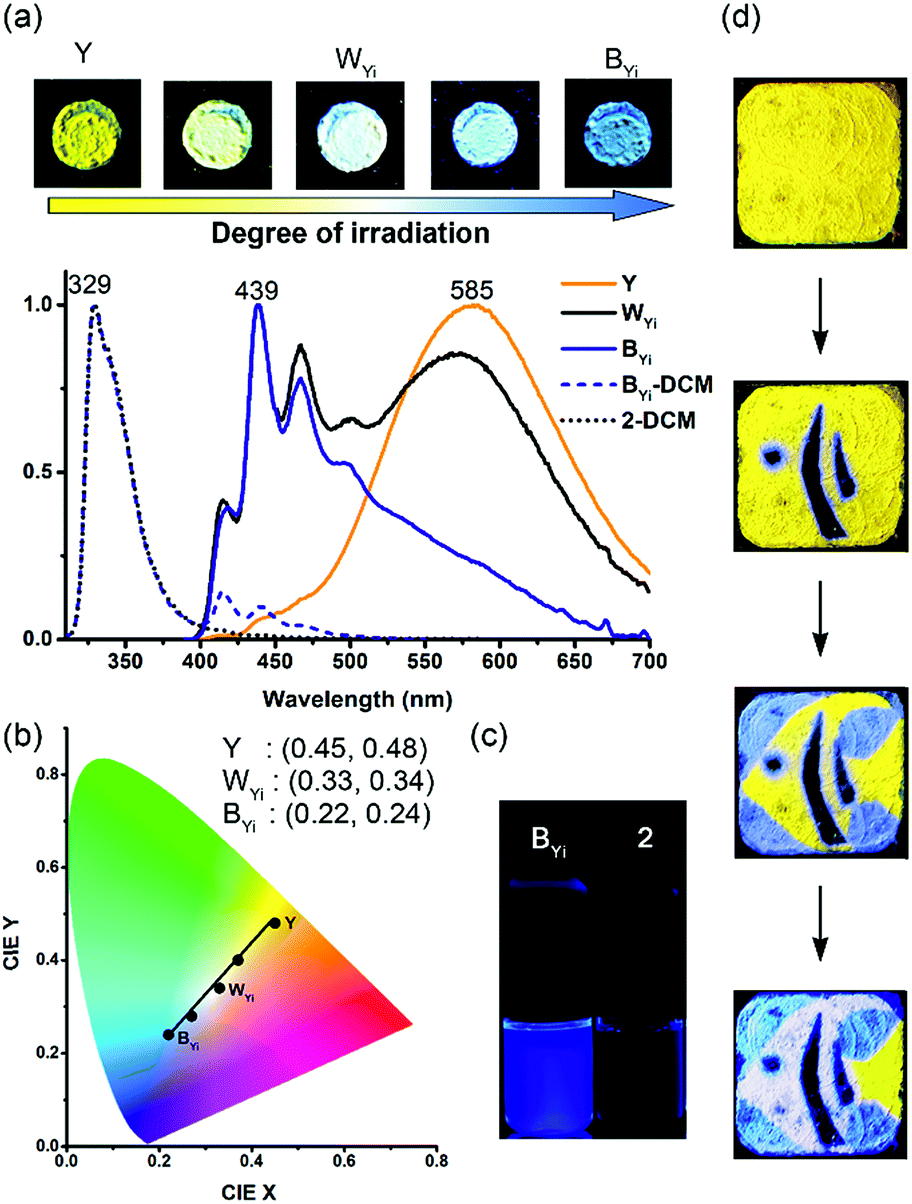 | ||
| Fig. 5 Photochromic fluorescence of the Y form of 1 on drop-cast films: (a) photographs of the fluorescence colour (λex = 365 nm) and the corresponding normalized fluorescence spectra (solid lines, λex = 380 nm; dashed lines, λex = 300 nm) obtained with different degrees of irradiation at 340–390 nm; (b) the corresponding coordinates and positions on the CIE chromaticity diagram; (c) photographs of BYi form and 2 dissolved in DCM (λex = 365 nm); (d) spatial and temporal control of irradiation (λex = 340–390 nm) to perform multicolour fluorescence patterning (1.0 cm × 1.0 cm) of an angelfish with the use of photomasks. The fluorescence spectra of the BYi form and 2 dissolved in DCM are also shown in (a) for comparison. | ||
The switching from excimeric yellow emission to monomeric blue emission of 1 along with the photodimerization reaction indicates the presence of a photomechanical effect. More specifically, when a molecular pair of 1 undergoes photodimerization, it simultaneously exerts mechanical stresses on its unreacted neighbours in a way that the pairwise anthracene stacking mode of excimeric interactions was interrupted to form monomers with blue emission. As the photomechanical stresses occur at the molecular level, the impact is localized and limited to the neighbouring molecules. Therefore, the degree of photomechanical excimer-to-monomer switching could be spatially or temporally controlled, as demonstrated by the formation of the WYi form, an intensity-balanced mixture of the unperturbed excimeric yellow emission and the perturbed monomeric blue emission in the film. While the WYi and BYi forms are stable in the dark at ambient temperature, we found that a near quantitative Y-to-2 conversion that led to a black (KYi) state is achievable upon prolonged irradiation at 419 nm.31 Evidently, the photomechanical excimer-to-monomer switching is a reversible process under the irradiation conditions such that the perturbed monomers are paired again through excited-state structural relaxation and eventually dimerized. A schematic representation of the photomechanical excimer-to-monomer switching and of the subsequent photoinduced recovery of π-stacking followed by photodimerization is depicted in Fig. 6a.
