
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHetero-metallic active sites coupled with strongly reductive polyoxometalate for selective photocatalytic CO2-to-CH4 conversion in water†
Shuai-Lei
Xie
a,
Jiang
Liu
*b,
Long-Zhang
Dong
 b,
Shun-Li
Li
b,
Ya-Qian
Lan
*b and
Zhong-Min
Su
*ac
b,
Shun-Li
Li
b,
Ya-Qian
Lan
*b and
Zhong-Min
Su
*ac
aInstitute of Functional Material Chemistry, Department of Chemistry, National & Local United Engineering Lab for Power Battery, Northeast Normal University, Changchun 130024, P. R. China. E-mail: zmsu@nenu.edu.cn
bSchool of Chemistry and Materials Science, Jiangsu Key Laboratory of Biofunctional Materials, Nanjing Normal University, Nanjing 210023, P. R. China. E-mail: liuj@njnu.edu.cn; yqlan@njnu.edu.cn
cSchool of Chemistry and Environmental Engineering, The Collaborative Innovation Center of Optical Materials and Chemistry, CUST, Changchun University of Science and Technology, Changchun 130028, P. R. China
First published on 2nd October 2018
Abstract
The photocatalytic reduction of CO2 to value-added methane (CH4) has been a promising strategy for sustainable energy development, but it is challenging to trigger this reaction because of its necessary eight-electron transfer process. In this work, an efficient photocatalytic CO2-to-CH4 reduction reaction was achieved for the first time in aqueous solution by using two crystalline heterogeneous catalysts, H{[Na2K4Mn4(PO4) (H2O)4]⊂{[Mo6O12(OH)3(HPO4)3(PO4)]4[Mn6(H2O)4]}·16H2O (NENU-605) and H{[Na6CoMn3(PO4)(H2O)4]⊂{[Mo6O12(OH)3(HPO4)3(PO4)]4[Co1.5Mn4.5]}·21H2O (NENU-606). Both compounds have similar host inorganic polyoxometalate (POM) structures constructed with strong reductive {P4Mo6V} units, homo/hetero transition metal ions (MnII/CoIIMnII) and alkali metal ions (K+ and/or Na+). It is noted that the {P4Mo6V} cluster including the six MoV atoms served as a multi-electron donor in the case of a photocatalytic reaction, while the transition metal ions functioned as catalytically active sites for adsorbing and activating CO2 molecules. Additionally, the presence of alkali metal ions was believed to assist in the capture of more CO2 for the photocatalytic reaction. The synergistic combination of the above-mentioned components in NENU-605 and NENU-606 effectively facilitates the accomplishment of the required eight-electron transfer process for CH4 evolution. Furthermore, NENU-606 containing hetero-metallic active sites finally exhibited higher CH4 generation selectivity (85.5%) than NENU-605 (76.6%).
Introduction
The massive discharge of CO2 resulting from the combustion of limited fossil fuels is leading to global warming, which has driven people to find effective ways to alleviate this crisis.1–4 The photocatalytic reduction of CO2 to valuable hydrocarbon fuels, such as methane (CH4), provides a promising ‘one stone two birds’ approach to realize the purpose of eliminating energy shortage and environmental challenges.5,6 Nevertheless, the accomplishment of this catalytic reaction means that chemically inert CO2 molecules adsorbed on the catalyst have to undergo a proton-assisted multi-electron transfer process; it is a rather challenging task for catalyst design because of the uncontrollable multi-electron supply and complicated photocatalytic reaction mechanism.7,8 Moreover, the heterogeneous nature of the catalyst in principle is also indispensable in view of its practical application.7,9–17 So far, the studies on heterogeneous catalysts, especially those applied in the photocatalytic reduction of CO2 to CH4, mainly focus on nano-sized semiconducting materials or composites18–21 because of their rich interfacial modification and structural stability that are beneficial for the photocatalytic CO2 reduction reaction (CO2RR) towards high activity and good photocatalytic durability.22–27 It is well-known that these inorganic nanomaterials are stable, but their indistinct active sites and even their electron donors are commonly difficult for the explanation of the photocatalytic reaction mechanism.28 Also, the introduction of noble metal co-catalysts or heterojunctions sometimes would raise the cost and/or cover the surface active sites to obstruct incident light absorption, which results in reduced photo-conversion efficiency. Consequently, the construction of crystalline inorganic heterogeneous catalysts with well-defined active sites and multi-electron sources would be a better choice for obtaining multi-electron reductive hydrocarbon fuels and investigating the mechanism of the photocatalytic CO2RR.29Polyoxometalate (POM) with inherent redox and semiconductor features is one kind of inorganic crystalline material, which has been surveyed extensively in different catalytic reaction types.30–35 A lot of classic POM units or POM-based derivatives often exhibit favourable responses to specific catalytic reactions, where the relevant active sites and electron transfer of the catalytic reaction can be explained by their crystal structures.36–40 However, the dissolvability of traditional POM clusters has been a major limiting factor for catalytic durability and needs to be primarily considered. So, the structural stability is the prerequisite for POM chemistry to be applied in any heterogeneous catalysis. As far as we know, high dimensional POM-containing inorganic architecture compared with the POM monomer usually shows good structural insolubility and an extended solar spectrum absorption range, which have been investigated in many photo-stimulated catalytic reactions such as water splitting, organic degradation, etc. In these reactions, the fast and reversible multi-electron transfer character of the POM subunit plays an extremely important role in promoting photocatalytic performance.41–44 In this context, a POM-assembled inorganic complex is also believed to have advantages in the field of the heterogeneous photocatalytic CO2RR. In particular, if the POM component within the structure can offer the CO2 molecule sufficient electrons, then multi-electron oriented reductive products would be achievable.
