
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid CuCoO–GO enables ultrasensitive detection of antibiotics with enhanced laser desorption/ionization at nano-interfaces†
Enhui
Wu‡
a,
Kun
Feng‡
a,
Rui
Shi‡
a,
Rui
Lv
a,
Fuzhong
Ouyang
a,
Shawn S. C.
Li
b,
Jun
Zhong
 *a and
Jian
Liu
*a
*a and
Jian
Liu
*a
aInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-based Functional Materials and Devices, Joint International Research Laboratory of Carbon-based Functional Materials and Devices, Soochow University, Suzhou, Jiangsu Province 215123, China. E-mail: jliu@suda.edu.cn; jzhong@suda.edu.cn
bDepartment of Biochemistry, Western University, London, Ontario N6A 5C1, Canada
First published on 8th October 2018
Abstract
The soaring concerns globally on antibiotic overuse have made calls for the development of rapid and sensitive detection methods urgent. Here we report that the hybrid CuCoO–GO matrix allows for sensitive detection of various antibiotics in combination with MALDI TOF MS. The new matrix is composed of few-layered GO nanosheets decorated with CuCoO nanoparticles with an average size of 10 nm, and exhibits excellent aqueous suspensibility. Accurate quantitation of the sulfonamide antibiotics in milk samples have been demonstrated using a CuCoO–GO matrix and a stable isotope (C13)-labeled analyte as the internal standard. Our experiments have achieved lower limits of detection (LOD) by several hundred fold for the detection of a panel of representative antibiotics, in comparison with the literature reports. Both intrabacterial and extrabacterial residual antibiotics can be sensitively detected with our method. We have further investigated the molecular mechanism of the enhanced desorption/ionization efficiency by the CuCoO–GO matrix with synchrotron radiation techniques for the first time. This work provides a sensitive matrix enabling MALDI-TOF MS to be applied in small molecular analysis, but also presents a distinct perspective on the mechanism behind the material functions.
Introduction
There are growing concerns globally over the inappropriate use of antibiotics which are polluting the environment, contributing to food contamination, and threatening the medical practice of public health.1–5 A report in the literature reveals that early exposure to antibiotics may lead to potentially long-term harmful impacts by changing gut microbiota, brain cytokines and behavior.1 It has become a more serious danger to human healthcare with the emergence of “superbugs” induced by the overuse of antibiotics.3 Increasing needs demand the development of rapid and sensitive techniques to detect various antibiotics in trace amounts. The traditional methods include microbial growth inhibition assays,6 chromatography,7–9 immunochemical detection,10–12 as well as receptor- or enzyme-based tests.13 However, they are subject to several critical limitations. Assays of microbial growth inhibition suffer from low sensitivity and many factors interfere in signal reading.6 Chromatographic methods require lengthy procedures for sample-pretreatment.7,9 Receptor-based screening methods, such as radio-receptor assay, enzyme-labelling receptor assay, colloidal gold-labelling receptor assay and enzyme colorimetry are limited by enzymatic activity fluctuations and labelling-reagent contamination.12,13Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is an indispensable tool for the analysis of biomolecules, with the advantages of high sensitivity, high throughput, and a fast procedure to obtain results.14–21 However, the conventional organic matrices (CHCA, DHB, SA, etc.) widely used in MALDI-TOF MS are subject to their own structural fragmentation by lasers, which generates intensive background noise below m/z 700 intrinsically. This overwhelms the true signals of small molecular analytes, thus severely limiting the applications of MALDI-TOF MS in the (m/z < 700) molecular range.22,23 Researchers have attempted to synthesize different types of organic molecules as replaceable matrix candidates to suppress background noise.24,25 Alternatively, various inorganic nanomaterials and nanostructures have been proposed as MALDI matrices to increase the signal to noise ratio in the detection of small molecules, including silicon, metal, metallic oxide and carbon-based nanomaterials.26–34 Among them, graphene oxide (GO) is attractive due to its large surface area, good light absorption, and excellent optoelectric properties.32 It has been suggested that graphene oxide/multi-walled carbon nanotube composites can promote small molecule detection by MALDI MS because of the improved wettability in water and enlarged surface area.32 However, many nanomaterial-based matrices still suffer from drawbacks such as the background noise generated by their inherent structural defects, or the relatively lower efficiency in assisting molecular ionization.34–36
Herein, we report an important discovery and mechanism revelation for the sensitive detection of various antibiotics using hybrid CuCoO–GO as the MALDI matrix. Previously we reported that tuning the ratio of copper and cobalt atoms in their metal oxide–graphene composite could maximize the electron transfer properties for a synergistic catalytic performance in the hydrolysis of ammonia borane.37 In this work, we hypothesize that the electron transfer properties of hybrid CuCoO–GO could promote the ionization of small molecules under laser irradiation, with significant improvement in the signal to noise ratios in MS spectral analysis. We have demonstrated that the CuCoO–GO matrix enables the detection of 7 representative antibiotics with lower limits of detection (LOD) by several hundred fold. Accurate results with absolute quantitation of the antibiotic in milk samples have been demonstrated using our approach integrated with a stable isotope-labelled analyte as the internal standard. Intrabacterial and extrabacterial antibiotic residues can be sensitively detected using our method. For the first time, we have revealed the mechanism of molecular deprotonation assisted by a CuCoO–GO matrix for its high performance in MALDI-TOF MS with synchrotron radiation techniques. This work provides a sensitive and robust matrix enabling MALDI-TOF MS applications in small molecule analysis, but also offers a distinctly important perspective on the molecular mechanism behind the material functions.