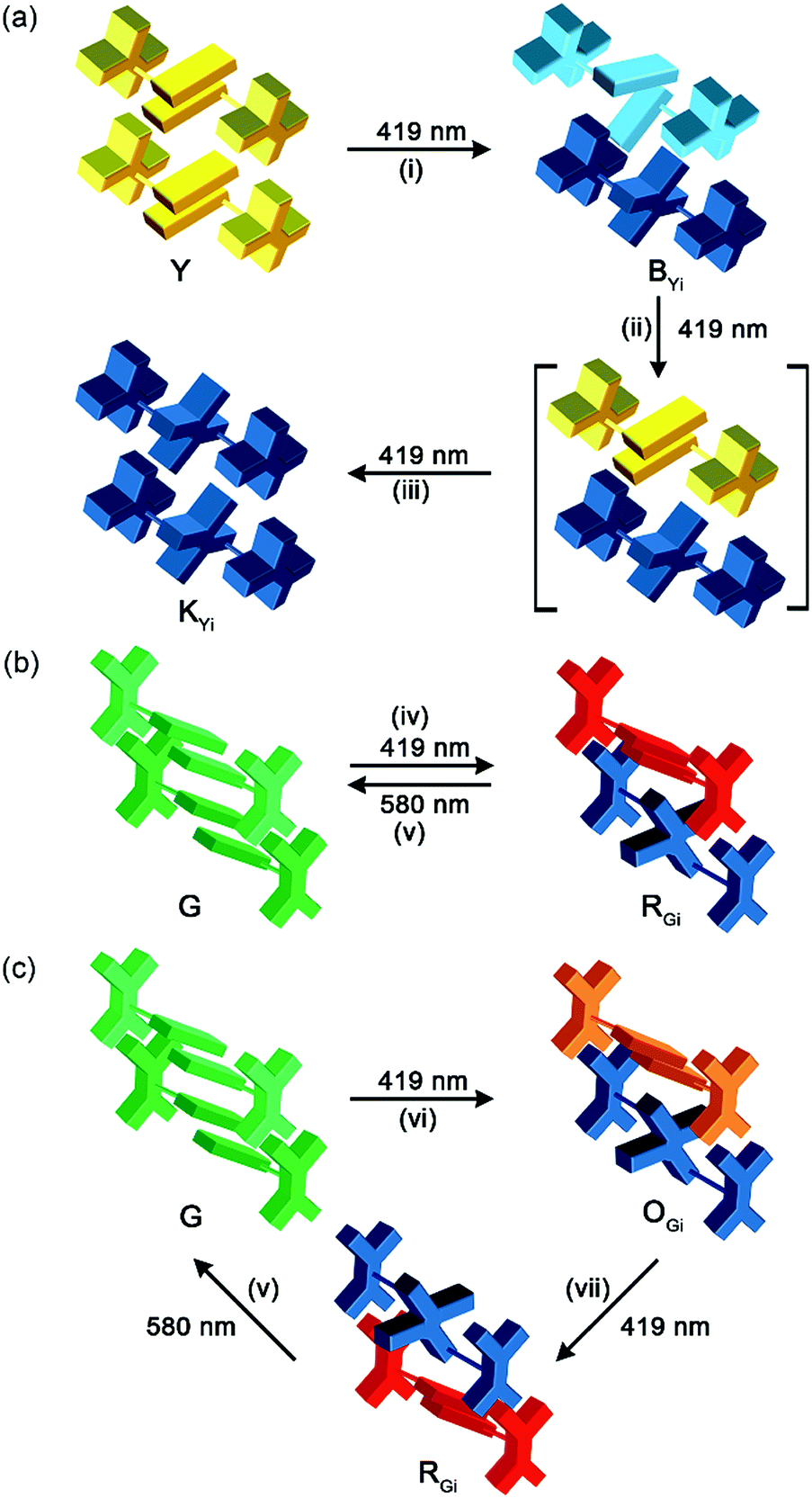 | ||
| Fig. 6 Schematic representations of the mechanistic concepts of photomechanofluorochromism of the (a) Y and (b and c) G forms of 1: (i) photodimerization-induced excimer-to-monomer switching; (ii) recovery of the excimer via structural relaxation of the monomers in the excited state; (iii) photodimerization of the recovered excimers; (iv) photodimerization-induced super dimer formation via compression; (v) structural relaxation of the super dimer in the excited state and the accompanied cycloreversion of the photodimer; (vi) photodimerization-induced formation of orange-emissive super dimers; (vii) photodimerization of the super dimer that triggers a breakdown of the photodimer and formation of the red-emissive super dimer. | ||
The PMFC behaviour of the Y form provides a unique opportunity for multicolour fluorescence patterning via irradiation. For example, a four-colour pattern of an angelfish could be created on a drop-cast film of the Y form using 419 nm light and paper-made photomasks (Fig. 5d). The sequence of patterning depends on the irradiation time, and the one that needed a longer irradiation time was created earlier: namely, the dark (KYi) stripes were first created followed by the blue (BYi) water and then the white (WYi) stripes, and no irradiation was applied on the yellow (Y) tail. Compared with the MFC and VFC-based pattern that shows visible contours of the objects under room light (Fig. S8†), the PMFC-based pattern could not be seen under room light (Fig. S11†), demonstrating the difference in the contact vs. non-contact patterning mode. The noncontact patterning mode feature of the PMFC might make the Y form a useful anti-counterfeiting dye. In addition, an independent creation of three common fluorescence colours (i.e., blue, black, and white) from the same substrate (i.e., the Y form) via different stimuli sources (i.e., photo vs. mechano–vapo mode) is to our knowledge unprecedented.
The photoinduced WYi, BYi, and KYi forms could be thermally reversed back to the Y form (Fig. S12†). A controlled experiment using pure photodimer 2 as the substrate shows that 2 has a half-life of 47.5 min at 165 °C in the recovery of the Y form (Fig. S13†), and there is no noticeable decomposition or formation of side products according to 1H NMR spectroscopy (Fig. S14†). The activation energy for the solid-state cycloreversion of 2 is 30.6 kcal mol−1 on the basis of the reaction rates determined in the temperature range of 160–190 °C (Fig. S15†), revealing good stability of 2 at ambient temperature.