Herein, we report two stable POM-containing inorganic compounds, H{[Na2K4Mn4(PO4) (H2O)4]⊂{[Mo6O12(OH)3(HPO4)3(PO4)]4[Mn6(H2O)4]}·16H2O (NENU-605) and H{[Na6CoMn3(PO4)(H2O)4]⊂{[Mo6O12(OH)3(HPO4)3(PO4)]4[Co1.5Mn4.5]}·21H2O (NENU-606), which have very similar host skeletons but different catalytically active species. It is noted that assembling {P4Mo6V} ([Mo6O12(OH)3(HPO4)3(PO4)]6−) units with strong reducibility into the structures endows NENU-605 and NENU-606 with efficient heterogeneous photocatalytic CO2-to-CH4 reduction ability in water. To our knowledge, this is the first report of {P4Mo6V}-based crystalline inorganic materials applied in the photocatalytic CO2RR. Photocatalytic analysis revealed that the synergistic combination of strong reductive {P4Mo6V} units (donating electrons) and the first-row transition metal active centres (MnII/CoIIMnII) in NENU-605 and NENU-606 effectively boosts the necessary eight-electron reduction process for CH4 evolution. Furthermore, the hetero-metallic active sites of NENU-606 finally exhibited a higher CH4 generation selectivity (85.5%) in the photocatalytic CO2RR than those of NENU-605 (76.6%). Besides, the coordination effect of alkali metal ions can also assist the accomplishment of the photocatalytic reaction by influencing the CO2 adsorption ability of the title compounds. At the same time, the crystalline and heterogeneous nature of these stable inorganic POM-included compounds also provides some insight into the photocatalytic CO2RR mechanism.
Results and discussion
NENU-605 and NENU-606 were prepared by a hydrothermal synthesis method with a slight difference in the use of metal salts and organic templates, and they can be clearly distinguished by the colour and shape of the crystals (Fig. S1†). Single crystal X-ray diffraction analysis reveals that NENU-605 and NENU-606 crystallize in the same I41/acd tetragonal space group and have very similar pure inorganic matrices composed of three kinds of independent MnII/CoII–MnII ions, Na+–K+/Na+ ions, {P4Mo6V} polyanions, phosphate anions and water molecules (Fig. S2†). The overall three-dimensional (3D) architecture of these two compounds is assembled with nesting subunits by using PO43− and O2− bridges. Such a nested substructure in NENU-605 can be described as a three-shell assembly where the PO43− group is the innermost shell (Fig. 1a). Four μ2-O atoms of PO43− connect with four six-coordinated Mn1 and four K1 atoms, and these Mn1 and K1 atoms establish two types of metal-based tetrahedrons respectively. The interpenetrating coupling of two tetrahedra forms a twisted hexahedron whose diagonal-linked vertexes are occupied by Mn1 or K1 atoms. Meanwhile, the top and bottom surfaces of this hexahedron are capped by two Na atoms to build the shuttle-shaped second shell (Fig. 1b). Through the 16PO43− and 8O2− groups, the second shell further communicates with the outer four Mn2 atoms and four {Mo6V} rings. Each Mn2 atom is coordinated by five O atoms from four PO43− anions and one O atom from the coordinated water molecule (Fig. S3†) and is surrounded by four {Mo6V} rings made of six edge-sharing {MoVO6} octahedrons. It is noted that every {Mo6V} ring supported by four PO43− anions constitutes a strong reductive component, P4Mo6V,45–49 which includes six MoV atoms (Fig. S4†). Interestingly, these