Experimental
Chemicals
All of the chemicals were purchased from Sigma Aldrich, Aladdin Industrial Corporation, or Shanghai Macklin Biochemical Co., Ltd. Sulfapyridine-13C was bought from WITEGA Laboratorien Berlin-Adlershof Gmbh (Germany). The milk samples were purchased from a local grocery store. Deionized water (18 MΩ cm−1) was used in the experiments.Preparation of CuCoO–GO
A modified Hummers’ method was used to prepare graphene oxide (GO) from natural graphite. A GO solution was prepared by re-suspending 80 mg GO in 80 mL isopropanol, followed by sonication for 20 min. 38.3 mg (0.192 mmol) Cu(CH3COO)2·H2O and 12 mg (0.048 mmol) Co(CH3COO)2·4H2O were mixed with the GO isopropanol solution. The mixture was heated to 80 °C for 1 h with a reflux set-up. After 80 mL of deionized water was added into the reaction, the mixture was kept at 80 °C for 30 min. The product was cooled to room temperature, then collected by centrifugation at 8000 rpm for 3 min with an ethanol rinse. This was dried in an oven at 70 °C for 12 h. The samples of CuCoO–GO with different Cu/Co atom ratios (including CuO–GO) were prepared similarly by referring to this procedure.37Characterization
High-resolution transmission electron microscopy (HRTEM) images were acquired with a FEI Tecnai G2 F20 S-TWIN TEM. Atomic force microscopy (AFM) images were obtained by using a Veeco Multimode V atomic force microscope. Raman spectra were collected using a HR 800 Raman spectroscope (J Y, France). The Fourier Transform Infrared (FT-IR) spectrum was obtained using a Bruker Fourier Transform Infrared Spectrometer (Hyperion). X-ray photoelectron spectroscopy (XPS) characterization was performed with a KRATOS Axis ultra DLD X-ray photoelectron spectroscope. X-ray diffraction (XRD) data were collected by a PANalytical B.V. Empyrean powder diffractometer equipped with a PIXcel3D detector. Scanning electron microscope (SEM) images of samples were acquired by an S-4700 SEM (Hitachi, Japan).Sample preparation for MALDI-TOF MS analysis
The conventional organic matrices of α-cyano-4-hydroxycinnamic acid (CHCA) and 3,5-dimethoxy 4-hydroxycinnamic acid (SA) were individually prepared by a standard method.38,39 Graphene oxide, CuO–GO, CuO, CuCoO, CuCoO–GO were all dispersed in water at a concentration of 1 mg mL−1. The analytes of 7 different antibiotics and 3 additional chemical analogs, including sulfadiazine (SDZ), sulfamethoxazole (SMX), norfloxacin, ampicillin, benzylpenicillin, streptomycin, tetracycline, sulfapyridine, sulfonamide and pyrimethanil, were dissolved in water or ethyl alcohol as the standard mother solutions. They were subsequently diluted to a series of different concentrations as specified in the experiments. The sample spots on the MALDI plate were prepared by a standard method of sandwiching the matrix and analyte solutions. The liquid volume in each pipetting step for one sample spot was 1 μL. The milk samples were mixed with different concentrations of the antibiotic solutions for the tests. They were defatted by mixing with 20% TFA and citrate buffer (0.4 M, pH = 4), followed by centrifugation (4000 rpm for 20 min) to collect the supernatants for MALDI-TOF MS analysis.40Mass Spectrometry
The MALDI-TOF MS data were acquired using a MALDI TOF/TOF Ultraflextreme mass spectrometer equipped with a SmartBeam II Nd:YAG/355 nm laser operating at 2000 kHz (Bruker Daltonics, Inc., Billerica, MA), either in the reflective positive-ion or negative-ion mode. The laser energy was optimized at 30% corresponding to 57 μJ per pulse (laser pulse duration: 3 ns). The laser spot size was approximately 50–100 μm. Each sample was tested with a repetition of n = 4. An accumulation of 3000 laser shots was applied for each spectrum.For the comparison of different matrices, the negative ion mode was carried out using CuCoO–GO, GO, CuO–GO, and CuCoO as nano-matrices for detection of SDZ and SMX. In addition, the noise baselines of traditional matrices of CHCA and SA were also included in the measurements under identical instrumental conditions.
Antimicrobial susceptibility testing
The minimal inhibitory concentrations (MICs) of sulfadiazine sodium (SDS) for E. coli and S. aureus were determined on the basis of microdilution assay.41 First, a single colony of E. coli and S. aureus on the solid Luria–Bertani (LB) agar plate was transferred into 5 mL of liquid LB culture medium and grown overnight at 37 °C. This preculture was diluted in 5 mL of liquid LB culture medium (dilution ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) and grown for 16 h at 37 °C. The bacterial suspension was then diluted with LB culture medium to 1 × 106 CFU mL−1 (colony-forming unit per milliliter). Different concentrations of sulfadiazine sodium from 2 to 0.125 mg mL−1 were mixed with an equal volume of the diluted bacterial suspension and incubated at 37 °C for 20 h. The tube without sulfadiazine sodium was included as a blank control. Finally, the optical density values at 600 nm (OD600) of the solutions were recorded by a Varioskan Flash (Thermo Scientific Company, USA).
:20) and grown for 16 h at 37 °C. The bacterial suspension was then diluted with LB culture medium to 1 × 106 CFU mL−1 (colony-forming unit per milliliter). Different concentrations of sulfadiazine sodium from 2 to 0.125 mg mL−1 were mixed with an equal volume of the diluted bacterial suspension and incubated at 37 °C for 20 h. The tube without sulfadiazine sodium was included as a blank control. Finally, the optical density values at 600 nm (OD600) of the solutions were recorded by a Varioskan Flash (Thermo Scientific Company, USA).
In parallel, sample aliquots of the bacteria incubated with antibiotics were centrifuged at 5000 rpm for 5 minutes. The supernatant solutions were collected for analysis by CuCoO–GO assisted MALDI MS. Simultaneously, the pellets of bacteria were resuspended in a Tris–HCL buffer and centrifuged again. After the washing steps, the bacteria were lysed by sonication in equal volume aliquots. The lysed bacteria were also tested by CuCoO–GO assisted MALDI MS.