PMFC of the G form
The G form also displays an intriguing PMFC behaviour, but it differs from the case of the Y form at least in three aspects. The first one is the direction of spectral shifts. Upon UV irradiation (340–390 nm), the fluorescence colour of the single crystalline G form (Φf = 0.09) turned first to orange (OGi form, Φf = 0.05) and then to red (RGi form, Φf = 0.02) (Fig. 7a), corresponding to the growth of a new emission band at 604 nm and a shoulder at ∼650 nm at the cost of the band at 534 nm (Fig. 7b). The possibility that the orange and red emissions are from photochemical products could be excluded by the fact that both the OGi and RGi forms contain only 1 and 2 and no other photoproducts could be detected. The percentages of photodimerization are 21% and 42% in the OGi and RGi forms, respectively, according to the analyses by UV-vis absorption spectroscopy. This again points to a photomechanical effect exerted by the [4 + 4] photodimerization. However, unlike the excimer-to-monomer transformation (Fig. 1b, mode I) in the case of the Y form, the photomechanical effect in the G form leads to an enhancement of intermolecular electronic couplings such that the emission is further red-shifted relative to the pristine excimeric green emission (Fig. 1b, mode II). The formation of a red fluorescent state for a blue-emissive anthracene derivative is unusual,32 because it requires intermolecular electronic couplings to an extent even stronger than that in the Y form and meanwhile a low efficiency of the [4 + 4] photodimerization reaction to prevent fluorescence quenching. Indeed, the fluorescence excitation spectrum monitored at 610 nm shows a mirror-imaged band at 580 nm (Fig. 7b), which is much more red-shifted than the 422 nm peak for the Y form (Fig. S4†), revealing the dimeric nature of the red-emissive state (i.e., strong ground-state electronic coupling). Such a “super dimer” state is unprecedented and expected to be located in a constrained environment created by the photodimerization reactions. A larger impact of the photodimerization reactions on the G vs. Y form is evidenced by the much more significant variations in the PXRD pattern on going from the G to RGi form than that from the Y to BYi form (Fig. S9†). Another difference in PMFC between the G and Y forms is the photochemical reversibility of the fluorescence switching. Both the photochemically induced RGi and BYi forms are stable in the dark at ambient temperature, but they are responsive to photoexcitation in different manners. When the super dimers in the RGi form were excited with 580 nm light for 50 h, the fluorescence colour turns to yellowish green (Fig. 7a), indicating the occurrence of excited-state structural relaxation of the super dimers back to the G form. However, to our surprise, this process is accompanied by a cycloreversion of 2 in the crystal form, as evidenced by the reduced fraction of 2 from 42% to 15%. Since 2 does not absorb the 580 nm light, the cycloreversion must result from the photomechanical effect exerted by the structural relaxation of the super dimers. This scenario is consistent with a constrained environment for the super dimers such that a release of the geometrical constraint is required for the reversion of the super dimers back to the crystalline G form. Regarding the good thermal stability of pure 2 in the solid state (vide supra), the photodimers in RGi must encounter significant structural strains such that the energy barrier for photomechanical cycloreversion of 2 is low. Indeed, control experiments show that no cycloreversion occurs for 2 in BYi at 70 °C for 50 h, but the same conditions lead to 14% cycloreversion of 2 in the RGi form. The presence of substantial structural strains for the photodimers in RGi but not in BYi is consistent with the unfavourable vs. ideal topochemical geometry for [4 + 4] photodimerization in the G vs. Y form (Fig. 4b and d). The photochemical reversibility of 2 in the crystalline G form is in contrast to the photochemically irreversible feature of 2 in the Y form and represents the first example of two-way photomechanofluorochromism.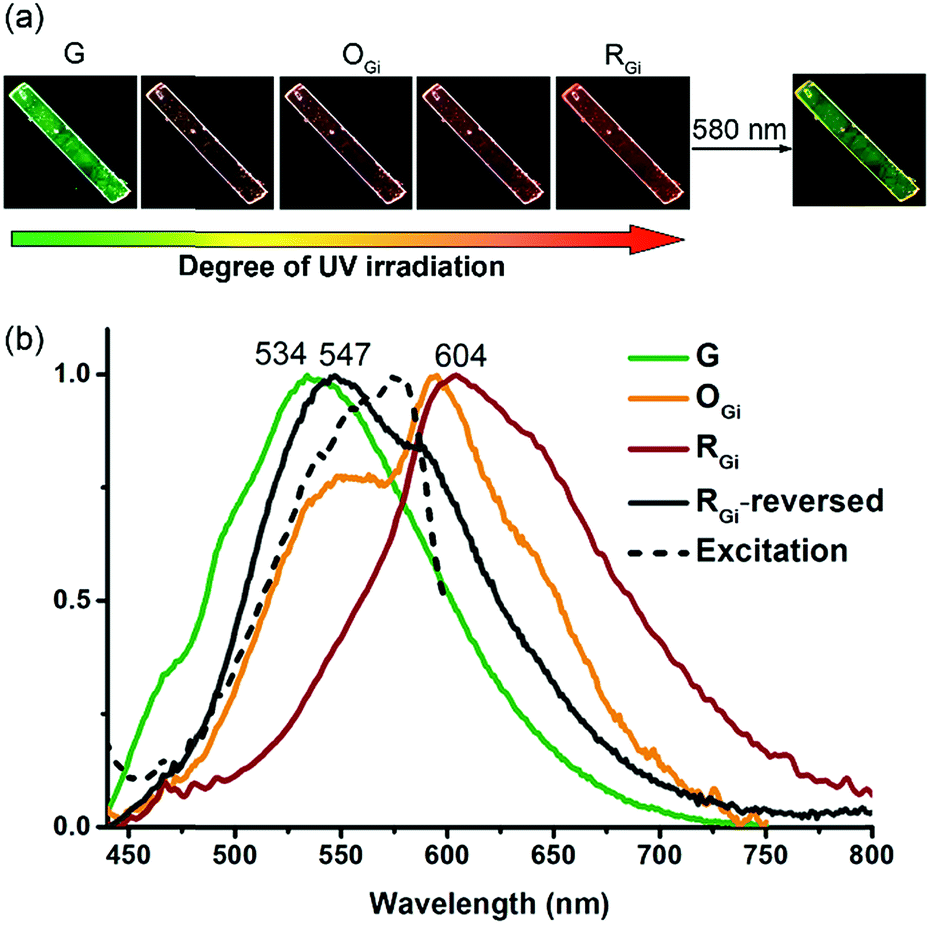 | ||
| Fig. 7 Photoinduced fluorescence colour changes of a single crystal (0.67 mm × 0.10 mm) of the G form of 1: (a) photographs obtained at different stages of photoirradiation at 340–390 nm followed by irradiation of the RGi form at 580 nm; (b) the corresponding normalized fluorescence emission (solid lines, λex = 430 nm) and excitation (black dashed line, λex = 610 nm) spectra. | ||
To account for the photomechanical formation of the red-emissive super dimers, two possible mechanisms are proposed. The first mechanism is a direct compression: that is, the super dimer is created via the squeezing and confinement by the surrounding photodimers, as is schematically depicted in Fig. 6b. The alternative one is a breakdown mechanism (Fig. 6c), in which the squeezing effect by the surrounding photodimers might not be sufficient to result in a red-emissive super dimer but an intermediate state of yellow or orange emission, and the subsequent photodimerization of the intermediate state triggers the cycloreversion but inhibits a full structural relaxation of the neighbouring photodimer to form the super dimer. Recently, a red-shifted absorption spectrum recorded during the breakdown of an intramolecular anthracene photodimer has been attributed to the partially relaxed structure of the cycloreversed product.33 Regardless of the exact mechanism of the super dimer formation, the common feature of both mechanisms is the presence of significant structural constraints for photodimerization.
The third difference between the PMFC behaviour of the G vs. Y form is the limitation of the sample nature. While the crystals and powder of Y form undergo the same PMFC process, the above-observed PMFC properties of the single crystals of G form do not apply to its powder. Under the same irradiation conditions, the G powder form did not lead to the OGi or the RGi form but a dark state (KGi form), which consists of not only 1 and 2 but also an additional side product 3 with an isolated yield of 21% (Scheme 1). Since the formation of endoperoxide 3 requires photosensitization of molecular oxygen to singlet oxygen34 followed by the [4 + 2] cycloaddition35 with 1, the significant yield of 3 indicates that the singlet excited state of 1 (11*) could undergo efficient intersystem crossing (ISC) to form the triplet excited state (31*) that functions as the sensitizer. This is corroborated by a control experiment, in which irradiation of 1 in DCM under an O2 atmosphere at 419 nm for 6 h led to the formation of 3 in 57% yield (Scheme S2†). In this context, the lack of formation of 3 in the single crystals of G form could be attributed to inefficient photosensitization, presumably due to a low surface area. In the case of Y form, a much more efficient [4 + 4]-photodimerization than the photosensitization and photooxidation in both crystals and powder might account for the absence of the oxygen adduct 3. Indeed, the Y form undergoes near quantitative photodimerization under prolonged irradiation at 419 nm (vide supra), but all the attempts to have a quantitative conversion of the G form to the photodimer 2 have failed; a maximum yield of 66% was obtained from the irradiation of the G form at 419 nm in Ar, in which formation of endoperoxide 3 was undetectable. Therefore, the formation of 3 is not the origin of incomplete photodimerization of the G form; instead, the unfavourable crystal packing mode and the poor stability of 2 in the G form (vide supra) are responsible. Note that even when the powders are irradiated in Ar, the dark KGi state remains to be the outcome, and the super dimer-based orange and red colours could be detected only using a camera with a longer exposure time (Fig. S16†). A possible explanation for the fluorescence quenching is the generation of quenchers during photoirradiation. Since the formation of endoperoxide 3 is inhibited and no other photoproducts could be detected, the nature of the quenchers could be attributed to photoinduced formation of nonradiative excimers or dimers. A similar mechanism but operated in a reverse direction has been proposed to explain the fluorescence turn-on of stilbenic π-dimer crystals via [2 + 2] photodimerization.36 The formation of nonemissive excimers/dimers in the powders but emissive super dimers in the crystals as a result of the same [4 + 4] photodimerization reactions might reflect the differences in the photomechanical effect in bulk crystals vs. the surface of the molecular solids.
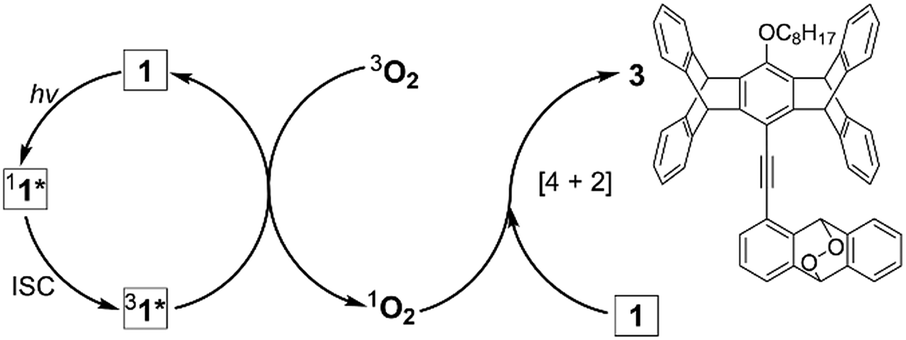 | ||
| Scheme 1 Mechanistic formation of endoperoxide 3 upon irradiation of 1 in DCM and in the G powder form in the presence of molecular oxygen. | ||
With the bimodal PMFC behaviour of the crystalline Y and G forms of 1 that results in various fluorescence colours, including red (R), green (G), and blue (B), the three basic colours of light, and the black- and white-light emission, a rainbow-coloured fluorescent mosaic could be created by temporal and spatial control of photoirradiation on a set of microcrystals of 1 in a mixed Y and G form. As shown in Fig. 8, the pre-assembled two-colour angelfish pattern of the Y and G crystals could be switched into a multicolour version by photoirradiation. To our knowledge, this is the first example of a photochromic seven-colour fluorescence image created solely using a single unichromophoric molecular solid.