Mn2 atoms and {Mo6V} rings can construct peripherally larger distorted hexahedrons in the same interpenetrating way as the second shell; the diagonal-linked vertexes are occupied by Mn2 atoms or {Mo6V} rings that can be treated as the third shell (Fig. 1c). In such a way, a three-shell nested substructure is formed (Fig. 1d). To better further understand the evolution of this nested subunit, it has been simplified as a distorted hexahedral entity whose eight vertexes are taken up by Mn2 atoms and {Mo6V} rings, as shown in Fig. 2a and b. These Mn2 atoms and {Mo6V} rings serve as nodes to come into contact with the eight adjacent subunits in Mn2-to-Mn2 and {Mo6V}–Mn3–{Mo6V} modes (Fig. 2c), which finally gives rise to the 3D inorganic structure of NENU-605 (Fig. 2d). The linkage of Mn2-to-Mn2 is achieved by two μ2-O atoms from two PO43−, while every six-coordinated Mn3 atom is shared by two {Mo6V} rings from two neighboring subunits (Fig. S5†). For NENU-606, the coordination environment of the Mn2 atom is crooked tetragonal pyramid structure consisting of five O atoms from four PO43− groups (Fig. S6†); moreover the K1 atoms are fully replaced by Na atoms (Fig. S7†). The most important difference between these two compounds is that the original Mn atom sites in the lattice of NENU-605 are partially substituted by Co atoms, and the doping ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) of Co/Mn in NENU-606 is determined by an Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES). Besides, if the nested substructure and the Mn2/Mn3 atoms are regarded as 8-connected nodes and linkers, respectively, then the skeletons of NENU-605 and NENU-606 feature a unimodal topology with the Schläfli symbol of 424·64 (Fig. S8†).
:3) of Co/Mn in NENU-606 is determined by an Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES). Besides, if the nested substructure and the Mn2/Mn3 atoms are regarded as 8-connected nodes and linkers, respectively, then the skeletons of NENU-605 and NENU-606 feature a unimodal topology with the Schläfli symbol of 424·64 (Fig. S8†).
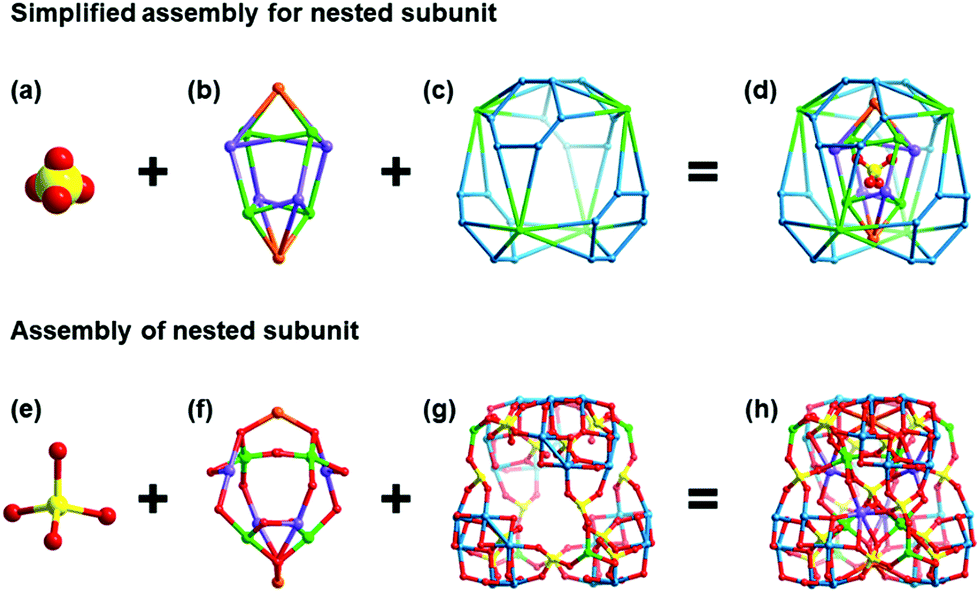 | ||
| Fig. 1 Simplified (a–d) and real (e and h) three-shell nested subunit for NENU-605. (a, e) The innermost shell (central PO43− group); (b, f) the shuttle-shaped second shell ({Na2K4Mn4}); (c, g) the cage-like outermost shell (Mn4({P4Mo6})4); (d, h) the overall three-shell nested substructure. Colour code: P yellow, O red, Na orange, K light purple, Mn green, and Mo light blue. | ||
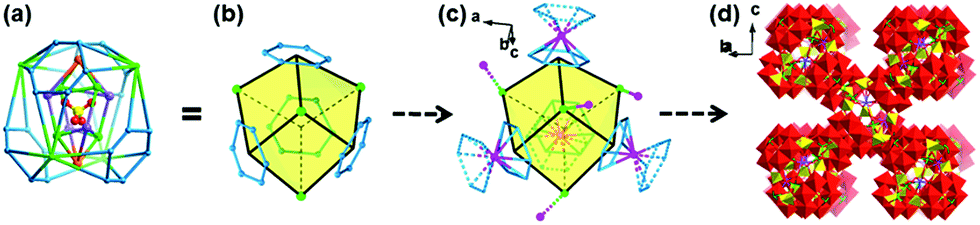 | ||
| Fig. 2 Graphical growth of the 3D architecture of NENU-605: (a) simplified three-shell nested substructure; (b) further simplified nested substructure as a distorted hexahedral entity (Mn2, green; {Mo6V} ring, light blue); (c) nested substructure with an 8-connected node in contact with the eight adjacent subunits (Mn2/Mn3, pink; {Mo6V} ring, dashed light blue); (d) real 3D inorganic structure of NENU-605. | ||
The phase purity and thermal stability of NENU-605 and NENU-606 were demonstrated using well-matched powder X-ray diffraction (PXRD) patterns and thermogravimetric analysis, respectively (Fig. S9 and S10†). As shown in Fig. S9a and b,† these two compounds can also remain stable when being soaked in aqueous solutions at different pH values for several days, which indicated that their structures have strong acid and alkali resistance.50 Besides, in order to confirm the heterogeneous catalytic nature, the structural stability of the title compounds was tested again under the conventional conditions of the photocatalytic CO2RR. It is obvious that the PXRD patterns of all the treated crystals remain intact, indicating that no phase transition or structural collapse occurred. A broad UV-vis absorption range of 200–600 nm for NENU-605 and NENU-606 revealed that they indeed have better light-harvesting ability than a single POM cluster, whose absorption mainly focuses on the ultraviolet region (200–400 nm).51–53 Based on this, the band gaps of 3.20 (NENU-605) and 2.57 eV (NENU-606) were evaluated by the Kubelka–Munk (KM) method (Fig. S11†), unveiling the potential for these two compounds to be used as semiconducting photocatalysts. At the same time, Mott–Schottky measurements at frequencies of 1000, 1500, and 2000 Hz were used to determine the LUMO positions of NENU-605 and NENU-606 such that the occurrence of the photocatalytic CO2RR and relevant reductive products can be simply inferred (Fig. S12 and S13†). As we can see, the LUMO locations of compounds are more negative than the reduction potentials required for producing CO (−0.53 V vs. NHE) and CH4 (−0.24 V vs. NHE), indicating that the electrons can be transferred to the CO2 molecule for further reduction.
Taking the above features of NENU-605 and NENU-606 into consideration, the photocatalytic CO2RR was conducted under a pure CO2 (1.0 atm, 293 K) atmosphere in an aqueous solution with triethanolamine (TEOA) as a sacrificial agent (TEOA/H2O = 2:28 mL, pH ≈ 10.5). In addition, [Ru(bpy)3]Cl2·6H2O (0.01 mmol) as an auxiliary photosensitizer (PS) was added into the reaction system for increased visible-light absorption.54 Because of the matched LUMO positions between the PS and catalysts (Fig. S14 and S15†), photo-generated electrons were allowed to migrate from the PS to the catalysts. During the whole photoreduction process, gaseous CH4 and CO were the main reaction products detected by gas chromatography, while trace amounts of HCOOH were produced in the aqueous solution as detected by ion chromatography. Moreover, no competitive H2 was produced during the whole reaction (Fig. S16†). With the increasing irradiation time, the yields of CO and CH4 increase simultaneously at different reaction rates (Fig. 3a and b); the amount of CH4 for NENU-605 reached up to 170 nmol (i.e., 894.7 nmol g−1 h−1) after 19 h. In contrast, the maximum production of CH4 achieved for NENU-606 was 402 nmol (i.e., 1747.8 nmol g−1 h−1) after 23 h (Fig. 3c). Moreover, they finally exhibit a very high selectivity (CH4 over CO) of 76.6% (NENU-605) and 85.5% (NENU-606) (Fig. 3c). It is significant that this is the first report of heterogeneous POM-based catalysts applied in the photocatalytic CO2RR that exhibited such a high selectivity towards CH4, although the corresponding CH4 outputs are still very low and need to be greatly improved. The CO amounts determined after the reaction were 267.0 nmol g−1 h−1 (NENU-605) and 295.7 nmol g−1 h−1 (NENU-606), and the relevant parameters including the TONs and TOFs of these photocatalytic systems were summarized in Table S1.†NENU-606 obviously has higher photocatalytic CH4 selectivity than NENU-605, which can be further proved by their distinguishing transient photocurrent responses and electrochemical impedance spectra (Fig. S17†). The higher photocurrent response and smaller size of the Nyquist plot of NENU-606 represented its better separation efficiency of photo-induced electron–hole pairs and faster interfacial charge transfer process compared to NENU-605. Such differences in the charge separation and the kinetics of charge transfer probably resulted from the different active sites in NENU-605 (homometallic MnII ions) and NENU-606 (heterometallic MnII/CoII ions). Additionally, a series of reference experiments were carried out to explore the importance of each component in the photocatalytic reaction system, and the results are summed up in Table S1.† As we can see, in the absence of POM-based catalysts, TEOA, PSs, CO2 or light illumination, no detectable products were observed in the reaction system. The photocatalytic durability of the compounds was tested to confirm their heterogeneous nature. From the time course plots of CH4 evolution, the POM-containing catalysts maintain almost unchanged activities even after three cycles (Fig. S18†). The slight decline in the CH4 evolution activity in subsequent runs is probably related to the small amount of mass loss of the samples used in the recovery process. Furthermore, there was no noticeable alteration in the PXRD patterns and IR and X-ray photoelectron (XPS) spectra obtained before and after the recycling experiments of the photocatalytic reaction that evidenced the structural robustness of NENU-605 and NENU-606 again (Fig. S9c, d, S19, S20 and S21†).
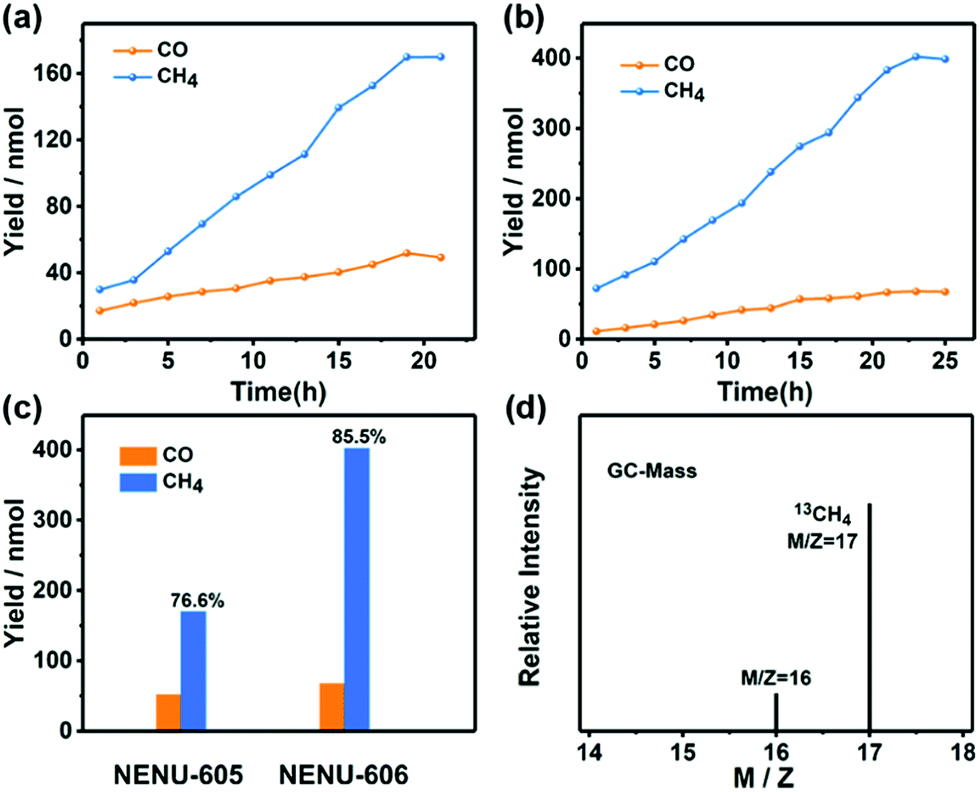 | ||
| Fig. 3 Amounts of CH4 and CO produced as a function of the time of visible-light irradiation using (a) NENU-605 and (b) NENU-606; (c) total product yield and the selectivity of gaseous products in the photocatalytic CO2RR; (d) the mass spectra of 13CH4 recorded under a 13CO2 atmosphere. The reaction with the catalyst (10 mg) in the H2O/TEOA (14:1 v/v, 30 mL) solution irradiated using a Xe lamp filtered to produce light in the range of 420–800 nm. | ||
To exclude the influence of possible active component decomposition on the photocatalytic activity, the reaction solution was filtrated after irradiating 11 hours and then the filtrate was detected. The fact that the generation of CH4 and CO would stop if the catalysts were removed from the reaction system clearly suggests that the photocatalytic activity comes from the catalysts themselves (Fig. S22†). Meanwhile, only trace amounts (NENU-605, 0.67%; NENU-606, 0.81%) of metal residue in the filtrate after the photocatalytic reaction were detected by ICP-AES. All these results pointed out that both NENU-605 and NENU-606 possess good photocatalytic durability towards the CO2RR under visible-light irradiation. To further validate the source of the produced CO and CH4, an isotope experiment using 13CO2 as the substrate was performed, and then the associated products were analysed by gas chromatography and mass spectrometry.55 As shown in Fig. S23† and 3d, the peaks at m/z = 29 and m/z = 17 were assigned to 13CO and 13CH4, respectively, providing solid proof that NENU-605 and NENU-606 are indeed active and capable of selectively converting CO2 to CH4 under visible-light irradiation.
To disclose the origin and difference of the photocatalytic performances of NENU-605 and NENU-606 in the CO2RR, the role of the {Mn[Mo6O12(OH)3(HPO4)3(PO4)]2} (abbr.{Mn(P4Mo6)2}) cluster (NENU-607) (Fig. S24–26†) was mainly considered in our system. NENU-607 is a dimer, with one {MnII} atom sandwiched between two {P4Mo6} units, displaying a similar connection mode to that of NENU-605 and NENU-606. When NENU-607 was synthesized and used as a catalyst applied under identical conditions of the photocatalytic reaction, only a small amount of CO (47 nmol) and CH4 (70 nmol) were detected, which demonstrated that the light-induced CO2 reduction process more likely occurred on the active MnII ions (NENU-605) or MnII/CoII ions (NENU-606). The strongly reductive P4Mo6V cluster including six MoV atoms in the case of redox reactions can theoretically offer multiple electrons for the CO2RR. Because each Lewis acid metal active site in title compounds is surrounded by four P4Mo6V units, the achieved photo-stimulated CO2-to-CH4 conversion seems to be understandable. As for the higher CH4 selectivity of NENU-606 than NENU-605, we speculated that the interaction between hetero-metallic MnII/CoII ions was more beneficial than that between homo-metallic MnII ions in terms of adsorption and activation of CO2 molecules. Besides, the reaction medium was also found to be an important factor in influencing the catalytic result. When the aqueous solution was replaced by dry MeCN (entry 9, Table S1†), no noticeable reduction products can be detected in the photocatalytic system. Additionally, the generation of reduction products relies on the participation of H2O as the solvent, as demonstrated in Fig. S27.† Moreover, the further increase of reduction products is also related to the increased amount of H2O. All the above cases have suggested the importance of H2O as the proton donor for the CO2-to-CH4 reduction reaction.
Based on the analysis of related experimental results, a speculative reaction mechanism with respect to the photocatalytic CO2-to-CH4 conversion using these two crystalline POM-containing compounds, as well as possible photo-generated electron transport pathways, was proposed (Fig. 4). First, the photosensitizer in the system absorbs sunlight to generate photo-excited electrons from the HOMO and then transports them to the catalyst through the matched LUMO positions; TEOA as the sacrificial reagent consumes the electron holes produced in the valence band. Second, strongly reductive {P4Mo6V} units enrich and offer electrons to the active metal centre under the stimulation of the photo-induced redox reaction. Third, the adsorbed CO2 molecule obtains electrons from the active metal sites, with the assistance of H2O as a proton donor, eventually undergoing the multi-electron transfer process of CO2-to-CH4 reduction. In addition, because the low local concentration of CO2 around the typical catalysts can make the reaction suffer from slow kinetics,56,57 the coordination of alkali metal cations in NENU-605 and NENU-606 architectures is helpful to effectively physically adsorb CO2 molecules through non-covalent interactions,58,59 which could lower the overpotential and Gibbs free energy ΔG of the chemical reduction of CO2.60–62
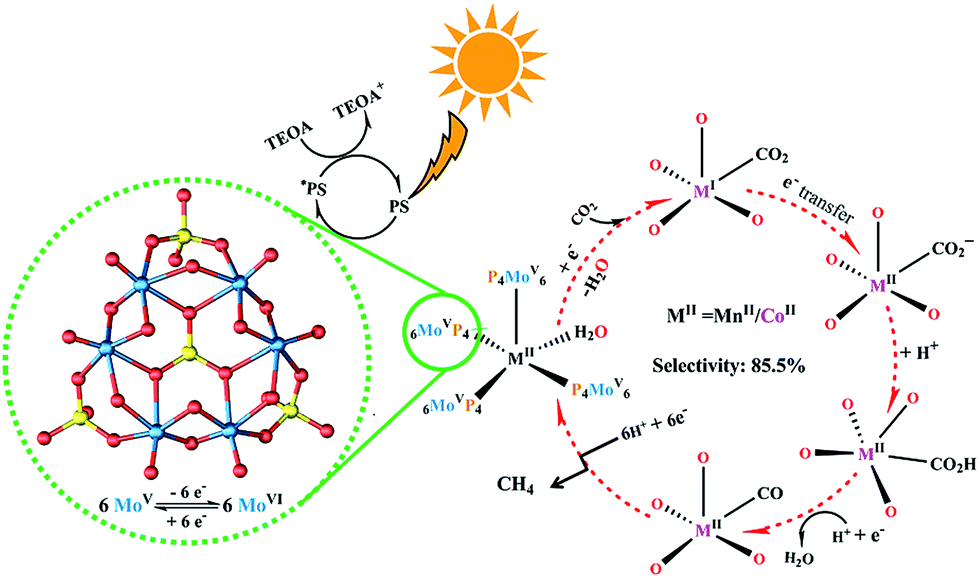 | ||
| Fig. 4 Proposed mechanism for the photocatalytic reduction of CO2 to CH4 using NENU-605 and NENU-606. | ||
Conclusions
In summary, this is the first report of inorganic POM-containing crystalline materials as heterogeneous catalysts applied in the photocatalytic CO2RR, and significant CO2-to-CH4 conversion in the aqueous phase is realized. Both NENU-605 and NENU-606 have almost identical 3D host structures composed of three-shell nested substructures, which were further composed of {P4Mo6} clusters, first-row transition metals and alkali metals. Because of the strong reducibility and electron-enrichment of the {P4Mo6} unit, sufficient electrons can be transported to active metal sites under the effect of the photo-stimulated redox reaction for the activation and reduction of CO2 molecules. In virtue of the synergistic coupling of structural components, these compounds finally exhibit high photocatalytic CH4 selectivity (76.6%, NENU-605; 85.5%, NENU-606). Especially for NENU-606 with hetero-metallic active sites, the interaction between MnII and CoII ions is found to be more efficient for the photocatalytic CO2RR than that between the homo-metallic MnII ions in NENU-605. Notably, the introduction of the {P4Mo6} building block not only endows NENU-605 and NENU-606 with favorable structural rigidity, but also, indeed, facilitates the accomplishment of the eight-electron transfer process of CH4 formation by delivering sufficient electrons. We anticipate that such a feasible strategy, embedding strongly reductive components into the visible-light sensitized catalyst architecture, could inspire more enthusiasm to construct stable inorganic networks for the effective reduction of CO2 to other high-valued hydrocarbons and further enhance their photocatalytic activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by NSFC (No. 21622104, 21471080, 21701085 and SBK2017040708), the NSF of Jiangsu Province of China (No. BK20171032), the Natural Science Research of Jiangsu Higher Education Institutions of China (No. 17KJB150025), Priority Academic Program Development of Jiangsu Higher Education Institutions and the Foundation of Jiangsu Collaborative Innovation Center of Biomedical Functional Materials.Notes and references
- C. Steinlechner and H. Junge, Angew. Chem., Int. Ed., 2018, 57, 44–45 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- A. Rubino, Nat. Energy, 2018, 3, 255–256 CrossRef CAS.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935–7947 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., 2014, 126, 1052–1056 CrossRef.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., 2016, 128, 14522–14526 CrossRef.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS.
- Y. Wang, N.-Y. Huang, J.-Q. Shen, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- W. Min, C. Lingjing, L. Tai-Chu and R. Marc, Angew. Chem., Int. Ed., 2018, 57, 7769–7773 CrossRef.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., 2017, 129, 4945–4949 CrossRef.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- Y. Fang and X. Wang, Chem. Commun., 2018, 54, 5674–5687 RSC.
- G. Chao, M. Qiangqiang, Z. Kun, Y. Huajie, W. Dawei, G. Jun, Z. Shenlong, C. Lin, H. Meng, L. Qunxiang, Z. Huijun, H. Xingjiu, G. Yan and T. Zhiyong, Adv. Mater., 2016, 28, 6485–6490 CrossRef PubMed.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624–1704632 CrossRef.