Synchrotron radiation experiments
X-ray absorption spectroscopy (XAS) experiments were performed at the Shanghai Synchrotron Radiation Facility (SSRF, 14W and 08U), the Beijing Synchrotron Radiation Facility (BSRF, soft X-ray beamline), and the National Synchrotron Radiation Laboratory (NSRL, XMCD beamline). The in situ XAS experiments at the Cu K-edge were performed at Beamline 14W at the SSRF, in which the samples of CuCoO–GO with or without SDZ was pressed on the 3M tape with the option of UV irradiation for measurements.Results and discussion
Synthesis and characterization of CuCoO–GO
CuCoO–GO was synthesized by the method of a refluxing reaction using Cu(CH3COO)2·H2O and Co(CH3COO)2·4H2O as the metal precursors in a mixture with the GO/isopropanol suspension.37 As shown in Fig. 1a, the TEM image indicates that the as-prepared nanocomposites are few-layered GO nanosheets decorated with nanoparticles with an average size of 10 nm. The elemental mapping of a localized area by random selection further reveals the distribution of O, Cu, Co atoms in the nanocomposites. The AFM image suggests that CuCoO–GO nanocomposites are uniform with a typical lateral dimension of several hundred nanometers (Fig. 1b). The height profiling by AFM demonstrates thicknesses of 1–2 nm, suggesting that the nanocomposites contain only a couple of GO layers (Fig. 1(b1 and b2)). In the Raman spectral analysis of CuCoO–GO, CuO–GO, and GO (the latter two as the control samples), there are two characteristic peaks at 1350 cm−1 (D-band) and 1580 cm−1 (G-band), which are respectively attributed to the vibration of sp3-bonded or sp2-bonded carbon atoms in the graphitic nanostructures (Fig. 1c). The appearance of a third peak at 2800 cm−1 (2D-band) in all of these three different samples suggests that the few-layered feature in the nanosheets is maintained for the CuCoO–GO nanocomposites after the synthetic procedure (Fig. 1c). The analysis by XRD also identifies the characteristic diffraction peaks contributed by GO in all of these three samples as specified (Fig. 1d). XPS spectral analysis of CuCoO–GO verifies the elements copper, cobalt, carbon, and oxygen in the composition of the hybrid nanomaterials (Fig. S1a†). In the Fourier Transform Infrared (FTIR) spectrum of CuCoO–GO, the peaks at 3400 cm−1 and 1570 cm−1 are attributed to the chemical groups O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (stretching mode). The peak at 1250 cm−1 is attributed to the epoxy groups (Fig. S1b†). In the contact angle measurements, CuCoO–GO (θ = 5.1°) exhibits a slightly enhanced hydrophilicity compared to GO (θ = 20.8°). This facilitates forming a relatively uniform film of nanoparticles on the substrate, which is favorable for the deposition of soluble analytes (Fig. S2†).
O (stretching mode). The peak at 1250 cm−1 is attributed to the epoxy groups (Fig. S1b†). In the contact angle measurements, CuCoO–GO (θ = 5.1°) exhibits a slightly enhanced hydrophilicity compared to GO (θ = 20.8°). This facilitates forming a relatively uniform film of nanoparticles on the substrate, which is favorable for the deposition of soluble analytes (Fig. S2†).
 | ||
| Fig. 1 Characterization of CuCoO–GO (CuO–GO and GO as the control samples). (a) TEM image; (a1) dark-field TEM image; (a2) zoomed-in view of the dark-field in the red box; (a3 and a4) corresponding elemental mapping in the red box: Cu (green) and Co (brown). Sample: CuCoO–GO; scale bars as specified. (b) AFM image of CuCoO–Go in the larger field of view; (b1) AFM of a single nanosheet of CuCoO–Go; (b2) height profile along the corresponding blue line of (b1). Scale bars: 5 μm. (c and d) Raman spectra and X-ray diffraction (XRD) patterns of GO, CuO–GO and CuCoO–GO. | ||
The performance of the CuCoO–GO matrix for MALDI-TOF
We compared the background baseline of the CuCoO–GO matrix without any analytes in MALDI-TOF against traditional organic matrices such as CHCA and SA under identical instrumental conditions. As shown in Fig. 2a, the organic matrices (CHCA or SA) produce intensive background noise in a low molecular weight range (m/z < 700). In contrast, the baseline of CuCoO–GO is ultraclean, at least 4 orders of magnitude lower than the traditional organic matrices in noise level. Its property of being free from the interference of fragmented species suggests a good potential for CuCoO–GO as a MALDI matrix to detect small molecules in the range of m/z < 700. The background baselines of the pristine nanomaterials of GO and CuO–GO are included in the ESI (Fig. S3†) for reference. | ||
| Fig. 2 High performance of the CuCoO–GO matrix in MALDI mass spectrometry for antibiotic analysis. (a) The background noise of the CuCoO–GO matrix in comparison with traditional organic matrices of CHCA and SA, in the negative-ion mode. (b) The mass spectra of SDZ and SMX in the positive-ion and negative-ion modes using the CuCoO–GO matrix. (c) Mass spectra of SDZ and sulfapyridine-13C (internal standard) in water solution or the milk sample, and the calibration curves for absolute quantitation of SDZ in the negative-ion mode (n = 4). Error bars: standard deviation. | ||
The performance of the CuCoO–GO matrix was evaluated by MALDI-TOF MS in both the positive-ion and negative-ion modes for the detection of small molecular antibiotics, including sulfadiazine (SDZ) and sulfamethoxazole (SMX). As shown in Fig. 2b, there are clear characteristic peaks for the analytes without the interference of background noise in both of these two modes. In the positive-ion mode, two characteristic peaks are attributed to SDZ (m/z 273 [M + Na]+, 289 [M + K]+) or SMX (m/z 276 [M + Na]+, 292 [M + K]+). In the negative-ion mode, each of the small molecule analytes are identified with the characteristic peak of the deprotonated ion [M − H]− (m/z 249 [M − H]− for SDZ; m/z 252 [M − H]− for SMX). All of these characteristic peaks, featured with a high signal-to-noise ratio, benefit from the high ionization efficiency and low-background noise of the CuCoO–GO matrix. The reproducibility of the CuCoO–GO matrix in MALDI MS has been evaluated by two different modes, including tests of different positions in one spot and tests of different spots in parallel from the same sample. The relatively low coefficient of variation (CV, ∼5%) in both of these two modes indicates a highly reproducible performance for the CuCoO–GO matrix in the ionization of small molecular antibiotics for detection (Fig. S4†).
We performed quantitative analysis on SDZ by including an isotope labelled analog of the analyte with reference to the standard method. Stable isotope labelled sulfapyridine-13C was selected as the internal standard due to its great similarity in chemical structure to SDZ. It was further validated for being a good choice by the linear calibration curves in the titration of SDZ concentrations in MALDI MS. As shown in Fig. 2c, the characteristic peaks for SDZ (m/z 249) and sulfapyridine-13C (m/z 254) are presented in the mass spectra. The intensities of these peaks are consistent with their spiking ratios in the water or the milk samples. There is excellent linearity for the calibration curve in the aqueous solution prepared with either water (R2 = 0.983) or milk (R2 = 0.982) between the MS signal intensity ratios (SDZ: sulfapyridine-13C) and the concentrations of SDZ (10–1000 μg L−1). In addition, the MS spectrum of the blank milk without any antibiotic addition is available in the ESI (Fig. S5†) as the negative control. It verifies that the characteristic peaks of the antibiotic analytes are not contaminated. These results provide good evidence to demonstrate the usefulness of our approach with the CuCoO–GO matrix for residual antibiotic quantification in milk samples.