 | ||
| Fig. 8 A fluorescence mosaic pattern (5.2 mm × 7.0 mm) of “an angelfish” created by photoirradiation (340–390 nm) of the pre-assembled Y and G forms of 1. | ||
Structure–property relationship
The correlations between the structure and the stress-responsive multicolour fluorescence properties of 1 deserve further discussion. Compound 1 consists of two key components, the planar anthracene and the nonplanar cogwheel-shaped pentiptycene scaffold, linked by the linear ethynylene group. The unichromophoric nature of 1 is evidenced by the results of TDDFT (B3LYP/6-31+G(d, p)) calculations, in which the configuration HOMO → LUMO accounts for the lowest excited singlet state (>98%), and both HOMO and LUMO are localized on the π-conjugated backbone of anthracene–ethynylene–central phenylene of pentiptycene (Fig. S17†). While the anthracene moiety plays an active role in determining the extent of intermolecular π-stacking interactions and in performing the [4 + 4] photodimerization reactions that are directly associated with the observed MFC and PMFC properties, the pentiptycene group is by no means a silent character. We believe that the volume-demanding pentiptycene scaffold plays a critical role in directing the crystal packing that leads to the two polymorphs of Y and G forms. For the Y form, the pentiptycene groups mesh with one another through the V-shaped notches to form a one-dimensional (1D) array, and the U-shaped cavities accommodate the anthracene pairs in one side and the octyl chains in the other side (Fig. 9a). The pentiptycene scaffolds in the G form also form a 1D array via the V-notches, but both sides of the U-cavities of pentiptycene are occupied by the octyl chains, leading to a distinct layer of anthracene pairs in a parallel arrangement (Fig. 9b). Since there is no inclusion of solvent molecules in both crystals, it seems that the competition between the alkyl-pentiptycene C–H⋯π interactions and the anthracene–pentiptycene π–π interactions during the nucleation processes determines whether the crystal packing is the Y or the G form. In conjunction with the conditions for selective preparation of the two forms (see the Experimental section), the C–H⋯π interactions appear to dominate at low temperatures, but the opposite is true for the π–π interactions at high temperatures.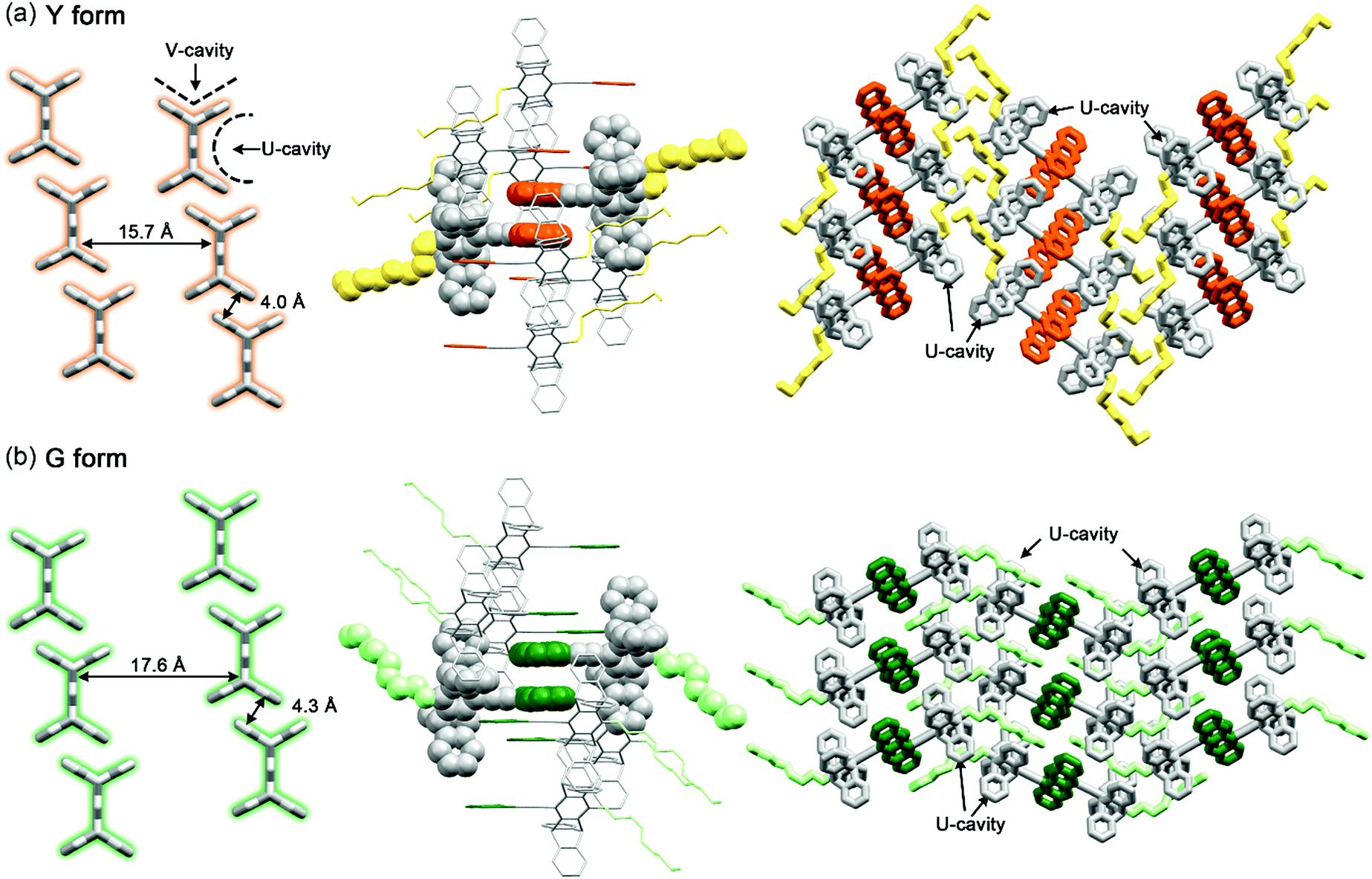 | ||
| Fig. 9 Three views of crystal packing modes of the (a) Y and (b) G forms of 1 that show the one-dimensional pentiptycene array stacked through the V cavities (left, the rest of the molecular groups are removed for clarity), the neighbourhood of the anthracene pairs (middle), and the groups located in the pentiptycene U cavities (right). Hydrogen atoms are omitted and the anthracene pairs are highlighted with orange and green colours for the Y and G forms, respectively, for clarity. | ||
The cogwheel pentiptycene scaffold should be also critical to the significant excimer emission (Φf = 0.17–0.19) of both the Y and G forms in spite of the photochemical activity of the anthracene pairs. We reason that the V-cavity-meshed packing of the pentiptycene cogwheels retards to some extent the molecular translational movements required to carry out the photodimerization reaction, giving room for the excimers to decay via the radiative route.
On the other hand, the gear-like meshing of the pentiptycene cogwheels19 might facilitate librational motions.13 This could explain the MFC of both the Y and G forms in response to gentle grinding forces, as a small amplitude of libration around the molecular long axis of 1 could readily remove the π-stacking interactions of the anthracene pairs to give monomer emission, in a way similar to the photomechanical effect depicted in Fig. 6a.
The different extent of MFC and VFC spectral shifts between the Y and G forms might reflect the different degrees of molecular freedom in the crystals. The fluorescence maximum shifts from 591 to 453 nm upon grinding and from 453 to 542 nm upon subsequent fuming with pMDMA of the Y form, but the corresponding shifts are from 538 to 482 nm and from 482 to 530 nm for the G form. Assuming that the degree of anthracene–anthracene and anthracene–pMDMA stacking interactions is responsible for the observed MFC and VFC properties, respectively, the relatively smaller MFC and VFC shifts for the G vs. Y form indicate a more rigid molecular packing and a larger steric shielding of the anthracene groups in the G form.
