
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrathin two-dimensional covalent organic framework nanoprobe for interference-resistant two-photon fluorescence bioimaging†
Peng
Wang
,
Fang
Zhou
,
Cheng
Zhang
,
Sheng-Yan
Yin
,
Lili
Teng
,
Lanlan
Chen
,
Xiao-Xiao
Hu
,
Hong-Wen
Liu
 ,
Xia
Yin
* and
Xiao-Bing
Zhang
*
,
Xia
Yin
* and
Xiao-Bing
Zhang
*
Molecular Science and Biomedicine Laboratory, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Life Sciences, Collaborative Innovation Center for Chemistry and Molecular Medicine, Hunan University, Changsha, Hunan 410082, China. E-mail: xbzhang@hnu.edu.cn
First published on 10th September 2018
Abstract
The complex environment of living organisms significantly challenges the selectivity of classic small-molecule fluorescent probes for bioimaging. Due to their predesigned topological structure and engineered internal pore surface, covalent organic frameworks (COFs) have the ability to filter out coexisting interference components and help to achieve accurate biosensing. Herein, we propose an effective interference-resistant strategy by creating a COF-based hybrid probe that combines the respective advantages of COFs and small-molecule probes. As a proof of concept, a two-photon fluorescent COF nanoprobe, namely TpASH-NPHS, is developed for targeting hydrogen sulfide (H2S) as a model analyte. TpASH-NPHS exhibits limited cytotoxicity, excellent photostability and long-term bioimaging capability. More importantly, compared with the small-molecule probe, TpASH-NPHS achieves accurate detection without the interference from intracellular enzymes. This allows us to monitor the levels of endogenous H2S in a mouse model of cirrhosis.
Introduction
Fluorescence imaging is a powerful technique to monitor physiological and pathological processes and to study the functions of biomolecules in their native environment by virtue of its high sensitivity, non-destructive and fast response, and capacity for real-time analysis.1–3 In the past few decades, small-molecule fluorescent probes have been widely applied for the detection and imaging of various biological targets. The fluorescent properties of probes could be tuned via structural adaptation.4,5 This allowed for the design of probes showing significant changes in their spectroscopic properties upon interaction with their target analytes. However, the effective biosensing performance of a given small-molecule fluorescent probe can turn out to be quite susceptible to the complex biological environment. In particular, intricate intracellular enzymes may challenge the selectivity and sensitivity of the designed probe in question. For example, cytochrome P450 enzymes (CYP450) can lead to the reduction of azide groups in live cells under hypoxic conditions, which impacts the availability of azide-containing probes for the detection of intracellular hydrogen sulfide (H2S).6 Moreover, some limitations of classic small-molecule fluorescent probes, such as unsatisfactory signal stability and/or heterogeneous intracellular distribution, may cause a biosensing application to fail, including any attempt for long-term bioimaging.7,8 Therefore, any strategy with the potential to overcome these limitations and realize interference-resistant and long-term imaging is worthy of exploration.We come to focus on covalent organic frameworks (COFs) as such a platform to solve the abovementioned problems. COFs are an emerging class of organic, porous and crystalline materials that exemplify the power of reticular chemistry and expand the range of materials for real-world applications.9–15 Due to their predesigned topological structure, high and regular porosity and large surface area, COFs could regulate the enzyme activity by pore-environment control.16 We speculate that COFs might also have the potential to circumvent the problems associated with the interference by enzymes as encountered by small-molecule fluorescent probes, because their nanoscale pores can filter biomacromolecules, including enzymes, rather than small biomolecular analytes. Furthermore, COFs have the potential to improve the stability of a given classic small-molecule fluorescent probe, thereby opening the path to long-term fluorescence imaging.
In this work, we envisaged combining the respective advantages of small-molecule probes and COFs by creating a hybrid probe. As a proof of concept, a reported imine-linked COF, TpASH,17 was selected and applied to the design of a fluorescent probe, termed TpASH-NPHS, that targets hydrogen sulfide (H2S) as a model analyte present in biological systems. In view of the micron-sized nature of COFs, there don't appear to exist any reports on their use for the detection and imaging of biological analytes in live cells.18–22 To obtain an ultrathin, two-dimensional (2D) COF nanoprobe by classic solvent-assisted liquid sonication,22–24 we take advantage of the subdued π–π stacking by sequential post-synthesis fluorescence modification. As we will show, this nanoprobe exhibited satisfactory cell uptake and allowed for interference-resistant, long-term imaging in live cells. We will further demonstrate that TpASH-NPHS is useful for the investigation of the role of endogenous H2S in counteracting liver cirrhosis.
Results and discussion
Synthesis of the COF “TpASH”
As the COF component, we selected the reported imine-linked TpASH. First, this type of COF exhibits irreversible enol–keto tautomerism (Scheme S6†) and is characterized by its exceptional chemical stability.17 Second, the p-toluenesulfonic acid (PTSA)-mediated synthesis of TpASH is mild and convenient (90 °C, 12 hours, water) compared to traditional solvothermal methods and could produce highly crystalline porous products and overcome the hurdles in their further bioapplications. Third, the phenolic hydroxyl present in TpASH serves well in the post-synthesis conjugation with a fluorescent probe. This post-synthesis modification could weaken the interlayer π–π stacking and benefit the formation of nanoscale COFs by solvent-assisted liquid sonication.
TpASH is prepared via Schiff-base condensation (Fig. 1A) from 1,3,5-triformylphloroglucinol (Tp) and the PTSA salt of 4-aminosalicylhydrazide (ASH) (Scheme S1 and S2†). The morphology and size of TpASH were first investigated by transmission electron microscopy (TEM) and field emission scanning electron microscopy (SEM). As shown in Fig. 1B and C, both the TEM and SEM images clearly showed the sheet-like morphology of TpASH. The high-resolution TEM (HRTEM) image showed an interlayer distance of 3.2 Å (Fig. S8†). This indicates a good agreement with the interlayer distance calculated from the (001) diffraction.17 PXRD patterns of TpASH showed the first peak at 2θ = ∼3.9°, which corresponds to the reflection from the (100) planes (Fig. S9†).17 The broad peak at 2θ = ∼26.7° could be indexed to the π–π stacked planes (001) of TpASH.17 Fourier transform infrared (FTIR) spectra of TpASH exhibited characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N stretching frequencies at 1569 cm−1 and 1250–1257 cm−1, respectively (Fig. 1D).
C and C–N stretching frequencies at 1569 cm−1 and 1250–1257 cm−1, respectively (Fig. 1D).
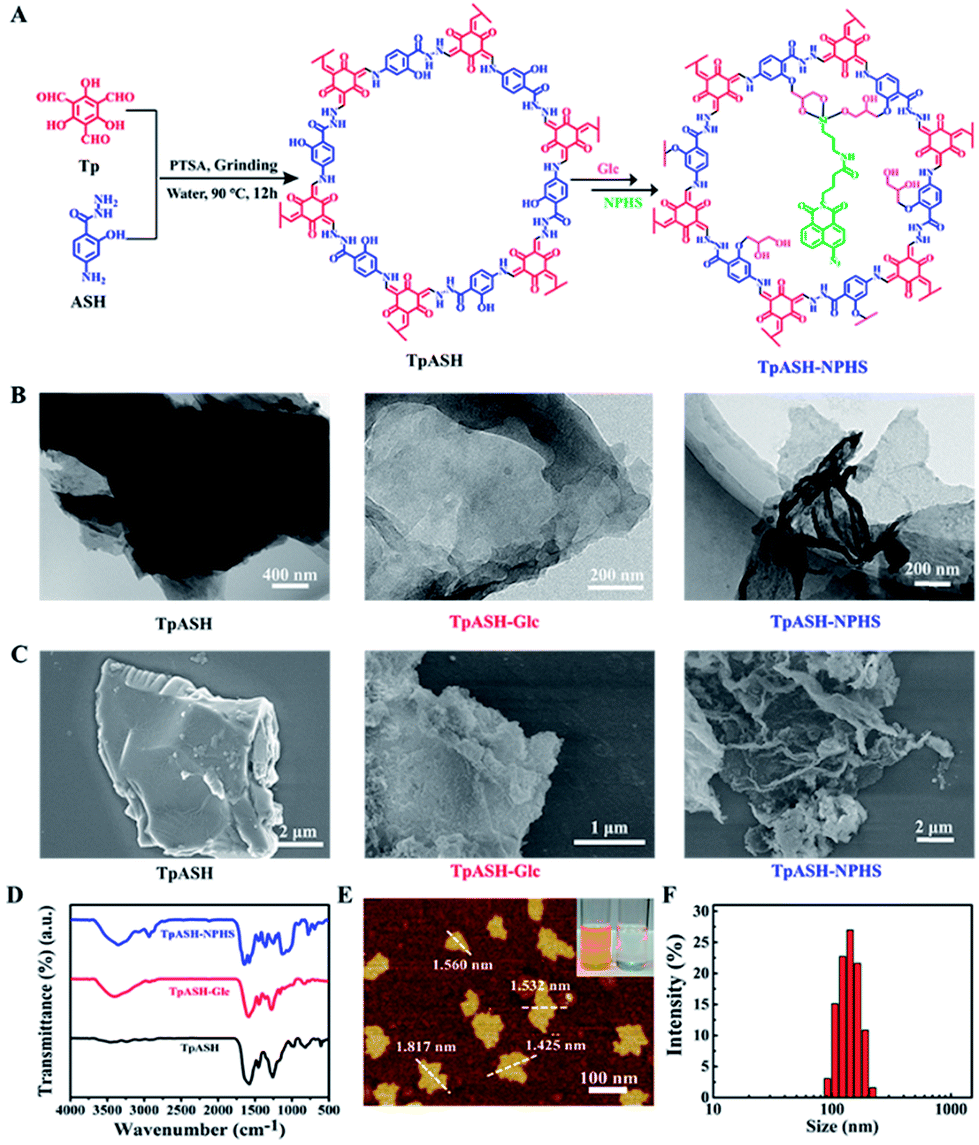 | ||
| Fig. 1 Construction and characterization of the two-photon fluorescent covalent organic framework nanoprobe TpASH-NPHS. (A) Synthesis of TpASH and its sequential post-synthesis modification. (B) TEM and (C) SEM images of TpASH, TpASH-Glc and TpASH-NPHS. (D) FTIR spectra of TpASH after sequential post-synthesis modification. (E) AFM image of the COF nanoprobe TpASH-NPHS. Inset: the image of the Tyndall effect of the TpASH-NPHS suspension (left: TpASH-NPHS and right: water; source of laser light on the left). (F) The size distribution of the COF nanoprobe TpASH-NPHS by DLS. | ||
Post-synthesis fluorescent probe conjugation
In order to demonstrate the utility of a COF-based biosensor, we selected H2S as a model analyte. As one of the vital gasotransmitters in living organisms, H2S plays a crucial role in many biological processes including neurodegenerative diseases, aging, and tumor proliferation.25–30 A number of studies have focused on H2S detection with fluorescent probes containing an azide group.31–33 Based on our previous work, we chose here to use a two-photon fluorescent probe to functionalize TpASH.31,34–37 Two-photon microscopy, which employs near-infrared (NIR) radiation as the excitation source, provides an opportunity for bioimaging with minimal background, reduced photo-damage, better three-dimensional localization and increased penetration depth (>500 μm).3,38,39 A 4-amino-1,8-naphthalimide derivative (NPHS) was chosen as the two-photon fluorescent probe by virtue of its large two-photon absorption cross-section, high quantum yield and straightforward synthesis (see Scheme S3† for an optimized synthetic path).37,40,41 We also synthesized a reported H2S fluorescent probe (CNPHS) for comparison (Scheme S4†).The phenolic hydroxyl group of TpASH turned out to be a welcome molecular anchor to allow for conjugation with NPHS (Scheme S5†). We thus alkylated TpASH by using glycidol (Glc) via epoxide ring opening. The thus established presence of multiple glycols in close vicinity then served to create a strong trivalent link between NPHS (containing a 3-aminopropyltriethoxysilane (APTES) unit) and the COF. In order to confirm the success of this modification, FTIR analysis was performed (Fig. 1D). Compared with TpASH, TpASH-NPHS showed additional peaks corresponding to C–H asymmetric and symmetric stretching frequencies at 2920 and 2853 cm−1, respectively, which were attributed to the methylene fragments of Glc. In TpASH-NPHS, additional peaks corresponding to Si–O stretching bands were also observed at 1095 cm−1 and 1020 cm−1. After fluorescent probe conjugation, the Brunauer–Emmett–Teller (BET) surface area of TpASH-NPHS showed a sharp reduction compared to that of TpASH (Fig. S10†). The PXRD pattern suggested that TpASH-NPHS maintained the structural integrity of COFs (Fig. S9†). Further analysis by TEM and SEM furnished a direct morphological comparison. As shown in the TEM and SEM images in Fig. 1B and C, TpASH-NPHS showed a rippled sheet-like morphology with lateral dimensions in the micron range. This difference between TpASH and TpASH-NPHS is in accord with the notion of post-synthesis weakening of interlayer π-π stacking. 13C cross-polarization magic-angle spinning (CP/MAS) solid state NMR spectra supported the conjugation of NPHS on the TpASH (Fig. S12†).17 Energy dispersive X-ray (EDX) analysis associated with TEM demonstrated the successful conjugation by proving the presence of silicon in TpASH-NPHS (Fig. S13†). The zeta potential value changed after sequential post-synthesis modification of the COF (Fig. S14†). The characteristic peak at 416 nm (two-photon fluorophore naphthalimide) in the UV-Vis spectra is further evidence for the successful conjugation (Fig. S15†).
Preparation of the COF nanoprobe TpASH-NPHS
For further bioapplication, nanoscale COFs are desired. However, the obtained COFs are usually micron-sized, which is attributed to the strong interlayer π–π stacking interactions.42,43 Fortunately, 2D nanosheets can be obtained directly from their bulk counterparts via solvent-assisted exfoliation. As a new member of the family of 2D materials, COF nanosheets may be easily prepared using this strategy.22–24 Hence, we take advantage of subdued π–π stacking by sequential post-synthesis modification to acquire nanoscale COFs by classic solvent-assisted liquid sonication. After exfoliation, the solution of TpASH-NPHS showed a uniform dispersion and a typical “Tyndall effect” (inset in Fig. 1E), confirming its nanoscale colloidal structure.22 The atomic force microscopy (AFM) images in Fig. 1E and S16† clearly showed a change between the bulk COFs and the nanoscale COFs. Our ultrathin 2D COF TpASH-NPHS showed a size of 87.35 ± 18.64 nm and a thickness of 1.58 ± 0.25 nm, while bulk TpASH-NPHS had a micron-scale size and thickness. The average hydrodynamic diameter of TpASH-NPHS was 144.55 ± 45.59 nm as determined by dynamic light scattering (DLS) (Fig. 1F). The FTIR spectrum of nanoscale TpASH-NPHS remains the same as that of the bulk material (Fig. S17†), further confirming the same structures for the bulk and the nanoscale TpASH-NPHS.Fluorescence sensing of the nanoprobe TpASH-NPHS
In order to explore the biosensing performance of the COF-based fluorescent probe, we report here the detection of the naturally occurring analyte H2S by our nanoprobe TpASH-NPHS described above. Its azide units suffer from reduction to amino groups in the presence of H2S, which enhances its fluorescence emission (Scheme S7†). We thus recorded the fluorescence spectral changes in TpASH-NPHS upon the gradual addition of H2S. Sodium hydrosulfide (NaHS) was used as the H2S source in all experiments. As shown in Fig. 2B and S18,† the addition of NaHS triggered a significant increase in fluorescence intensity at 535 nm. This response was linearly proportional to NaHS in the concentration range of 0–20 μM (Fig. 2C). The detection limit (S/N = 3) for NaHS was calculated to be 0.11 μM, a competitive value compared to previous reports of intracellular H2S detection.31 The signal reached a plateau after about 40 min (Fig. S19†). The loading concentration of NPHS in TpASH-NPHS (100 μg mL−1) was calculated to be 1.05 μM (Fig. S20†).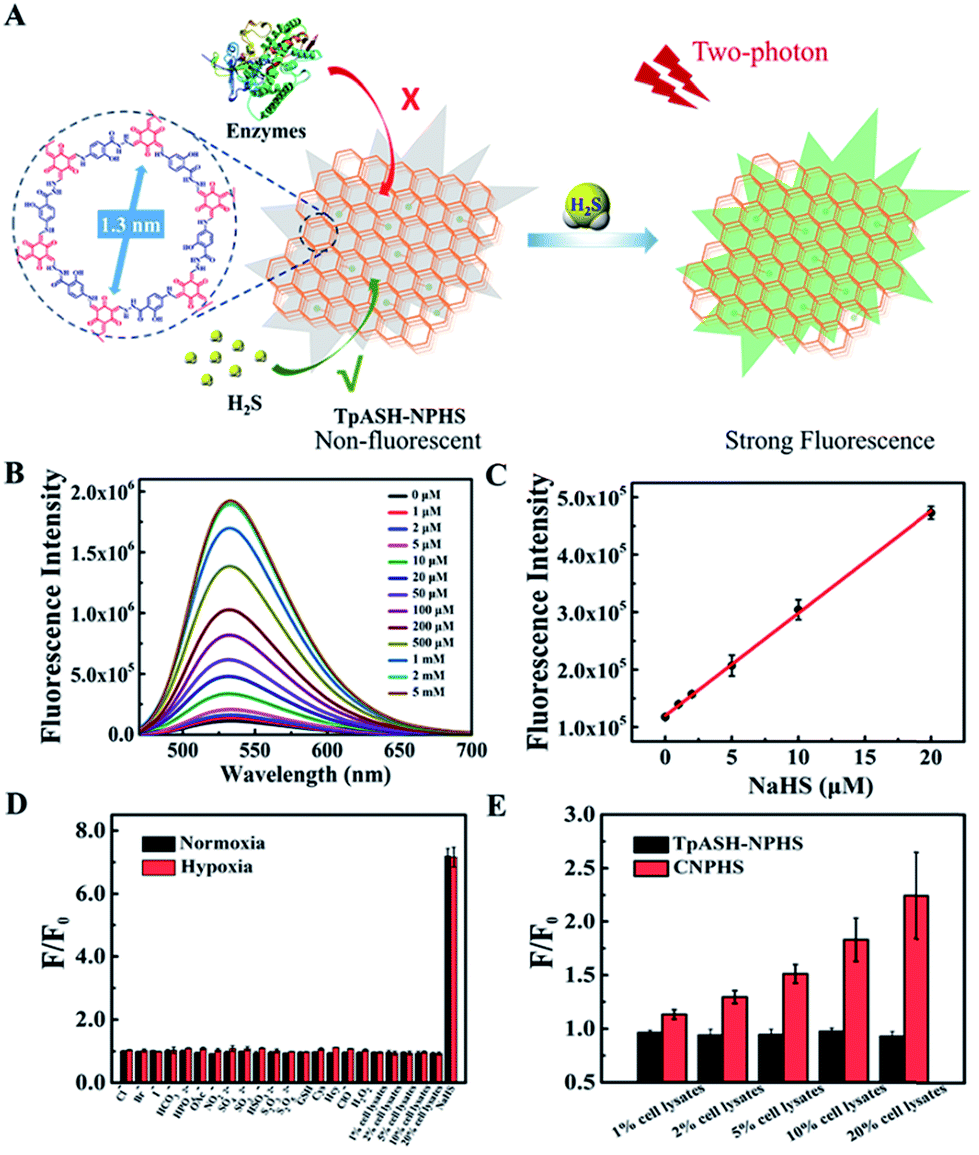 | ||
Fig. 2 Interference-resistant fluorescence sensing of the COF nanoprobe TpASH-NPHS. (A) Constitution and mode of action of the COF-based nanoprobe TpASH-NPHS. (B) The fluorescence emission spectra of TpASH-NPHS in the presence of different concentrations of NaHS. (C) Linear response at low NaHS concentrations, R2 = 0.998. The standard curve of the TpASH-NPHS response to NaHS is y = 119![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 908.37448 + 17831.54373x. λex = 440 nm. (D) Fluorescence response of TpASH-NPHS to various substances under normoxic (21% O2) and hypoxic (<0.1% O2) conditions. (E) The comparison of fluorescence response between TpASH-NPHS and CNPHS to different concentrations of HepG2 cell lysates under hypoxia. 908.37448 + 17831.54373x. λex = 440 nm. (D) Fluorescence response of TpASH-NPHS to various substances under normoxic (21% O2) and hypoxic (<0.1% O2) conditions. (E) The comparison of fluorescence response between TpASH-NPHS and CNPHS to different concentrations of HepG2 cell lysates under hypoxia. | ||
The selectivity of the COF nanoprobe for H2S was first investigated in vitro. As can be seen in Fig. 2D and S23,† the fluorescence of TpASH-NPHS and CNPHS was insensitive to the presence of common anions (Cl−, Br−, I−, HCO3−, HPO42−, OAc−, NO2−, SO42−, SO32−, HSO3−, S2O32−, and S2O52−), biothiols (glutathione (GSH), cysteine (Cys), and homocysteine (Hcy)) and reactive oxygen species (ClO− and H2O2) under normoxic (21% O2) and hypoxic (<0.1% O2) conditions, a result that is in agreement with reported conclusions.31,33
However, a recent study reported that intracellular enzymes are able to reduce the azide group under hypoxic conditions.6 We suggest here that COFs (pore size of TpASH = 1.3 nm) can act as the host material for such a probe, thus denying the access of macromolecular enzymes (usually size > 3 nm) to sequestered responsive probes within the COF (Fig. 2A).16,17 To check the interference-resistant ability of our nanoprobe, we investigated the detection performance of TpASH-NPHS in the lysates from HepG2 cells under hypoxic and normoxic conditions. For comparison, experiments with the small-molecule probe (CNPHS) were also conducted. As shown in Fig. 2D, E and S23,†TpASH-NPHS displayed no obvious fluorescence enhancement in either case, while CNPHS showed strong fluorescence enhancement only for the hypoxic case. This supports the notion that intracellular enzymes are able to cause a false positive signal for H2S.6 In addition, both TpASH-NPHS and CNPHS maintained the capacity to respond to the presence of H2S under hypoxic conditions. Thus, we conclude that our COF nanoprobe is able to solve the problem of biomacromolecular interference to which most classic small-molecule fluorescent probes are susceptible.
Stability and cytotoxicity of TpASH-NPHS
Due to the irreversible enol–keto tautomerism found in the structure of TpASH (Scheme S6†), this type of COF promises elevated chemical stability, which is necessary for bioapplications. DLS profiles taken from PBS (pH = 7.4) and DMEM (cell culture medium) samples support this notion. No significant change in size was found after 24 h (Fig. S24†), thus demonstrating that the structure of TpASH-NHS remains unaltered in a physiological environment. Next, we investigated the photostability of the nanoscale COFs by monitoring the fluorescence change (Fig. S25†). The results showed that the prepared nanoprobe TpASH-NPHS had good photostability in PBS or DMEM for long periods of time. When compared to CNPHS (Fig. S26†), the intensity of TpASH-NPHS remains virtually untouched over 3600 s, while that of CNPHS has decreased by 26 percent in just a short period of real time laser scanning. We interpret this to be the fruit of the sheltering effect exercised by the framework architecture of COFs. Identical conclusions were obtained when TpASH-NPHS was tested for fluorescence variations with changing pH (from 3.0 to 10.0, see Fig. S27†). Importantly, we also determined the cytotoxicity of TpASH-NPHS by an MTS assay using human cervical carcinoma cells (HeLa) and human liver cancer cells (HepG2). As shown in Fig. S28,† both the HeLa and HepG2 cells maintained high viability (>90%) after incubation with TpASH-NPHS for 24 h, even at high concentrations (200 μg mL−1), clearly indicating the limited cytotoxicity of this COF nanoprobe and suggesting its suitability for intracellular biosensing.Two-photon fluorescence imaging of H2S in live cells
The favorable properties of our COF-based probe technology as elucidated above encouraged us to explore its performance during two-photon fluorescence imaging of H2S in live cells. As shown in Fig. 3A, incubation of HepG2 or HeLa cells with TpASH-NPHS afforded a weak green fluorescence signal with two-photon excitation at 820 nm, thereby demonstrating sufficient cell permeability and the promising prospect of endogenous H2S sensing with low background fluorescence in cells. To confirm this, HepG2 and HeLa cells were pretreated with PMA for 30 min, which favors a decrease of endogenous H2S by inducing phagocytosis-associated ROS generation, and then co-incubated with the probe before fluorescence imaging. As shown in Fig. 3A, almost no fluorescence was observed. Conversely, the presence of TpASH-NPHS generates a gradually enhanced fluorescence when the cells are co-incubated with NaHS. The mechanism of internalization of TpASH-NPHS was investigated with HepG2 cells. The confocal microscopy images of treatments at 4 °C (to inhibit energy-dependent endocytosis), or with genistein (a caveolae-dependent endocytosis inhibitor), with chlorpromazine hydrochloride (CPZ, a clathrin-dependent endocytosis inhibitor), and with methyl-β-cyclodextrin (MBC, a lipid raft-mediated endocytosis inhibitor) are shown in Fig. S29.† The images demonstrate that internalization of TpASH-NPHS is significantly slowed down after the treatment at low temperature or with CPZ, suggesting that the uptake of TpASH-NPHS is mainly through clathrin-dependent endocytosis. We also used the bulk COF TpASH-NPHS for imaging. Poor internalization was evident (Fig. S30†).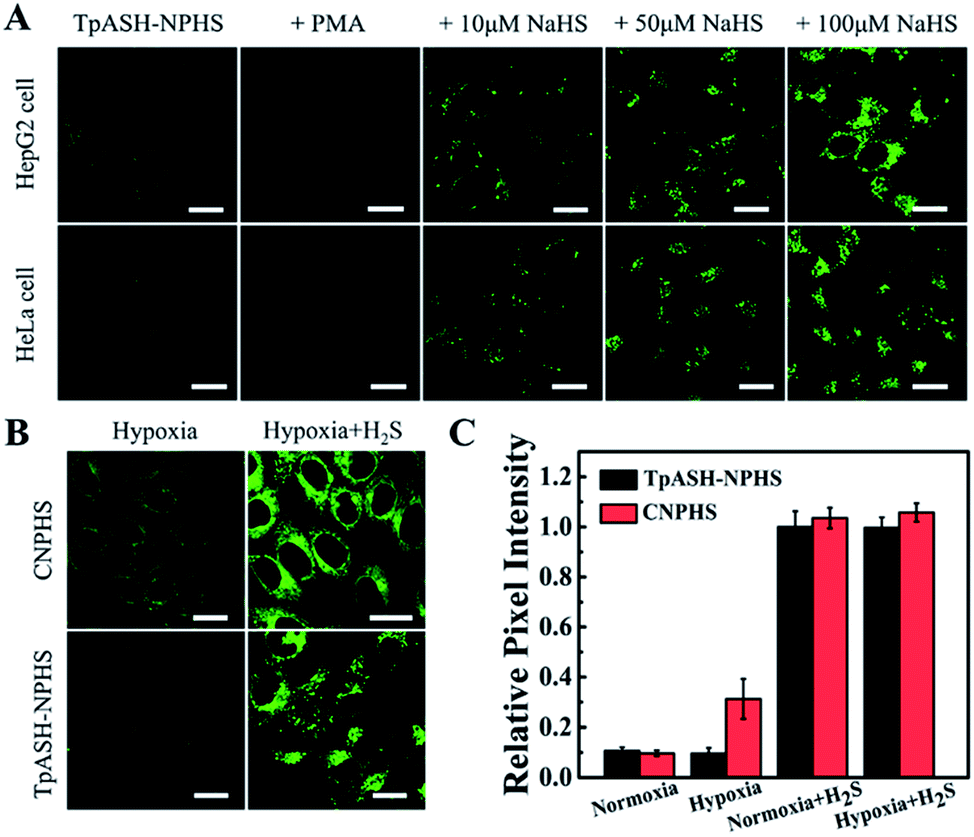 | ||
| Fig. 3 Interference-resistant imaging of the COF nanoprobe TpASH-NPHS in live cells. (A) Two-photon confocal fluorescence imaging of TpASH-NPHS in HepG2 and HeLa cells. Scale bar: 20 μm. (B) Confocal fluorescence imaging of CNPHS or TpASH-NPHS in HepG2 cells under hypoxic conditions without or with the addition of exogenous H2S. Scale bar: 20 μm. (C) The relative pixel fluorescence intensity of HepG2 cell images. The pixel intensity from TpASH-NPHS incubated with H2S in HepG2 cells under normoxic conditions is defined as 1.0. | ||
Next on our agenda was to determine whether components of the complex intracellular environment could interfere in the sensing and imaging of H2S. As shown in Fig. 3B, CNPHS generates dramatically enhanced fluorescence under hypoxic conditions, while TpASH-NPHS does not, which is consistent with the fluorescence response in HepG2 cell lysates under similar conditions. When exogenous H2S is added, both TpASH-NPHS and CNPHS show strong fluorescence under normoxic and hypoxic conditions (Fig. 3C and S31†). This proves that the use of TpASH-NPHS over small-molecule responsive probes can prevent enzymes from interfering with the biosensing and bioimaging of H2S. To illustrate the difference in the signal stability of fluorescent COF nanoprobes and small-molecule fluorescent probes, we conducted real-time imaging of HepG2 cells in the presence of TpASH-NPHS and CNPHS. Indeed, TpASH-NPHS exhibited enhanced photostability over CNPHS, suggesting that the COF scaffold not only shelters the fluorophore from enzymes but also from photo-degradation (Fig. S32†). We now turned to investigating the long-term cellular tracking capability of TpASH-NPHS and CNPHS after different incubation periods. As shown in Fig. S33,†CNPHS tended to translocate out of cells within one hour, thereby limiting its application to short-term imaging experiments. On the other hand, TpASH-NPHS showed strong intracellular fluorescence at every time point for over six hours.
Two-photon fluorescence imaging of H2S in deep tumor tissues
The two major advantages of two-photon over single-photon fluorescence imaging are the minimization of tissue auto-fluorescence and enhanced penetration depth. Fig. S34† reports the data obtained for a two-photon imaging experiment of TpASH-NPHS monitoring H2S in HeLa tumor tissues. Signal intensity vs. scan depth was recorded with a two-photon microscope in the Z-scan mode. The control samples without COFs exhibited hardly any auto-fluorescence, demonstrating that two-photon excited confocal images indeed avoid the depiction of auto-fluorescence. On the other hand, tumor tissues incubated with TpASH-NPHS showed a weak fluorescence signal, thus demonstrating that two-photon fluorescent COF nanoprobes are capable of imaging endogenous H2S. When such tissue is pretreated with 100 μM NaHS, a dramatic fluorescence increase is observed, even at a depth of 50–250 μm, which confirms our design objective that TpASH-NPHS is capable of H2S imaging at greater penetration depths. Fig. 4 shows the data of imaging experiments on HepG2 tumor tissues. TpASH-NPHS showed no two-photon fluorescence intensity changes under normoxic and hypoxic conditions at a depth of 150 μm, while CNPHS caused an increase in intensity under hypoxic conditions. The penetration depth of TpASH-NPHS-emitted light was also probed in HepG2 tumor tissues (Fig. S35†).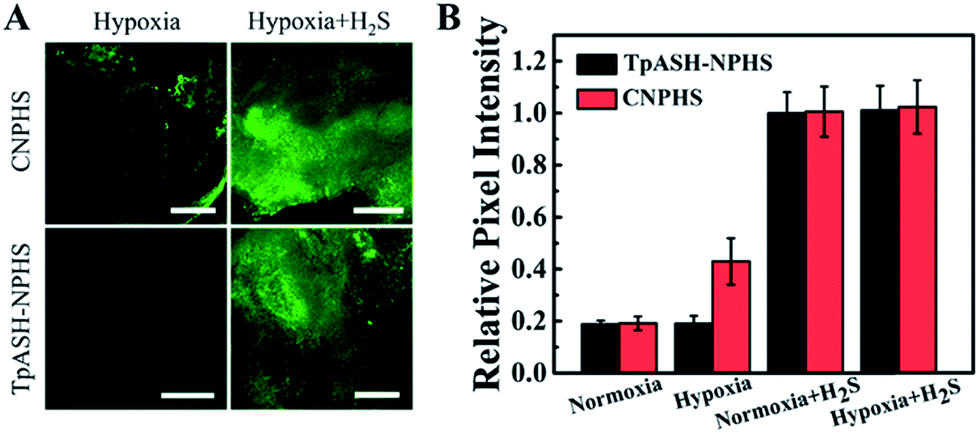 | ||
| Fig. 4 Interference-resistant two-photon fluorescence imaging of the COF nanoprobe TpASH-NPHS in deep tumor tissues. (A) Two-photon confocal fluorescence images of TpASH-NPHS and CNPHS under hypoxia in HepG2 tumor tissues without or with the addition of exogenous H2S. Scale bar: 300 μm. (B) The relative pixel fluorescence intensity of HepG2 tumor tissues. The pixel signal intensity from TpASH-NPHS incubated with H2S in HepG2 tumor tissues under normoxic conditions is defined as 1.0. | ||
Monitoring of endogenous H2S in a cirrhotic liver model
As a gasotransmitter with mediating vasodilatory function, endogenous H2S has a significant effect on antagonizing chronic liver injury or cirrhosis.44 The levels of endogenous H2S in the liver are a potential physiological index for evaluating the cirrhosis degree. Especially the up-regulated H2S expression at an early stage of cirrhosis benefits the early detection of this liver disease.45 However, the liver, as a vital metabolic organ of mammals, possesses the most complex enzyme networks and other components, which impact the biosensing performance of small-molecule fluorescent probes. Besides, a protocol to build a reliable cirrhosis mouse model is needed to subcutaneously inject tetrachloromethane (CCl4) to induce hepatotoxicity by CYP2E1 (a member of the CYP450 system) activation.46 Therefore, a responsive probe truly specific to H2S sensing would be more than welcome for the bioimaging of the mouse liver with early stage of cirrhosis. The liver tissue slices were harvested at different CCl4-treated periods and stained with TpASH-NPHS (Fig. 5A). As shown in Fig. 5B and S37,† the two-photon fluorescence intensity steadily increased from the third day (termed 3 day) to the fifteenth day (termed 15 day). This is supported by the observation that the level of endogenous H2S is up-regulated to prevent acute liver injury.44,45 In addition, a liver self-repairing process is indicated by the increased expression levels of alanine transaminase (ALT) and aspartate transaminase (AST) in hepatocytes (Fig. 5C). The expression levels of the H2S-generating enzyme cystathionine γ-lyase (CSE) initially decrease, which reveals that the hepatic CSE is expressed to compensate for H2S depletion (Fig. 5D). We also observed that the H2S level drops significantly with the progression of liver injury, while ALT and AST levels are down-regulated.This means that liver injury has gradually entered late stage and hepatocytes fail to selfrepair, eventually leading to cirrhosis. Histological changes from hematoxylin and eosin (H&E) stain indicate liver rupture, cellular shrinkage, and neutrophil infiltration (Fig. S38†). | ||
| Fig. 5 Monitoring of endogenous H2S in a cirrhotic liver model. (A) The liver cirrhosis mouse model was induced by subcutaneous injection of 40% CCl4. The liver tissue slices were harvested at different CCl4-treated periods. (B) Two-photon confocal fluorescence images of TpASH-NPHS in liver tissues of the cirrhotic mouse model at different CCl4-treated periods. Scale bar: 300 μm. (C) The enzyme activity of ALT and AST in mice serum. (D) Quantification of CSE levels in normal and liver cirrhosis models by ELISA. | ||
Conclusions
In summary, we developed the first example of a two-photon fluorescent COF nanoprobe, TpASH-NPHS, that shows favorable chemical stability and photostability, is conveniently synthesised, allows for versatile fluorescence modification, and exhibits a desirable nanoscale size. We have demonstrated the utility of TpASH-NPHS as a sensitive and selective fluorescent sensor of the gasotransmitter H2S. Our two-photon fluorescent COF serves to detect and image H2S in live cells and deep tissues under NIR excitation, thereby greatly minimizing cellular autofluorescence, reducing tissue injury and increasing tissue penetration depth. Furthermore, the detection process is not jeopardized by haphazard interaction with endogenous enzymes, which holds great promise as an indicator for clinical applications; initial results with a cirrhotic mouse model confirm this. We are confident that our results encourage future research on employing fluorescent COF nanoprobes for biosensing and nanomedicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21521063, 21325520, 21327009, and J1210040) and the Science and Technology Project of Hunan Province (2016RS2009 and 2016WK2002).Notes and references
- Z. Yang, A. Sharma, J. Qi, X. Peng, D. Y. Lee, R. Hu, D. Lin, J. Qu and J. S. Kim, Chem. Soc. Rev., 2016, 45, 4651–4667 RSC.
- X. Wu, A. Shao, S. Zhu, Z. Guo and W. Zhu, Sci. China: Chem., 2016, 59, 62–69 CrossRef.
- H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef PubMed.
- L. J. O'Connor, I. N. Mistry, S. L. Collins, L. K. Folkes, G. Brown, S. J. Conway and E. M. Hammond, ACS Cent. Sci., 2017, 3, 20–30 CrossRef PubMed.
- Z. Wang, S. Chen, J. W. Lam, W. Qin, R. T. Kwok, N. Xie, Q. Hu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 8238–8245 CrossRef PubMed.
- J. Liang, G. Feng, R. T. K. Kwok, D. Ding, B. Tang and B. Liu, Sci. China: Chem., 2016, 59, 53–61 CrossRef.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 1585 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef.
- J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef PubMed.
- Y. Zhang, J. Duan, D. Ma, P. Li, S. Li, H. Li, J. Zhou, X. Ma, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 16313–16317 CrossRef PubMed.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef PubMed.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef PubMed.
- H. Ding, Y. Li, H. Hu, Y. Sun, J. Wang, C. Wang, C. Wang, G. Zhang, B. Wang, W. Xu and D. Zhang, Chem.–Eur. J., 2014, 20, 14614–14618 CrossRef PubMed.
- Q. Sun, C. W. Fu, B. Aguila, J. Perman, S. Wang, H. Y. Huang, F. S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef PubMed.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Diaz Diaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef PubMed.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef PubMed.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef PubMed.
- Y. Zhang, X. Shen, X. Feng, H. Xia, Y. Mu and X. Liu, Chem. Commun., 2016, 52, 11088–11091 RSC.
- H. L. Qian, C. Dai, C. X. Yang and X. P. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 24999–25005 CrossRef PubMed.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef PubMed.
- I. Berlanga, M. L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, J. L. Fierro, R. Mas-Balleste and F. Zamora, Small, 2011, 7, 1207–1211 CrossRef PubMed.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef PubMed.
- Z. Du, B. Song, W. Zhang, C. Duan, Y. L. Wang, C. Liu, R. Zhang and J. Yuan, Angew. Chem., Int. Ed., 2018, 57, 3999–4004 CrossRef PubMed.
- G. Xu, Q. Yan, X. Lv, Y. Zhu, K. Xin, B. Shi, R. Wang, J. Chen, W. Gao, P. Shi, C. Fan, C. Zhao and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 3626–3630 CrossRef PubMed.
- Y. L. Pak, J. Li, K. C. Ko, G. Kim, J. Y. Lee and J. Yoon, Anal. Chem., 2016, 88, 5476–5481 CrossRef PubMed.
- J. Kang, Z. Li, C. L. Organ, C. M. Park, C. T. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef PubMed.
- X. Jiao, Y. Xiao, Y. Li, M. Liang, X. Xie, X. Wang and B. Tang, Anal. Chem., 2018, 90, 7510–7516 CrossRef PubMed.
- Y. Ma, C. Zhang, P. Yang, X. Li, L. Tong, F. Huang, J. Yue and B. Tang, Nanoscale, 2018, 10, 15793–15798 RSC.
- G. J. Mao, T. T. Wei, X. X. Wang, S. Y. Huan, D. Q. Lu, J. Zhang, X. B. Zhang, W. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2013, 85, 7875–7881 CrossRef PubMed.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef PubMed.
- M. D. Hammers, M. J. Taormina, M. M. Cerda, L. A. Montoya, D. T. Seidenkranz, R. Parthasarathy and M. D. Pluth, J. Am. Chem. Soc., 2015, 137, 10216–10223 CrossRef PubMed.
- L. Zhou, X. Zhang, Q. Wang, Y. Lv, G. Mao, A. Luo, Y. Wu, Y. Wu, J. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 9838–9841 CrossRef PubMed.
- H. W. Liu, X. B. Zhang, J. Zhang, Q. Q. Wang, X. X. Hu, P. Wang and W. Tan, Anal. Chem., 2015, 87, 8896–8903 CrossRef PubMed.
- P. Wang, C. Zhang, H. W. Liu, M. Xiong, S. Y. Yin, Y. Yang, X. X. Hu, X. Yin, X. B. Zhang and W. Tan, Chem. Sci., 2017, 8, 8214–8220 RSC.
- S. Xu, H. W. Liu, X. X. Hu, S. Y. Huan, J. Zhang, Y. C. Liu, L. Yuan, F. L. Qu, X. B. Zhang and W. Tan, Anal. Chem., 2017, 89, 7641–7648 CrossRef PubMed.
- H. W. Liu, Y. Liu, P. Wang and X. B. Zhang, Methods Appl. Fluoresc., 2017, 5, 012003 CrossRef PubMed.
- J. Li, C. S. Lim, G. Kim, H. M. Kim and J. Yoon, Anal. Chem., 2017, 89, 8496–8500 CrossRef PubMed.
- Z. R. Dai, G. B. Ge, L. Feng, J. Ning, L. H. Hu, Q. Jin, D. D. Wang, X. Lv, T. Y. Dou, J. N. Cui and L. Yang, J. Am. Chem. Soc., 2015, 137, 14488–14495 CrossRef PubMed.
- B. Zhang, X. Yang, R. Zhang, Y. Liu, X. Ren, M. Xian, Y. Ye and Y. Zhao, Anal. Chem., 2017, 89, 10384–10390 CrossRef PubMed.
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef PubMed.
- D. A. Vazquez-Molina, G. S. Mohammad-Pour, C. Lee, M. W. Logan, X. Duan, J. K. Harper and F. J. Uribe-Romo, J. Am. Chem. Soc., 2016, 138, 9767–9770 CrossRef PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef PubMed.
- T. B. Ren, W. Xu, Q. L. Zhang, X. X. Zhang, S. Y. Wen, H. B. Yi, L. Yuan and X. B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7473–7477 CrossRef PubMed.
- G. Tan, S. Pan, J. Li, X. Dong, K. Kang, M. Zhao, X. Jiang, J. R. Kanwar, H. Qiao, H. Jiang and X. Sun, PLoS One, 2011, 6, 25943 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03393e |
| This journal is © The Royal Society of Chemistry 2018 |