The limit of detection (LOD) using our approach was determined by titrating the analytes and linear extrapolation of the signal-to-noise ratio (S/N = 3), according to the standard method.42 As previously demonstrated in Fig. 2, the CuCoO–GO matrix is featured with a clean baseline in MALDI MS, which is important for improved LOD in the detection of antibiotics. The detailed values of LOD are summarized in Table S1 and Fig. S6 in the ESI,† including SDZ, SMX, and the other five representative antibiotics. For instance, the LOD for SDZ antibiotic is 0.5 μg L−1 by our approach, which is nearly 500 fold lower than the previously reported results in the literature. This value is also far lower than the maximum residue limit (MRL, 250 μg L−1 for SDZ) in the milk regulated by the China National Standards. The superior performance of the CuCoO–GO matrix was validated by testing the other antibiotics in the list (Table S1†), and comparing with the results using different matrices (including graphene, graphene oxide, and other common matrices) in the literature.15,40,43–45 This suggests that our approach of using the CuCoO–GO matrix can sufficiently meet the technical requirement for the sensitive detection of antibiotics.
Antibiotic susceptibility testing (AST) has been widely used in clinical microbiology labs to determine sensitiveness or the resistance of individual bacterial isolates to any specific antimicrobial agents. The methods for AST include broth dilution test, antimicrobial gradient method, disk diffusion test, or AST stripes, which typically rely on optical measurements or empirical naked-eye observations.41 Recently an important advance of TiO2-assisted MALDI TOF MS was developed to detect antimicrobial resistance-relevant proteins while minimizing sample preparation steps, which promotes simultaneous species identification and antimicrobial resistance analysis.46 Inspired by this work, we attempted to explore the utility of the CuCoO–GO matrix for the application of antimicrobial resistance analysis by measuring both the intrabacterial and extrabacterial antibiotic residues (Fig. 3a). We performed AST experiments to test the antibiotic SDS on E. coli and S. aureus, using a standard microdilution assay (Fig. S7†). The morphological changes of E. coli and S. aureus induced by the antibiotic treatments were characterized by scanning electron microscopy (SEM, Fig. S8†). The bacterial samples treated with different concentrations of SDS were centrifuged to separate the supernatants and bacterial pellets, which were subsequently processed to analyze the antibiotic residues inside or outside the bacteria by CuCoO–GO assisted MALDI MS. As shown in Fig. 3b and c, in the mass spectra of E. coli and S. aureus lysates, the characteristic peaks of SDS (m/z 273), sulfapyridine-13C (m/z 278), and a potential metabolite (4-OH-SDS, m/z 289) can be clearly identified. The calibration curve in the plot (Fig. 3d) according to the standard isotope internal control method exhibits a good linearity (R2 = 0.982), indicating a reliable performance of our approach for the antibiotic quantification using the samples of bacterial cultivation. As shown in Fig. 3e and f, the residual SDS outside and inside the E. coli or S. aureus after antibiotic treatment can be quantitatively analyzed. It suggests a trend that the proportion of SDS penetrating and leftover inside the bacteria increases, driven by the increase of the initial antibiotic dosages. Aligned to the standard AST experiments, our method can provide more detailed information on the uptake and potential metabolic changes of antibiotics, which are essential for the understanding of bacterial resistance mechanisms.28,47
 | ||
| Fig. 3 (a) Schematic diagram of antibiotic detection comparing our method to standard antibiotic susceptibility testing (AST). (b) Mass spectrometry data of E. coli lysates. (c) Mass spectrometry data of S. aureus lysates. O, Δ and # represent peaks of SDS, sulfapyridine-13C and a potential SDS metabolite in (b) and (c). (d) The calibration curves for absolute quantitation of sulfadiazine sodium using sulfapyridine-13C as the internal standard (n = 4). (e) Extrabacterial and (f) intrabacterial SDS residue detected by our method, after the treatment of bacteria for 20 h with different antibiotic dosages: 2 mg mL−1, 1 mg mL−1, 0.5 mg mL−1 light blue: E. coli; green: S. aureus. Data points were collected from 2 separate batches. Each of the batches contained a repetition of 4 in parallel. Error bar: standard deviation. | ||
Our design of hybridizing CuCoO nanoparticles on GO nanosheets is critical for the development of a high performance matrix. Indeed, GO plays an important role in this hybrid nanomaterial. GO nanosheets contain aromatic structures and chemical groups such as hydroxyl, epoxy, and carboxyl groups, enhancing molecular interactions with analytes and serving as a matrix choice.32,48 We designed experiments to compare the performance of CuO or CuCoO nanoparticles without GO, and the CuCoO–GO hybrid matrix in the detection of SDZ and SMX molecules. In contrast to the high efficiency of CuCoO–GO in the ionization of small molecules, CuO or CuCoO nanoparticles as the matrix can only produce very low signals for the detection of SDZ or SMZ. The difference in the signal intensity is nearly two orders of magnitude (Fig. S9†). This suggests that bare CuO or CuCoO nanoparticles without GO as the matrix are limited when promoting small molecule ionization. In addition, we compared the efficiency in laser desorption ionization (LDI) of the analytes using different types of graphene oxide-based matrices including GO, CuO–GO, CuCoO–GO under identical conditions. As shown in Fig. 4, both SDZ and SMX can be detected by using any of the above matrices. However, there appears to be prominent differences among these GO-based matrices in terms of the signal-to-noise (S/N) ratios for the detection of the antibiotic analytes. The CuCoO–GO matrix allows for the detection of either SDZ or SMX with the highest S/N ratio, which is approximately 2 fold higher than the CuO–GO matrix, or 8 fold higher than the bare GO matrix. The significantly enhanced S/N ratio by the CuCoO–GO matrix suggests a synergistic effect on LDI of the analytes, contributed to by the hybrid nanosystem of metal oxides and GO.