Although the photomechanical effect of a solid-state photoreaction is generally complicated,37 the crystal packing modes of the Y and G forms provide a clue to the observed PMFC behaviour. In principle, the major steric impact of the photodimerization would be the puckering of the planar anthracene π-moiety, which expands the lateral ring-to-ring distance by 0.4–0.7 Å from 3.40–3.69 Å to 4.11–4.13 Å according to the X-ray crystal structures shown in Fig. 4. The photo-driven puckering motion is expected to exert mechanical stresses on the neighbouring molecules, particularly the groups nearby. In this context, such photomechanical forces in the Y form would be on the pentiptycene groups, as the lateral anthracene pairs are embedded in the U-cavities of pentiptycene (Fig. 9a, right). Consequently, the stresses trigger a librational motion of the pentiptycene groups and lead to the excimer-to-monomer switching, resembling the external grinding effect. However, unlike the external grinding forces that cause a massive perturbation of the molecular packing and loss of crystallinity (Fig. S5†), which requires solvent molecule deposition for “recrystallization” to recover the Y form, the internal local photomechanical effect does not destroy the crystallinity but causes some local disorders of the molecular packing (Fig. S9†). Such a local perturbation could be fixed by photoexcitation of the monomers via excited-state structural relaxation, which finally leads to a complete photodimerization (Fig. 6a). In contrast, the lateral rings of the anthracene pairs are sandwiched between neighbouring anthracene pairs in the G form (Fig. 9b, middle), and the puckering stresses would be imposed on the unreacted anthracene pairs via a squeezing effect (Fig. 6b) or on the adjacent photodimers via a constrained structural breakdown (Fig. 6c) to form the super dimers. Note that the contribution of the adjacent pentiptycene groups to the formation of super dimers could not be ignored, considering the presence of short contacts between neighbouring anthracene and pentiptycene groups. Since the topochemical geometry is unfavourable for photodimerization in the G form (Fig. 4b and d), the resulting photodimers could put structural strains not only on the neighbours but also on the photodimers themselves. This might account for the reversed photomechanical effect in the super dimers that pushes the photodimers to convert back to monomers. The distinct photomechanical effect caused by the common [4 + 4] photodimerization reaction observed for the Y vs. G form highlights the importance of crystal engineering in the design of PMFC materials.
Conclusions
We have demonstrated that the unichromophoric system 1 displays rich and unprecedented multicolour photomechanochromic as well as mechanochromic and mechano-activated vapochromic fluorescence, which is summarized in Fig. 10. The fluorochromicity is governed by the variable extent of intermolecular electronic interactions in response to external or internal mechanical forces. The [4 + 4] photodimerization of the anthracene pairs serves as the internal photomechanical source that triggers an excimer-to-monomer or an excimer-to-super dimer transformation, depending on the molecular packing mode. Additional fluorescence colours could be created by linear colour mixing of the two interconverting states as a result of large differences in emission wavelengths and fraction-tunable phase transformation. Multicolour fluorescence patterning of the sample films could be achieved through spatial and temporal control of the mechanical forces. Our results illustrate the potential utility of photomechanical stresses in the design of organic photochromic luminescent materials.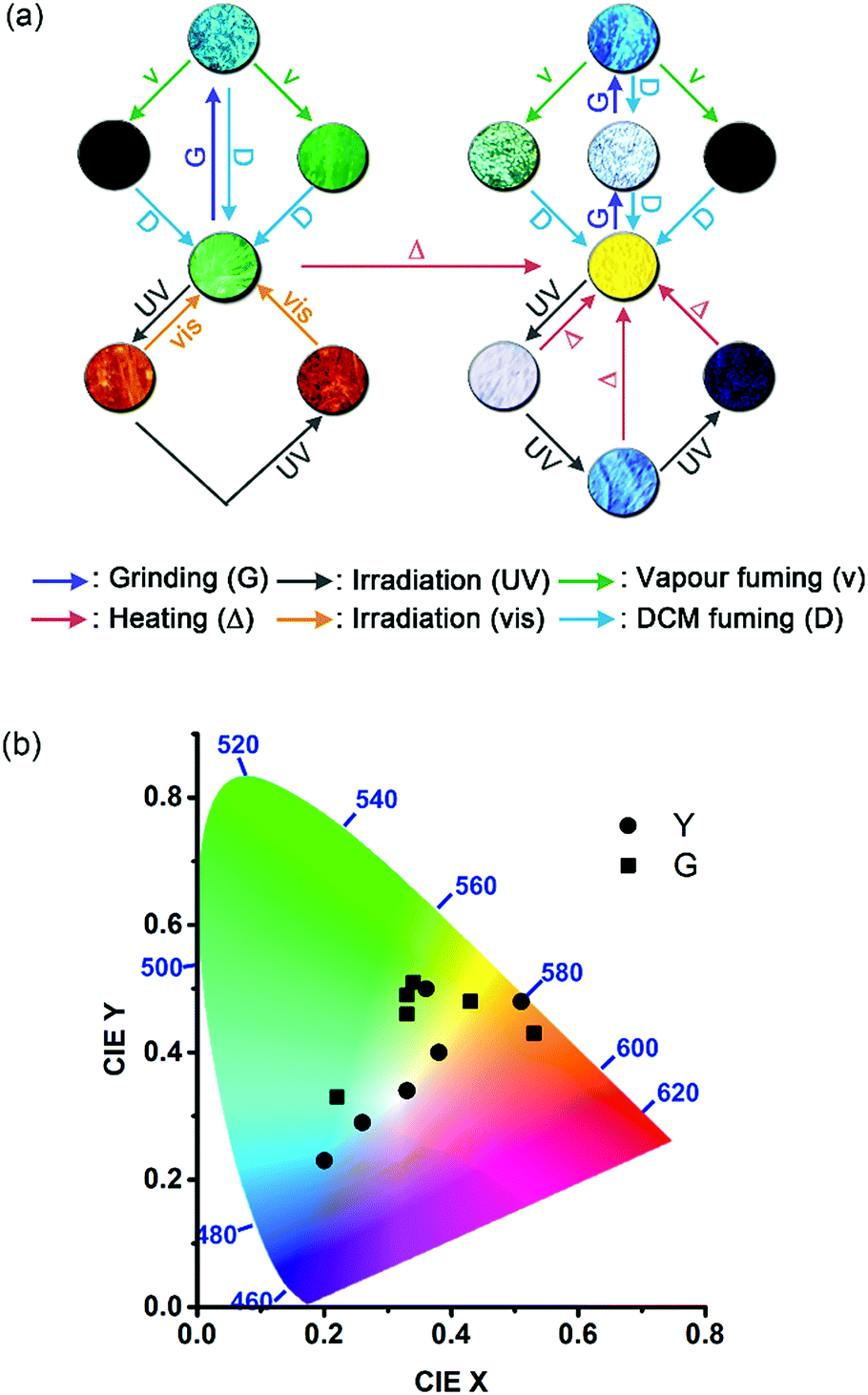 | ||
| Fig. 10 A summary of (a) the multistimuli, multiresponsive, and multicolour fluorescence and (b) the locations of the fluorescent states on the CIE chromaticity diagram for molecular solid 1. Different colours of the arrows in (a) represent different types of stimuli. | ||
Experimental
Materials
All commercial reagents, catalysts, solvents (HPLC grade for photophysical measurements) were used as received. Column chromatography was carried out on silica gel (Geduran® SI 60). Detailed synthetic procedures and compound characterization data for compounds 1–3 are provided as ESI.†The Y and G forms of 1 in powder or crystals were selectively prepared from the DCM/MeOH mixed solution via the control of temperature and nucleation time. For the powders, a concentrated DCM solution of 1 (1.3 × 10−2 M, 2.0 mL) was added dropwise to MeOH (50 mL) for 180 s at 50 °C or −70 °C to afford the Y and G forms, respectively. For crystals, the solutions were prepared by dropwise addition of MeOH (0.5 mL) to a warm (40 °C) DCM solution of 1 (4.9 × 10−3 M, 1.5 mL) until precipitates started to form. A few drops of DCM were then added to dissolve the precipitates (3.7 × 10−3 M, DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 3:1). The resulting pale yellow solution was kept at room temperature for crystal growth, in which the Y form crystals dominate. When the solution was placed in a fridge at 4 °C, the majority of crystals are of the G form.