- S. Yongfu, W. Ju, L. Xiaodong, S. Wen, L. Peiquan, J. Xingchen, G. Shan, L. Liang, X. Jiaqi, Y. Wensheng, W. Chengming and X. Yi, Angew. Chem., 2018, 130, 8855–8859 CrossRef.
- S. Wang, B. Y. Guan, Y. Lu and X. W. D. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, J. Am. Chem. Soc., 2018, 140, 6648–6655 CrossRef CAS PubMed.
- J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2011, 133, 188–190 CrossRef CAS.
- J. Dong, J. Hu, Y. Chi, Z. Lin, B. Zou, S. Yang, C. L. Hill and C. Hu, Angew. Chem., 2017, 129, 4544–4548 CrossRef.
- S. Mukhopadhyay, J. Debgupta, C. Singh, A. Kar and S. K. Das, Angew. Chem., 2018, 130, 1936–1941 CrossRef.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- P. Yang and U. Kortz, Acc. Chem. Res., 2018, 51, 1599–1608 CrossRef CAS.
- M. Zhao, X.-W. Zhang and C.-D. Wu, ACS Catal., 2017, 7, 6573–6580 CrossRef CAS.
- L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS.
- W. Guo, H. Lv, Z. Chen, K. P. Sullivan, S. M. Lauinger, Y. Chi, J. M. Sumliner, T. Lian and C. L. Hill, J. Mater. Chem. A, 2016, 4, 5952–5957 RSC.
- T. Ishizuka, S. Ohkawa, H. Ochiai, M. Hashimoto, K. Ohkubo, H. Kotani, M. Sadakane, S. Fukuzumi and T. Kojima, Green Chem., 2018, 20, 1975–1980 RSC.
- B. B. Sarma, I. Efremenko and R. Neumann, J. Am. Chem. Soc., 2015, 137, 5916–5922 CrossRef CAS.
- H. Shi, Y. Yu, Y. Zhang, X. Feng, X. Zhao, H. Tan, S. U. Khan, Y. Li and E. Wang, Appl. Catal., B, 2018, 221, 280–289 CrossRef CAS.
- S. Li, S. Liu, S. Liu, Y. Liu, Q. Tang, Z. Shi, S. Ouyang and J. Ye, J. Am. Chem. Soc., 2012, 134, 19716–19721 CrossRef CAS.
- M. Natali, I. Bazzan, S. Goberna-Ferrón, R. Al-Oweini, M. Ibrahim, B. Bassil, H. Dau, F. Scandola, J. Galán-Mascarós and U. Kortz, Green Chem., 2017, 19, 2416–2426 RSC.
- A. Rubinstein, P. Jiménez-Lozanao, J. J. Carbó, J. M. Poblet and R. Neumann, J. Am. Chem. Soc., 2014, 136, 10941–10948 CrossRef CAS PubMed.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356–5360 CrossRef CAS.
- Y. Dong, Z. Dong, Z. Zhang, Y. Liu, W. Cheng, H. Miao, X. He and Y. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 22088–22092 CrossRef CAS.
- D.-Y. Du, J.-S. Qin, Y.-G. Li, S.-L. Li, Y.-Q. Lan, X.-L. Wang, K.-Z. Shao, Z.-M. Su and E.-B. Wang, Chem. Commun., 2011, 47, 2832–2834 RSC.
- K. Gong, W. Wang, J. Yan and Z. Han, J. Mater. Chem. A, 2015, 3, 6019–6027 RSC.
- Z. Han, X. Xin, R. Zheng and H. Yu, Dalton Trans., 2018, 47, 3356–3365 RSC.
- D.-Y. Du, J.-S. Qin, T.-T. Wang, S.-L. Li, Z.-M. Su, K.-Z. Shao, Y.-Q. Lan, X.-L. Wang and E.-B. Wang, Chem. Sci., 2012, 3, 705–710 RSC.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, Y.-S. Zhu, L.-Y. Liu, Y.-Y. Sun, X. Bu, J. Zhang and Q. Lin, Adv. Mater., 2018, 30, 1704388–1704395 CrossRef.
- E. Haviv, L. J. W. Shimon and R. Neumann, Chem.–Eur. J., 2017, 23, 92–95 CrossRef CAS.
- C. Li, K. Suzuki, N. Mizuno and K. Yamaguchi, Chem. Commun., 2018, 54, 7127–7130 RSC.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- J. Zhao, Q. Wang, C. Sun, T. Zheng, L. Yan, M. Li, K. Shao, X. Wang and Z. Su, J. Mater. Chem. A, 2017, 5, 12498–12505 RSC.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242–3247 CrossRef.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and supporting figures and tables. CCDC 1855992–1855994. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03471k |
| This journal is © The Royal Society of Chemistry 2019 |