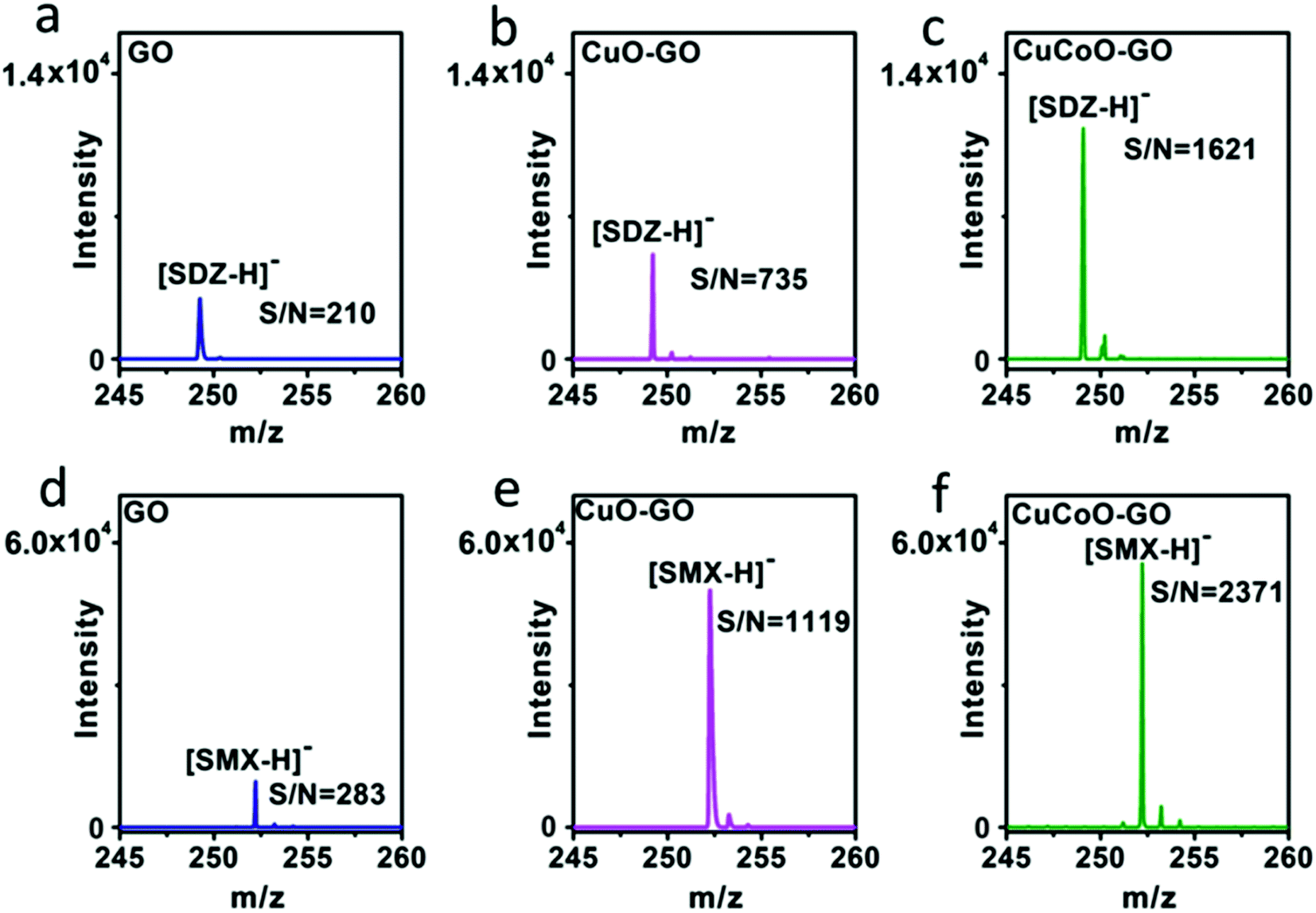 | ||
| Fig. 4 Comparison of performance of GO, CuO–GO and CuCoO–GO matrices in the MALDI-TOF MS detection of antibiotics. (a–c) SDZ; (d–f) SMX, under identical conditions (in the negative-ion mode, accumulated to 3000 shots, concentration: SDZ with 0.05 μg mL−1, SMX with 1 μg mL−1). | ||
Mechanistic studies by synchrotron radiation-based techniques
Synchrotron radiation-based X-ray absorption spectroscopy (XAS) has been widely used to reveal the chemical state information of elements in various materials.37,49,50 Here we employed this technique to explore the absorption changes of the specific elements in the CuCoO–GO matrix with/without small molecule analytes for mechanistic studies. The C K-edge XAS spectra (Fig. S10†) validate the interactions between SDZ molecules and CuCoO–GO nanosheets accompanying adsorption. The pristine sample of CuCoO–GO exhibits a XAS spectrum similar to typical GO-based materials as reported in the literature.37 The sample CuCoO–GO after addition of SDZ presents clear signals for its adsorption with the evidence the shifted peak A and the intensity increase in peak B. After the addition of the analyte (SDZ), there is no significant shift in the absorption peak of the Cu L-edge in CuO–GO (black, red curves in Fig. 5a and b). In contrast, the absorption peak of the Cu L-edge in CuCoO–GO is shifted to a higher energy level (ΔE ≈ 0.3 eV), suggesting a change for the higher chemical state of the Cu element (blue, purple curves in Fig. 5a and b). This change can be attributed to the interactions between SDZ and the Cu element, which induce an effect of partial electron shift from Cu to O in SDZ. Compared to CuO–GO, CuCoO–GO with Co doping may facilitate a delocalized electron effect by promoting interactions between CuCoO and GO. Furthermore, in the in situ Cu K-edge XAS experiments (Fig. 5c and d and S11†), addition of SDZ results in an intensity increase for the absorption peak of CuCoO–GO, corresponding to a chemical state change of Cu due to a partial electron shift to atoms with higher electronegativity such as the oxygen atom. These results are consistent with the previous measurements of Cu L-edge absorption in Fig. 5a and b. Interestingly, there is an inverse change for the absorption peak intensity at the Cu K-edge when the sample mixture of CuCoO–GO and SDZ is illuminatedin situ by UV light. This suggests recovery of the chemical state of copper, accompanying the removal of SDZ and a drop in the interactions between SDZ and Cu due to UV irradiation. | ||
| Fig. 5 Investigation on the interactions between SDZ and CuCoO–GO by synchrotron radiation-based techniques. (a) Hard X-ray XAS spectra of CuO–GO and CuCoO–GO with and without addition of SDZ. (b) Zoomed-in view of the peaks highlighted in the blue circles of (a). (c) In situ hard X-ray XAS spectra of CuCoO–GO at the Cu K-edge (d) Zoomed-in view of the peaks highlighted in the blue circle of (c). | ||
We propose a mechanism for the ionization of small molecules with the CuCoO–GO matrix, based on the above experimental evidence collectively (Fig. 6). The hybrid nanomaterial CuCoO–GO is able to ionize the small molecular analyte (SDZ) with high efficiency assisted by clustered Cu, Co, O atoms on the GO nanosheets. SDZ molecules can be adsorbed on the surface of CuCoO–GO by hydrogen bonds and van der Waals forces. The Cu and O sites on the matrix can respectively interact with the SO bond and the –NH– group in the SDZ molecule. The presence of Co as the doping element in the matrix enables enhanced interactions between the atom clusters and GO, which could promote charge transfer between the CuCoO–GO matrix and SDZ in the ionization process. The energy input by laser may facilitate the deprotonation of SDZ, allowing for the ionized small molecule (SDZ) to be detected by the mass analyzer in the MALTI-TOF MS instrument. The hybrid CuCoO–GO nanomaterial tends to exhibit a high proton affinity due to the delocalized pi-electron system across the GO surface. Due to the synergistic effect of the Co and Cu bimetallic oxides on the GO nanosheets, this hybrid matrix turns out to improve the ionization efficiency of small molecules, thus promoting their MS signal-to-noise ratios in detection.