:MeOH = 3:1). The resulting pale yellow solution was kept at room temperature for crystal growth, in which the Y form crystals dominate. When the solution was placed in a fridge at 4 °C, the majority of crystals are of the G form.
The powdered sample films for fluorochromic experiments were prepared by spreading the as-prepared powders (1.0–1.5 mg) with a stainless steel spatula on a quartz plate (1.0 cm × 1.0 cm or 1.5 cm × 1.5 cm) and then fuming with DCM vapour for 1 min. For the preparation of drop-cast films of the Y form for multicolour fluorescence patterning, a quartz plate (1.0 cm × 1.0 cm) was heated at 185 °C, and then a 200 μL DCM solution of 1 (31.6 mM) was added dropwise to the plate to form a thin solid film.
Methods
Steady-state fluorescence spectra, fluorescence quantum yield, and fluorescence lifetimes were measured on an Edinburgh FLSP920 spectrometer equipped with an integrating sphere (150 mm diameter, BaSO4 coating) and a gated hydrogen arc lamp. For the fluorescence lifetime measurements, the instrument response function was profiled using a scatter solution, and the goodness of the nonlinear least-squares fit of the decay profiles was judged by the autocorrelation function, the randomness of the residuals, and the reduced χ2 value. Photographs of the fluorescent samples were taken using an Olympus E-M1 camera or an Olympus IX73 inverted microscope equipped with a DP73 colour camera. The light source of the microscope is a halogen lamp equipped with an Olympus U-FUW filter, which passed light in the range of 340–390 nm. Differential scanning calorimetry (DSC) was performed on a premium differential scanning calorimeter, Netzsch DSC 204 F1 Phoenix, with a heating rate of 5 °C min−1. Single crystals were determined using a Rigaku Oxford Diffraction diffractometer (measurement device type: Xcalibur, Atlas, Gemini). The powder X-ray diffraction (PXRD) patterns were collected on a Bruker D2 Phaser diffractometer using nickel-filtered Cu Kα radiation (λ = 1.5413 Å) at a voltage of 30 kV and a current of 10 mA. Scanning electron microscopy (SEM) images were taken with a JEOL JSM-7600F field emission scanning electron microscope with INCA X-Max EDS. Photochemical reactions were carried out in a photochemical reactor equipped with light tubes of 419 nm. DFT and TDDFT calculations were performed using the Gaussian 09 program38 package using a density functional theory method with the B3LYP functional39 and the 6-31+G(d,p) basis sets. The vibration analyses were carried out for the optimized geometries at the same computational level to ensure that the obtained structures correspond to a minimum.The mechanofluorochromic tests were carried out by gentle hand grinding of the sample films with a spatula until the fluorescence colour was uniform and no longer changed. For vapofluorochromic tests, the sample films were placed in a 20 mL vial bottle that contains a piece of cotton soaked with 0.5 mL of the target chemicals or solvents for 1 min. The light sources for the photofluorochromic experiments are provided either from the halogen lamp of the optical microscope (340–390 nm) or from the light tubes of the photochemical reactor (419 and 580 nm) mentioned above.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Taiwan University (NTU-107L880101) and the Ministry of Science and Technology of Taiwan (MOST-104-2113-M-002-004-MY3) for financial support. We also thank Prof. Hao-Ming Chen and Ms Su-Jen Ji for the assistance in the PXRD and SEM measurements, respectively.Notes and references
- (a) J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef PubMed; (b) A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- (a) S. N. Corns, S. M. Partington and A. D. Towns, Color. Technol., 2009, 125, 249–261 CrossRef CAS; (b) Y. Inagaki, Y. Kobayashi, K. Mutoh and J. Abe, J. Am. Chem. Soc., 2017, 139, 13429–13441 CrossRef CAS PubMed; (c) C. M. Sousa, J. Berthet, S. Delbaere, A. Polònia and P. J. Coelho, J. Org. Chem., 2017, 82, 12028–12037 CrossRef CAS PubMed.
- I. Tochitsky, M. A. Kienzler, E. Isacoff and R. H. Kramer, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.7b00723.
- (a) A. Perrier, F. Maurel and D. Jacquemin, Acc. Chem. Res., 2012, 45, 1173–1182 CrossRef CAS PubMed; (b) M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC; (c) D. Genovese, A. Aliprandi, E. A. Prasetyanto, M. Mauro, M. Hirtz, H. Fuchs, Y. Fujita, H. Uji-I, S. Lebedkin, M. Kappes and L. De Cola, Adv. Funct. Mater., 2016, 26, 5271–5278 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- (a) H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC; (b) M. Dong, A. Babalhavaeji, C. V. Collins, K. Jarrah, O. Sadovski, Q. Dai and A. G. Woolley, J. Am. Chem. Soc., 2017, 139, 13483–13486 CrossRef CAS PubMed.
- (a) S.-Z. Pu, Q. Sun, C.-B. Fan, R.-J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4, 3075–3093 RSC; (b) R. Kashihara, M. Morimoto, S. Ito, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2017, 139, 16498–16501 CrossRef CAS PubMed; (c) G. Liu, Y.-M. Zhang, L. Zhang, C. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 12135–12140 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- (a) I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC; (b) C. Li, Y. Zhang, J. Hu, J. Cheng and S. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS PubMed; (c) G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CrossRef CAS PubMed; (d) S. Mo, Q. Meng, S. Wan, Z. Su, H. Yan, B. Z. Tang and M. Yin, Adv. Funct. Mater., 2017, 27, 1701210 CrossRef; (e) Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- Q. Chen, D. Zhang, G. Zhang, X. Yang, Y. Feng, Q. Fan and D. Zhu, Adv. Funct. Mater., 2010, 20, 3244–3251 CrossRef CAS.
- S. Kim, S.-J. Yoon and S. Y. Park, J. Am. Chem. Soc., 2012, 134, 12091–12097 CrossRef CAS PubMed.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- A. Gavezzotti and M. Simonetta, Chem. Rev., 1982, 82, 1–13 CrossRef CAS.
- (a) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC; (b) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed; (c) M. Jin, T. Seki and H. Ito, J. Am. Chem. Soc., 2017, 139, 7452–7455 CrossRef CAS PubMed.
- (a) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed; (b) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed; (c) M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci., 2017, 8, 2677–2686 RSC; (d) A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- (a) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed; (b) S. Hatanaka, T. Ono and Y. Hisaeda, Chem.–Eur. J., 2016, 22, 10346–10350 CrossRef CAS PubMed.
- Y. Matsunaga and J.-S. Yang, Angew. Chem., Int. Ed., 2015, 54, 7985–7989 CrossRef CAS PubMed.
- C.-J. Lin, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2017, 56, 4978–4989 CrossRef CAS PubMed.
- (a) J.-S. Yang and J.-L. Yan, Chem. Commun., 2008, 1501–1512 RSC; (b) C.-Y. Kao, Y.-T. Hsu, H.-F. Lu, I. Chao, S.-L. Huang, Y.-C. Lin, W.-T. Sun and J.-S. Yang, J. Org. Chem., 2011, 76, 5782–5792 CrossRef CAS PubMed.
- (a) T. Kim, L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, ChemPhysChem, 2014, 15, 400–414 CrossRef CAS PubMed; (b) P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed; (c) L. Zhu, R. O. Al-kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2016, 55, 7073–7076 CrossRef CAS PubMed; (d) H. Xie, M.-J. He, X.-Y. Deng, L. Du, C.-J. Fan, K.-K. Yang and Y.-Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9431–9439 CrossRef CAS PubMed; (e) F. Tong, W. Xu, M. Al-haider, D. Kitagawa, R. O. Al-kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2018, 57, 7080–7084 CrossRef CAS PubMed.
- T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed.
- J.-S. Yang, J.-L. Yan, C.-Y. Hwang, S.-Y. Chiou, K.-L. Liau, H.-H. Gavin Tsai, G.-H. Lee and S.-M. Peng, J. Am. Chem. Soc., 2006, 128, 14109–14119 CrossRef CAS PubMed.
- H. Yang, K. Ye, J. Sun, P. Gong and R. Lu, Asian J. Org. Chem., 2017, 6, 199–206 CrossRef CAS.
- (a) Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed; (b) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed; (c) B. Xu, Y. Mu, Z. Mao, Z. Xie, H. Wu, Y. Zhang, C. Jin, Z. Chi, S. Liu, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lian and M. R. Bryce, Chem. Sci., 2016, 7, 2201–2206 RSC; (d) B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- (a) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed; (b) A. Kobayashi, K. Komatsu, H. Ohara, W. Kamada, Y. Chisna, K. Tsuge, H.-C. Chang and M. Kato, Inorg. Chem., 2013, 52, 13188–13198 CrossRef CAS PubMed; (c) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed; (d) L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160–9164 CrossRef CAS PubMed.
- (a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed; (b) Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef CAS PubMed.
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433–481 CrossRef CAS.
- I. Zouev, D.-K. Cao, T. V. Sreevidya, M. Telhensky, M. Botoshansky and M. Kaftory, CrystEngComm, 2011, 13, 4376–4381 RSC.
- Z.-A. Huang, C. Chen, X.-D. Yang, X.-B. Fan, W. Zhou, C.-H. Tung, L.-Z. Wu and H. Cong, J. Am. Chem. Soc., 2016, 138, 11144–11147 CrossRef CAS PubMed.
- Long-term irradiation results in fragmentation of the crystals, which are not suitable for the crystallographic analysis of the single-crystal-to-single-crystal transformation.
- (a) B. Dong, M. Wang, C. Xu, Q. Feng and Y. Wang, Cryst. Growth Des., 2012, 12, 5986–5993 CrossRef CAS; (b) S. Hisamatsu, H. Masu, M. Takahashi, K. Kishikawa and S. Kohmoto, Cryst. Growth Des., 2015, 15, 2291–2302 CrossRef CAS; (c) H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC; (d) H. Liu, D. Cong, B. Li, L. Ye, Y. Ge, X. Tang, Y. Shen, Y. Wen, J. Wang, C. Zhou and B. Yang, Cryst. Growth Des., 2017, 17, 2945–2949 CrossRef CAS.
- T. Nishiuchi, S.-Y. Uno, Y. Hirao and T. Kubo, J. Org. Chem., 2016, 81, 2106–2112 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010, pp. 1001–1042 Search PubMed.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS PubMed.
- (a) J.-M. McBride, B. E. Segmuller, M. D. Hollingsworth, D. E. Mills and B. A. Weber, Science, 1986, 234, 830–834 CrossRef CAS PubMed; (b) T. Luty and C. J. Eckhardt, J. Am. Chem. Soc., 1995, 117, 2441–2452 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, M. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, characterization data, and 1H and 13C NMR spectra for compounds 1–3, procedures and kinetic plots for the thermal cycloreversion of 2, photophysical and PXRD data of 1 and 2, the DSC diagram of 1, and photos of the multicolour fluorescence patterning at different stages. CCDC (1) [G (1863069), Y (1863071)] and (2) [1863072]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03740j |
| This journal is © The Royal Society of Chemistry 2018 |