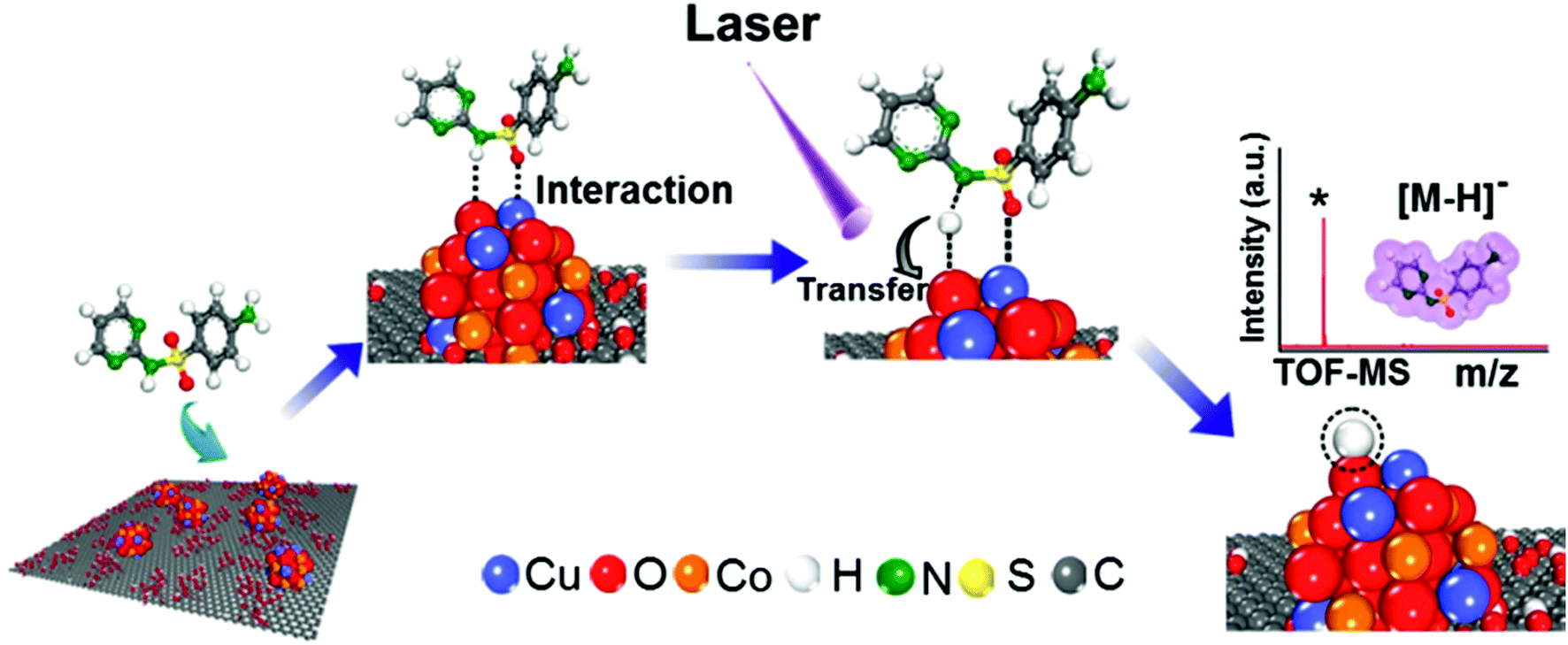 | ||
| Fig. 6 Schematic diagram of the mechanism of SDZ ionization using the CuCoO–GO matrix for MALDI TOF mass spectrometry. | ||
The proposed mechanism is suitable for the negative-ion mode in MALDI MS, by taking advantage of the enhanced interactions between small molecules and the CuCoO–GO matrix at the nano-interfaces. Distinctly different from the conventional matrices of organic molecules, the total atom number of the hybrid nanomaterial-based matrix is higher by several orders of magnitude. The nanomaterial-based matrix itself is resistant to desorption from the MALDI plate into the gas phase. Our experiments have also demonstrated that the CuCoO–GO matrix produces much fewer fragmented species (low-noise background, Fig. 2) in comparison to conventional matrices (CHCA, and SA) under irradiation with a UV laser. Therefore, the mechanism above mainly describes the dynamic interactions of matrix/small molecules at the condensed phase-gas interfaces. However, ion reactions may also occur in the gas phase by complicated in-plume processes, which are important scenarios for examining the complete picture of MALDI ionization.51–53 There are several representative types of secondary ion reactions such as proton-transfer reactions, cationization reactions, electron-transfer reactions, and electron-capture reactions.51,52,54
The gas-phase proton-transfer processes include matrix–matrix reactions and matrix–analyte reactions.49 As for matrix–matrix reactions,
| M+˙ + M → MH+ + (M − H)˙ | (1) |
| (M − H)˙ + e− → (M − H)− | (2) |
| M + e− → (M − H)− + H˙ | (3) |
As for matrix–analyte reactions,
| MH+ + A → M + AH+ | (4) |
| (M − H)− + A → M + (A − H)− | (5) |
As for the gas-phase cationization processes or the electron-transfer reactions such as:
| M+˙ + A → M + A+˙ | (6) |
| A + e− → (A − H)− + H˙ | (7) |
Herein we attempt to discuss several representative categories of secondary ion reactions. The actual processes may involve more complicated mechanisms of generating secondary ions. It is noteworthy that the distinctive properties of hybrid nanomaterials may foster mechanistic studies significantly different from conventional organic matrices. The current mechanism that we propose highlights the dynamic interactions between the hybrid nanomaterial and small molecules at the condensed phase-gas interfaces. A further detailed investigation on the mechanisms of primary and secondary ion generation will be necessary in the future.
In order to examine whether the proposed mechanism is limited by any particular chemical groups of the analytes, we have added three molecules, including sulfapyridine, sulfonamide, and pyrimethanil, in the experiments using the CuCoO–GO matrix. Sulfapyridine can be viewed as a derivative of SDZ, with a slight difference in the number of nitrogen atoms. The chemical structure of sulfonamide is similar to SDZ, except for the removal of the nitrogen-containing heterocyclic ring structure. Pyrimethanil also contains some structural characteristics of SDZ, but without the SO bond. The mass spectra of these molecules in comparison with SDZ have been updated in the ESI (Fig. S12†). The spectral data support that these three types of molecules can be efficiently ionized in MALDI TOF MS by using the CuCoO–GO matrix. In addition, our new matrix has been demonstrated to assist the ionization of different categories of antibiotics with an enhanced limit of detection. Several representative antibiotics such as tetracycline and norfloxacin are featured with other functional groups, primarily including CO, H2N–CO, HO–CO, and –OH, instead of SO and –NH–. Based on the experimental results, we propose that the mechanism of molecular interactions may be adapted to a relatively wide range of small molecules with different chemical groups. To the best of our knowledge, this is the first time investigation of the nanomaterial-assisted ionization mechanism of small molecules using synchrotron radiation-based techniques.
Conclusion
In summary, we have developed the hybrid CuCoO–GO as a new MALDI matrix for the sensitive detection of antibiotics, and revealed the molecule–nanomaterial interactions for the ionization mechanism. The CuCoO–GO matrix allows for the detection of various antibiotics with LOD improvement of several hundreds of fold compared with the literature reports. We have validated high reproducibility of this approach, and demonstrated its performance in the quantification of sulfonamides in milk samples. Our approach can provide quantitative analysis of extrabacterial and intrabacterial concentrations of antibiotics as well as their potential metabolites, which are difficult to obtain by conventional AST assays. A panel of commonly-used antibiotics and several additional chemical analogs have been successfully detected with our method. We expect that the CuCoO–GO matrix shall be able to allow sensitive detection of a wide range of biomolecules containing functional groups such as carboxyl, amine, amide, carbonyl, sulfonic, and hydroxyl groups. By taking advantage of synchrotron radiation-based techniques, we have investigated the interactions between the molecule SDZ and the CuCoO–GO matrix for the ionization mechanism of SDZ with the assistance of CuCoO–GO under UV irradiation. The pivotal understanding of the matrix/molecule interactions and the ionization mechanism may create knowledge for matrix development and the ultrasensitive detection of small molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Major State Basic Research Development Program (2013CB932702), and by the National Natural Science Foundation of China (21874096, 21575095); a project supported by the Collaborative Innovation Center of Suzhou Nano Science and Technology, and by the Joint International Research Laboratory of Carbon-Based Functional Materials and Devices; a 111 project; the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); J. L. is supported by the “1000 Youth Talents” plan of the Global Expert Recruitment Program.References
- S. Leclercq, F. M. Mian, A. M. Stanisz, L. B. Bindels, E. Cambier, H. Ben-Amram, O. Koren, P. Forsythe and J. Bienenstock, Nat. Commun., 2017, 8, 1–12 CrossRef.
- J. M. Mitchell, M. W. Griffiths, S. A. McEwen, W. B. McNab and A. J. Yee, J. Food Prot., 1998, 61, 742–756 CrossRef CAS.
- X. Z. Li, P. Plesiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef.
- R. Laxminarayan, A. Duse, C. Wattal, A. K. M. Zaidi, H. F. L. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057–1098 CrossRef.
- J. M. C. Cernadas, Arch. Argent. Pediatr., 2017, 115, 306–307 Search PubMed.
- A. M. Saviano, F. L. Francisco and F. R. Lourenco, Talanta, 2014, 127, 225–229 CrossRef CAS.
- B. Shaikh and W. A. Moats, J. Chromatogr. A, 1993, 643, 369–378 CrossRef CAS.
- M. Dubois, D. Fluchard, E. Sior and P. Delahaut, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 753, 189–202 CrossRef CAS.
- B. Blanchaert, E. P. Jorge, P. Jankovics, E. Adams and A. Van Schepdael, Chromatographia, 2013, 76, 1505–1512 CrossRef CAS.
- I. K. Cigic and H. Prosen, Int. J. Mol. Sci., 2009, 10, 62–115 CrossRef CAS.
- I. Y. Goryacheva, S. De Saeger, I. S. Nesterenko, S. A. Eremin and C. Van Peteghem, Talanta, 2007, 72, 1230–1234 CrossRef CAS.
- H. Y. Zhang and S. O. Wang, J. Immunol. Methods, 2009, 350, 1–13 CrossRef CAS.
- S. Ahmed, J. Ning, G. Y. Cheng, I. Ahmad, J. Li, M. Y. Liu, W. Qu, M. Iqbal, M. A. B. Shabbir and Z. H. Yuan, Talanta, 2017, 166, 176–186 CrossRef CAS.
- B. F. Cravatt, G. M. Simon and J. R. Yates, Nature, 2007, 450, 991–1000 CrossRef CAS.
- Y. C. Ling, L. N. Lin and Y. T. Chen, Rapid Commun. Mass Spectrom., 1998, 12, 317–327 CrossRef CAS.
- L. S. Cai, L. F. Sheng, M. C. Xia, Z. P. Li, S. C. Zhang, X. R. Zhang and H. Y. Chen, J. Am. Soc. Mass Spectrom., 2017, 28, 399–408 CrossRef CAS.
- R. Shi, H. Li, E. H. Wu, L. P. Xiong, R. Lv, R. C. Guo, Y. Liu, G. Q. Xu, Z. H. Kang and J. Liu, Nanoscale, 2017, 9, 8410–8417 RSC.
- J. J. Hu, F. Liu and H. X. Ju, Angew. Chem., Int. Ed., 2016, 55, 6667–6670 CrossRef CAS.
- D. Figeys, L. D. McBroom and M. F. Moran, Methods, 2001, 24, 230–239 CrossRef CAS.
- Y. M. Zhao, T. W. Muir, S. B. H. Kent, E. Tischer, J. M. Scardina and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4020–4024 CrossRef CAS.
- S. A. Stopka, C. Rong, A. R. Korte, S. Yadavilli, J. Nazarian, T. T. Razunguzwa, N. J. Morris and A. Vertes, Angew. Chem., Int. Ed., 2016, 55, 4482–4486 CrossRef CAS.
- A. N. Krutchinsky and B. T. Chait, J. Am. Soc. Mass Spectrom., 2002, 13, 129–134 CrossRef CAS.
- T. W. Jaskolla, W. D. Lehmann and M. Karas, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12200–12205 CrossRef CAS.
- R. Chen, S. M. Chen, C. Q. Xiong, X. L. Ding, C. C. Wu, H. C. Chang, S. X. Xiong and Z. X. Nie, J. Am. Soc. Mass Spectrom., 2012, 23, 1454–1460 CrossRef CAS.
- E. Gemperline, S. Rawson and L. J. Li, Anal. Chem., 2014, 86, 10030–10035 CrossRef CAS.
- W. T. Chen, C. K. Chiang, Y. W. Lin and H. T. Chang, J. Am. Soc. Mass Spectrom., 2010, 21, 864–867 CrossRef CAS.
- T. R. Northen, O. Yanes, M. T. Northen, D. Marrinucci, W. Uritboonthai, J. Apon, S. L. Golledge, A. Nordstrom and G. Siuzdak, Nature, 2007, 449, 1033 CrossRef CAS PubMed.
- L. Huang, J. J. Wan, X. Wei, Y. Liu, J. Y. Huang, X. M. Sun, R. Zhang, D. D. Gurav, V. Vedarethinam, Y. Li, R. P. Chen and K. Qian, Nat. Commun., 2017, 8, 222 CrossRef.
- Y. Gholipour, S. L. Giudicessi, H. Nonami and R. Erra-Balsells, Anal. Chem., 2010, 82, 5518–5526 CrossRef CAS.
- R. Shi, X. Dai, W. F. Li, F. Lu, Y. Liu, H. H. Qu, H. Li, Q. Y. Chen, H. Tian, E. H. Wu, Y. Wang, R. H. Zhou, S. T. Lee, Y. Lifshitz, Z. H. Kang and J. Liu, ACS Nano, 2017, 11, 9500–9513 CrossRef CAS.
- S. M. Chen, H. Z. Zheng, J. N. Wang, J. Hou, Q. He, H. H. Liu, C. Q. Xiong, X. L. Kong and Z. X. Nie, Anal. Chem., 2013, 85, 6646–6652 CrossRef CAS.
- Y. K. Kim, H. K. Na, S. J. Kwack, S. R. Ryoo, Y. Lee, S. Hong, S. Hong, Y. Jeong and D. H. Min, ACS Nano, 2011, 5, 4550–4561 CrossRef CAS.
- B. Gulbakan, E. Yasun, M. I. Shukoor, Z. Zhu, M. X. You, X. H. Tan, H. Sanchez, D. H. Powell, H. J. Dai and W. H. Tan, J. Am. Chem. Soc., 2010, 132, 17408–17410 CrossRef CAS.
- S. M. Chen, C. Q. Xiong, H. H. Liu, Q. Q. Wan, J. Hou, Q. He, A. Badu-Tawiah and Z. X. Nie, Nat. Nanotechnol., 2015, 10, 176–182 CrossRef CAS.
- M. S. Khan, M. L. Bhaisare, S. Pandey, A. Talib, S. M. Wu, S. K. Kailasa and H. F. Wu, Int. J. Mass Spectrom., 2015, 393, 25–33 CrossRef.
- W. J. Lu, Y. Li, R. J. Li, S. M. Shuang, C. Dong and Z. W. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 12976–12984 CrossRef CAS.
- K. Feng, J. Zhong, B. H. Zhao, H. Zhang, L. Xu, X. H. Sun and S. T. Lee, Angew. Chem., Int. Ed., 2016, 55, 11950–11954 CrossRef CAS.
- I. P. Smirnov, X. Zhu, T. Taylor, Y. Huang, P. Ross, I. A. Papayanopoulos, S. A. Martin and D. J. Pappin, Anal. Chem., 2004, 76, 2958–2965 CrossRef CAS.
- R. F. Gao, X. Chen, C. T. Chen, R. Shi, F. Z. Ouyang, J. Z. Yang, D. P. Sun and J. Liu, Cellulose, 2016, 23, 2475–2485 CrossRef CAS.
- J. Y. Liu, Y. Liu, M. X. Gao and X. M. Zhang, J. Am. Soc. Mass Spectrom., 2012, 23, 1424–1427 CrossRef CAS.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef CAS.
- J. I. Kim, J. M. Park, J. Y. Noh, S. J. Hwang, M. J. Kang and J. C. Pyun, Chemosphere, 2016, 143, 64–70 CrossRef CAS.
- V. Springer, J. Jacksen, P. Ek, A. G. Lista and A. Emmer, Chromatographia, 2015, 78, 285–290 CrossRef CAS.
- S. Notari, C. Mancone, M. Sergi, F. Gullotta, N. Bevilacqua, M. Tempestilli, R. Urso, F. N. Lauria, L. P. Pucillo, M. Tripodi and P. Ascenzi, IUBMB Life, 2010, 62, 387–393 CAS.
- J. J. A. van Kampen, P. C. Burgers, R. de Groot and T. M. Luider, Anal. Chem., 2006, 78, 5403–5411 CrossRef CAS PubMed.
- Y. D. Zhu, N. Gasilova, M. Jovic, L. Qiao, B. H. Liu, L. T. Lovey, H. Pick and H. H. Girault, Chem. Sci., 2018, 9, 2212–2221 RSC.
- R. T. Zhang, Q. Qin, B. H. Liu and L. Qiao, Anal. Chem., 2018, 90, 3863–3870 CrossRef CAS PubMed.
- Q. Liu, M. T. Cheng, J. Wang and G. B. Jiang, Chem.–Eur. J., 2015, 21, 5594–5599 CrossRef CAS PubMed.
- K. Ghanbari, A. S. Elahi and M. Ghoranneviss, J. X-Ray Sci. Technol., 2017, 25, 15–23 CAS.
- L. Chen, Y. Mi, X. Wan, B. Yin, Z. Yuan, L. He and Q. Li, Appl. Ecol. Env. Res., 2017, 15, 335–341 CrossRef.
- R. Zenobi and R. Knochenmuss, Mass Spectrom. Rev., 1998, 17, 337–366 CrossRef CAS.
- R. Knochenmuss and R. Zenobi, Chem. Rev., 2003, 103, 441–452 CrossRef CAS.
- A. G. Harrison, Mass Spectrom. Rev., 1997, 16, 201–217 CrossRef CAS.
- H. Ehring, M. Karas and F. Hillenkamp, Org. Mass Spectrom., 1992, 27, 472–480 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03692f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |