
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational synthesis of benzimidazole[3]arenes by CuII-catalyzed post-macrocyclization transformation†‡
Shohei
Tashiro
 *,
Tsutomu
Umeki
,
Ryou
Kubota§
and
Mitsuhiko
Shionoya
*
*,
Tsutomu
Umeki
,
Ryou
Kubota§
and
Mitsuhiko
Shionoya
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shionoya@chem.s.u-tokyo.ac.jp
First published on 5th September 2018
Abstract
A new series of calix[n]arene analogues, benzimidazole[3]arenes, was rationally synthesized by CuII-catalyzed post-macrocyclization transformation of a tris(o-phenylenediamine) macrocycle, and fully characterized by NMR, MS, and single-crystal X-ray diffraction (XRD) analyses. The resulting syn- and anti-benzimidazole[3]arenes have a bowl-shaped and a warped structure, respectively, in their crystalline states, and both display a dynamic inversion behavior in solution. This modification resulted in strong fluorescence due to the generated benzimidazole moieties. The mechanistic study of the post-macrocyclization transformation demonstrated that the formation of both benzimidazole[3]arenes was catalyzed, via triimine intermediates, by CuII ions in air through oxidation and cyclization of the tris(o-phenylenediamine) macrocycle.
Introduction
Since the discovery of calix[4]arene and its analogues such as cyclotriveratrylene, pyrogallol[n]arenes, and resorcin[n]arenes,1 macrocyclic compounds with several aromatic rings circularly arranged through methylene linkers have attracted much attention in both fundamental and applied chemistry. Significant efforts have been devoted to the synthesis of their new series including pillar[n]arenes,2a pillar[n]quinones,2b asar[n]arenes,2c biphen[n]arenes,2d oxatub[n]anrenes,2e and others.2f–i These macrocycles have a well-defined hydrophobic cavity enclosed by a relatively flexible ring framework due to the presence of methylene linkers. It is also worth noting that some macrocycles are chiral when they have a circumferentially and axially anisotropic three-dimensional structure.3 Their structure and function depend heavily on the type and sequence feature of the building unit of the macrocycles.N-Heterocycles are typical building blocks of macrocycles, as N-heterocycle-based calix[n]arene analogues such as calix[n]pyrroles,4a calix[n]imidazolium,4b calix[n]pyridines or pyridine[n]arenes,4c–e ExBox,4f Texas-sized box,4g and others4h–j exhibit strong fluorescence and ion recognition ability through non-covalent interactions. However, as far as we know, benzimidazole-based calix[n]arenes5,6 have not been reported so far due to the lack of rational synthetic strategies to circularly arrange benzimidazole moieties in macrocyclic skeletons, despite the fact that benzimidazole is known to show strong fluorescence, bioactivity, metal binding and ion recognition abilities.7 Here we report the CuII-catalyzed facile synthesis, structures, and properties of two isomeric syn- and anti-benzimidazole[3]arenes (syn-2 and anti-3) with C3- and C1-symmetry, respectively, based on the different permutations of three benzimidazole units. The molecular structures and their dynamic inversion behaviors were evaluated by single-crystal X-ray diffraction (XRD) and variable-temperature (VT) NMR analyses. We also found that three benzimidazole moieties in the macrocycle led to strong fluorescence.
A key step in the synthesis of syn-2 and anti-3 is post-macrocyclization transformation of macrocyclic tris(o-phenylenediamine) (1), which can be easily prepared from o-phenylenediamine and terephthalaldehyde in two steps of macrocyclization and hydrogenation.8a As in our previous report,8 macrocycle 1 reacted with PdII ions to form a trinuclear PdII-macrocycle. However, we found that a reaction with CuII ions produced not trinuclear CuII-macrocycles of 1, but two isomeric syn- and anti-benzimidazole[3]arenes (syn-2 and anti-3) through a CuII-catalyzed transformation reaction (Fig. 1). In general, calix[n]arene and its analogues can be synthesized by simple macrocyclization of each building block.1,2 However, it is not always suitable to apply this direct macrocyclization to an unsymmetrical arrangement of building blocks for lower-symmetry macrocycles. In contrast, post-macrocyclization transformation9 is a powerful tool to prepare lower-symmetry macrocycles as proven by the intensive synthetic studies of diverse porphyrinoids, because a macrocyclic skeleton is asymmetrically folded by the macrocyclic transformation reaction.10
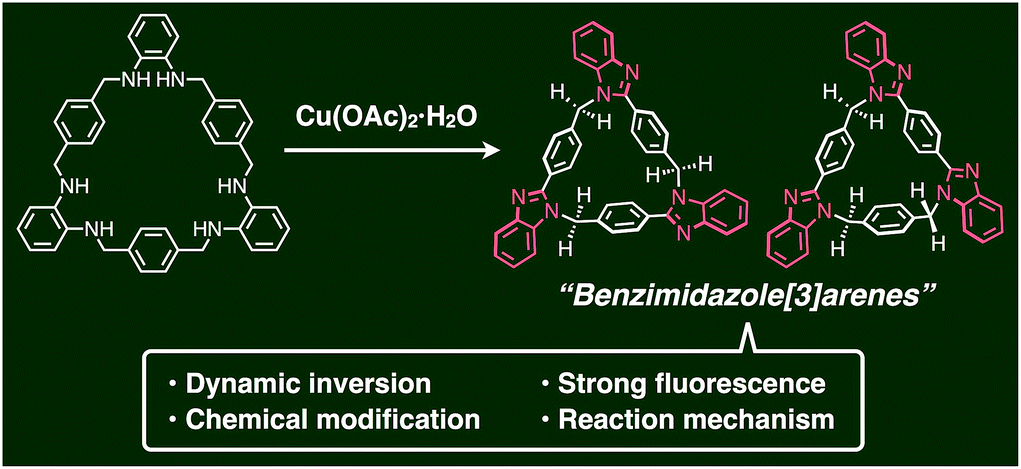 | ||
| Fig. 1 Schematic representation of the facile synthesis of benzimidazole[3]arenes through post-macrocyclization transformation. | ||
Results and discussion
Macrocycle 1 was reacted with an equimolar amount of Cu(OAc)2·H2O in a mixed solvent of CHCl3/CH3CN = 1/2 (v/v) at 55 °C in air ([1] = 5.0 mM). The reaction mixture became brown after 15 min, and then turned green after one day. The 1H NMR spectrum of the greenish reaction mixture showed broadened proton signals, indicating that CuII ions interacted with products. The 1H NMR analysis of the crude product after removing CuII ions by a liquid separating operation indicated that 1 was completely consumed to form the target compounds, syn- and anti-benzimidazole[3]arenes (syn-2 and anti-3), in the molar ratio of ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (Fig. 2a and b). The mixture was separated by silica gel chromatography and then purified by recrystallization to afford syn-2 and anti-3 in 3.3% and 47% isolated yields, respectively (Fig. 2c and d). This selectivity is discussed later. The products were fully characterized by high-resolution electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) (m/z = 619.2593 and 619.2593 for [syn-2 + H]+ and [anti-3 + H]+, respectively), single-crystal X-ray diffraction (XRD), and NMR analyses as shown below.
:10 (Fig. 2a and b). The mixture was separated by silica gel chromatography and then purified by recrystallization to afford syn-2 and anti-3 in 3.3% and 47% isolated yields, respectively (Fig. 2c and d). This selectivity is discussed later. The products were fully characterized by high-resolution electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) (m/z = 619.2593 and 619.2593 for [syn-2 + H]+ and [anti-3 + H]+, respectively), single-crystal X-ray diffraction (XRD), and NMR analyses as shown below.
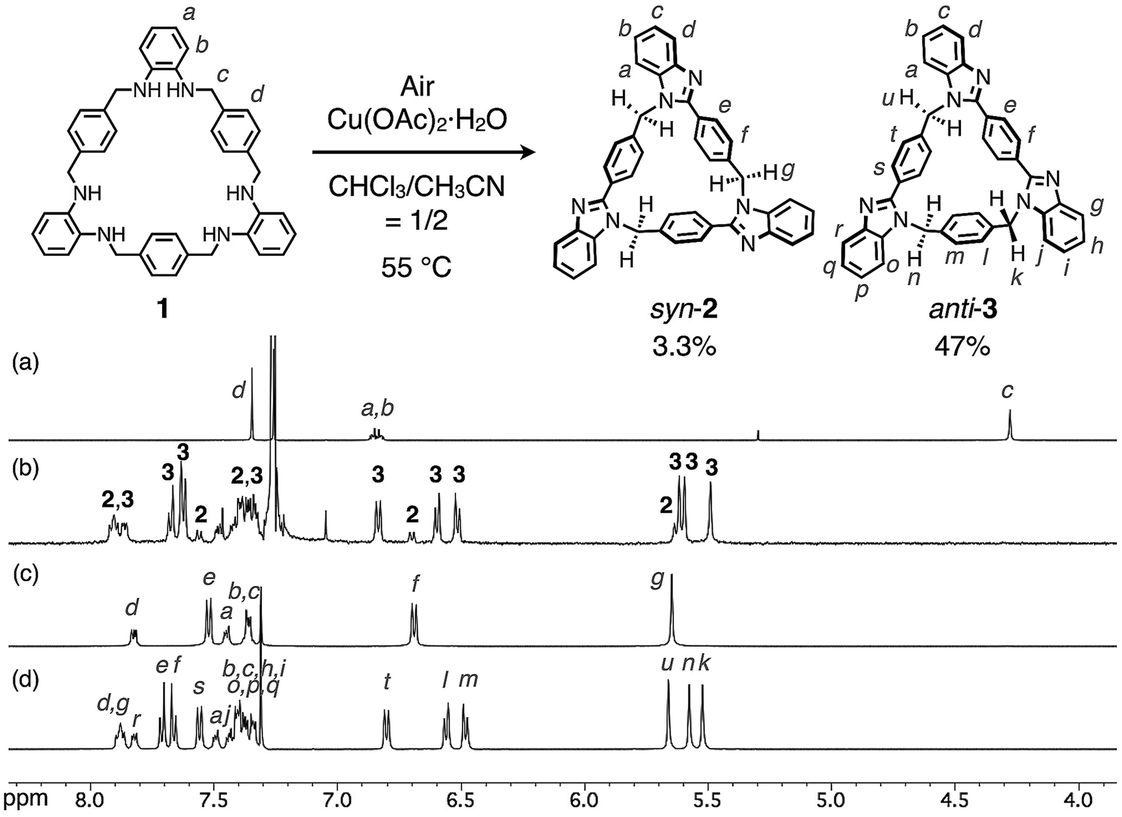 | ||
| Fig. 2 Reaction conditions of the post-macrocyclization transformation and 1H NMR spectra (500 MHz, 300 K, CDCl3 for (a) and (b) or CDCl3/CD3OD = 20/1 for (c) and (d)) of (a) 1, (b) crude product, (c) syn-2 and (d) anti-3. | ||
The molecular structures of both compounds syn-2 and anti-3 have been determined by single-crystal XRD analyses. It was confirmed that both compounds are 21-membered macrocycles composed of three benzimidazole units. In the molecular structure of syn-2, three 2-phenylbenzimidazole moieties are symmetrically arranged in the same direction to form a C3-symmetry structure in which all three methylene linkers are oriented toward the convex face of a folded bowl-shaped conformation (Fig. 3a). On the other hand, two benzimidazole moieties of anti-3 are directly connected to a p-phenylene moiety to form a 1,4-bis(benzimidazol-2-yl)benzene moiety with an extended conjugation system, which gives a warped C1-symmetry structure (Fig. 3b) in combination with the rest of the structure containing a 2-phenylbenzimidazole and a benzene moiety. It is worth noting here that both structures are chiral, as evident from the fact that both (P)- and (M)-stereoisomers coexist in the ratio of 1 to 1 in each crystal of syn-2 and anti-3.11 For instance, two molecules of syn-2 formed a homochiral dimer sandwiching one CH2Cl2 molecule between two small cavities of syn-2, and the other enantiomeric dimers were lined up in parallel (Fig. 3c). For anti-3, (P)- or (M)-stereoisomers of anti-3 were stacked to form homochiral columns. The interstitial spaces formed between enantiomerically paired columns were occupied by CHCl3 molecules (Fig. 3d).
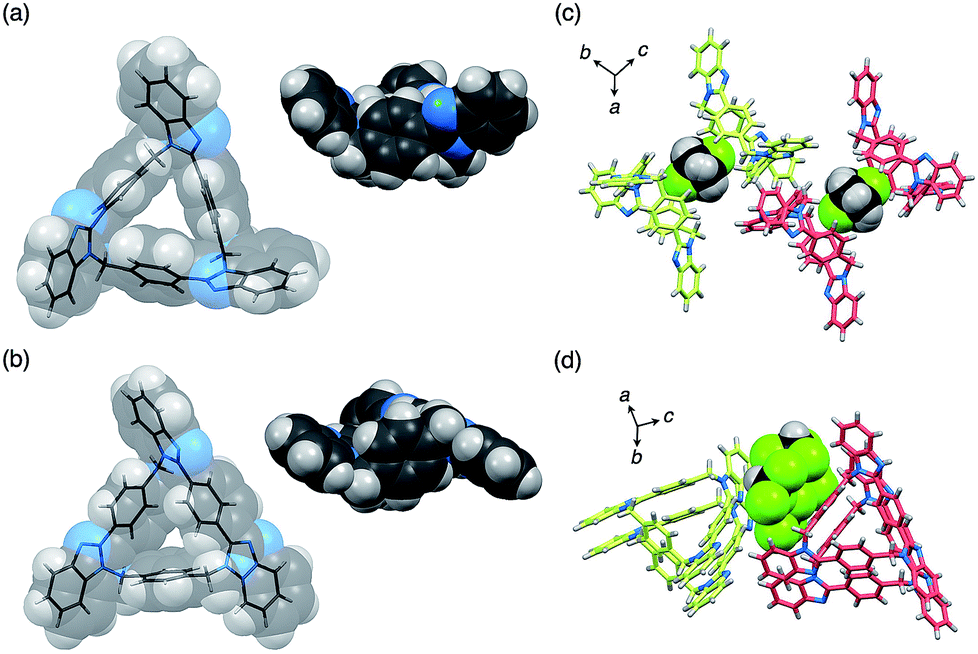 | ||
| Fig. 3 Molecular structures of (a) syn-2 and (b) anti-3 (top and side views), and crystal packing structures of (c) syn-2 and (d) anti-3. In (c) and (d), macrocycles and solvents are represented by stick and space-filling models, respectively, and colors for the carbon atoms of (P)- and (M)-stereoisomers are yellow-green and pale-red, respectively. | ||
We next evaluated the solution-state structures of syn-2 and anti-3 in CDCl3 based on 1H NMR spectroscopy (Fig. 2c and d). First, the signals of some p-phenylene protons next to methylene groups were observed in the range from 6.5 to 6.8 ppm, significantly upfield shifted from typical p-phenylene signals. These characteristic p-phenylene signals suggest that the folded conformations of syn-2 and anti-3 observed in the crystal state, in which the p-phenylene protons face benzimidazole moieties, are maintained also in CDCl3. On the other hand, the 1H NMR spectra of syn-2 and anti-3 in CDCl3 indicate the formation of C3h- and Cs-symmetry structures at 300 K, respectively. This higher symmetry in solution suggests that the rate of racemization of syn-2 and anti-3 between (P)- and (M)-isomers is significantly faster than the NMR timescale in CDCl3.
The dynamic behaviors of syn-2 and anti-3 in solution were then examined by VT 1H NMR measurements in CD2Cl2. When the temperature of syn-2 in CD2Cl2 was lowered to 203 K, the methylene signals around 5.65 ppm split into geminally coupled two doublet signals. In contrast, the other aromatic signals did not split except for the appearance of small broad signals probably due to the presence of a minor conformational isomer at 203 K (Fig. 4). The desymmetrization of only the methylene protons indicates that the temperature-dependent dynamic behavior is mainly derived from the racemization of (P)- and (M)-stereoisomers by the bowl-to-bowl inversion that is slower than the NMR timescale at 203 K. To estimate the coalescence temperature (Tc) and inversion barrier ΔG‡c of syn-2 at Tc, the Eyring plot was drawn based on the dynamic 1H NMR line-shape simulation (Fig. S24‡). As a result, the Tc and ΔG‡c values were estimated to be 231 K and 11.2 ± 0.6 kcal mol−1, respectively, and ΔH‡ and ΔS‡ were also calculated to be 17.8 ± 0.3 kcal mol−1 and 28.5 ± 1.4 cal mol−1 K−1, respectively. Macrocycle anti-3 also exhibited similar behaviors, and VT 1H NMR spectra were similarly analyzed to estimate Tc, ΔG‡c, ΔH‡, and ΔS‡, which were found to be 230 K, 11.0 ± 0.4 kcal mol−1, 13.8 ± 0.2 kcal mol−1, and 12.3 ± 0.9 cal mol−1 K−1, respectively (Fig. S25 and S26‡). The resultant inversion barriers of syn-2 (11.2 kcal mol−1) and anti-3 (11.0 kcal mol−1) were found to be almost the same, and it should be noted that the inversion barriers are comparable with that of corannulene (11.5 kcal mol−1),12 which is a representative bowl-shaped hydrocarbon.
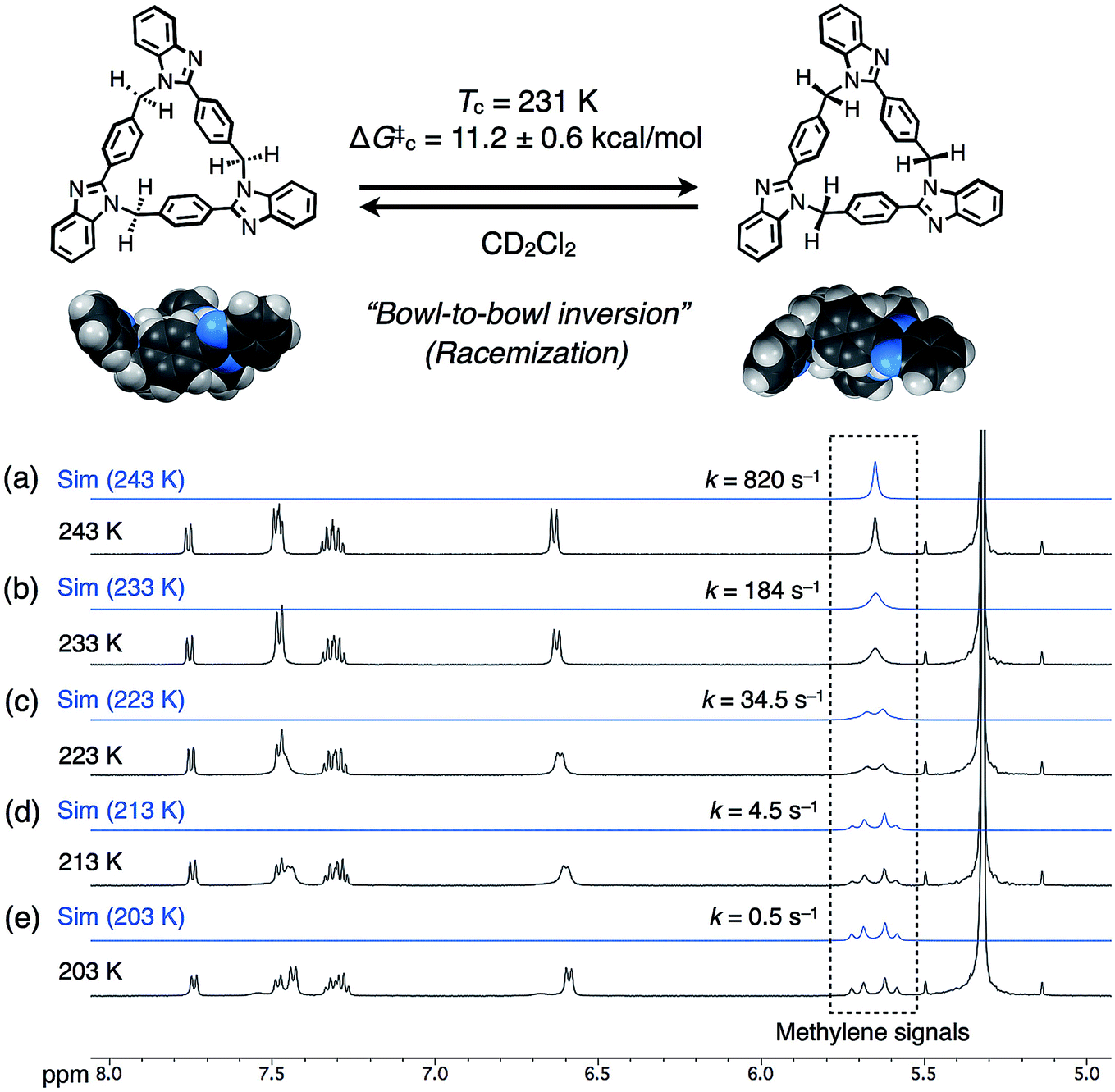 | ||
| Fig. 4 Schematic representation of the bowl-to-bowl inversion of syn-2, and simulated (blue lines, 5.5–5.8 ppm) and obtained (black lines, 5.0–8.0 ppm) 1H NMR spectra (500 MHz, CD2Cl2) of syn-2 at (a) 243, (b) 233, (c) 223, (d) 213, and (e) 203 K. | ||
The optical properties of syn-2 and anti-3 were examined by UV-vis absorption and fluorescence spectroscopies (Fig. 5). The UV-vis spectra of syn-2 and anti-3 in CDCl3 at 298 K showed absorption bands at λmax = 296 nm and 312 nm, respectively. The red-shifted absorption of anti-3 can be best explained by the presence of the 1,4-bis(benzimidazol-2-yl)benzene moiety with an extended conjugation system. This structural feature of anti-3 also gave fluorescence at longer wavelength (λmax = 390 nm) than that of syn-2 (λmax = 339, 354, 369 nm) in CHCl3 at 293 K. Quantum yields of syn-2 and anti-3 in degassed CHCl3 were determined to be 0.60 and 0.71, respectively, which are comparable to the quantum yield of 1-methyl-2-phenylbenzimidazole (ϕFL = 0.70), the structural unit of syn-2 and anti-3.13
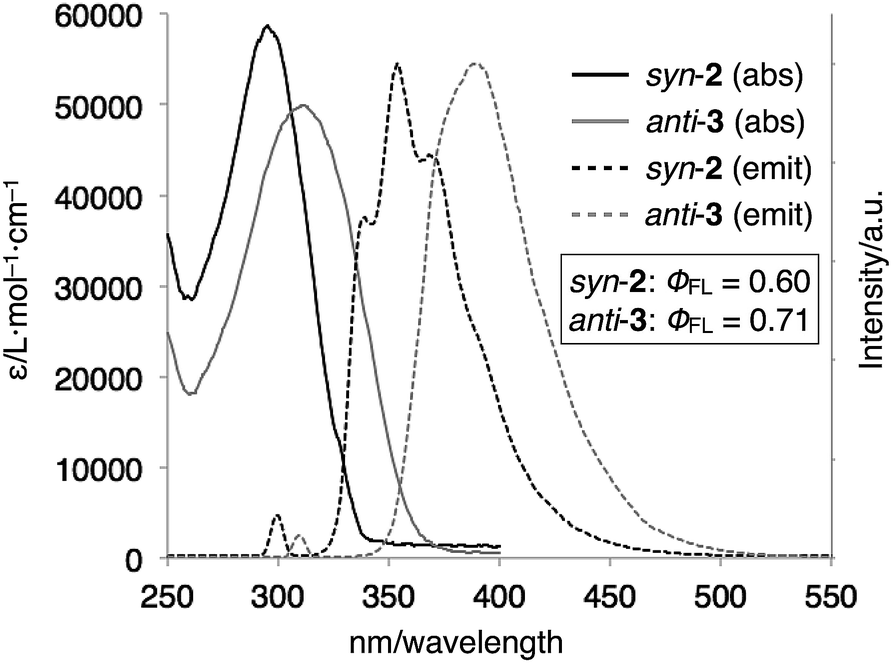 | ||
| Fig. 5 UV-vis absorption and normalized fluorescence spectra (CHCl3, 298 K) of syn-2 (1.5 μM, λex = 300 nm) and anti-3 (3.0 μM, λex = 310 nm). | ||
As benzimidazole[3]arenes possess three imidazole nitrogen atoms on the periphery of the macrocyclic skeletons, these nitrogen atoms were expected for further functionalization (Fig. 6a). For instance, anti-3 underwent tri-methylation by reaction with CH3I, and the resulting tri-methylated product was found to be soluble in water, maintaining similar fluorescence properties (Fig. S27–S29‡). In addition, anti-3 was fully protonated by mixing with p-toluenesulfonic acid monohydrate as confirmed by single-crystal XRD analysis. In the resultant crystal structure of [H3(anti-3)(p-TsO)3]·(H2O)3, all peripheral nitrogen atoms were protonated, interacting with p-TsO− anions (Fig. 6b). The p-TsO− anions further formed a hydrogen bonding network with water molecules along the b-axis. As a result, the crystal adopted a layer-by-layer structure composed of the macrocycle layers of anti-3 and the hydrated layers of p-TsO− anions (Fig. S32‡).
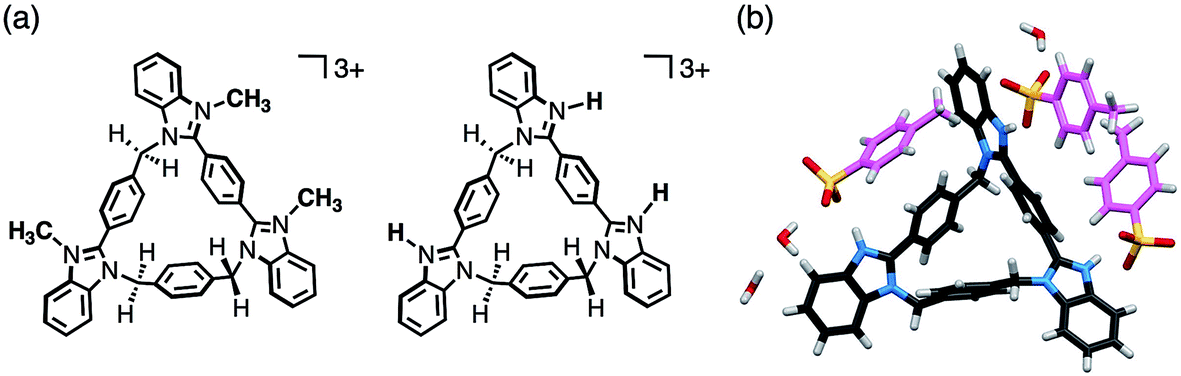 | ||
| Fig. 6 (a) Chemical structures of tri-methylated and tri-protonated anti-3. (b) Crystal structure of [H3(anti-3)(p-TsO)3]·(H2O)3. Disordered water molecules are omitted for clarity. The carbon atoms of p-TsO are shown in pink. | ||
Finally, to discuss the reaction mechanism of this post-macrocyclization transformation, a diluted CD3CN solution of 1 (0.34 mM) and Cu(OAc)2·H2O (1.1 mM) was mixed at room temperature to monitor the reaction by 1H NMR measurements. We found that a yellow clear solution obtained after 20 min contained two different intermediates. Based on the chemical shifts, integral ratios, 1H–1H COSY, and NOESY correlations (Fig. S33–S35‡), the intermediates were most likely to be macrocyclic triimine compounds 2′ and 3′ (Fig. 7). For instance, singlet signals around 8.7–8.8 ppm and broad signals around 5.3–5.6 ppm can be assigned to imine and amine protons, respectively. The remaining methylene protons were also observed around 4.3–4.6 ppm. The signal patterns suggested that the main species was Cs-symmetrical macrocyclic triimine 3′, and the minor one appeared to be C3h-symmetrical triimine 2′. The molar ratio of 2′ to 3′ was ca. 1:10, and further reaction of this solution for 8 days resulted in the formation of syn-2 and anti-3 in the molar ratio of ca. 1:10, which was almost the same as that under the synthetic conditions at 55 °C (Fig. 2b). This result indicated that the final ratio of syn-2 to anti-3 was determined at the first partial oxidation of 1 into triimine 2′ and 3′ (Fig. 7). The formation of the triimine intermediates was also confirmed by the ESI-TOF mass spectrometry measurement of a similarly prepared CH3CN reaction solution after 20 min (m/z = 647.26 for [1–6H + Na]+) (Fig. S36–S38‡).
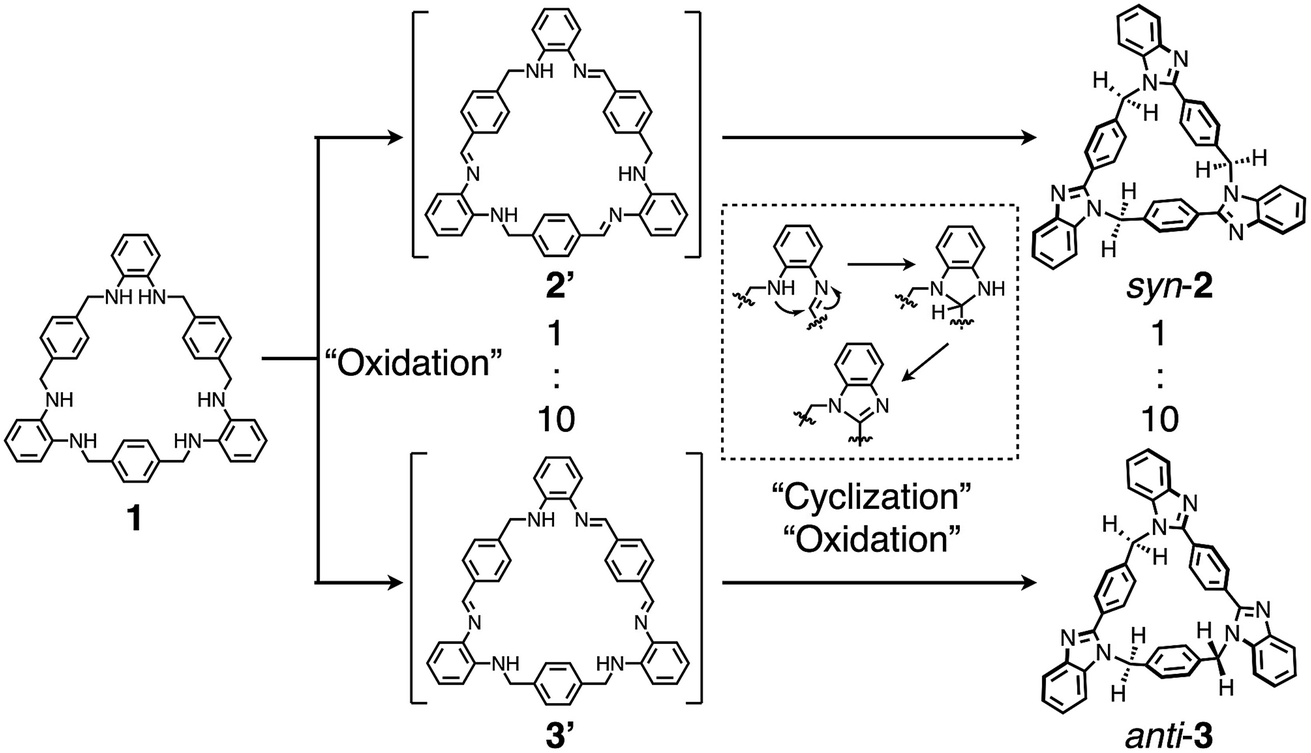 | ||
| Fig. 7 Plausible reaction mechanism of the formation of syn-2 and anti-3via intermediates 2′ and 3′. | ||
The molar ratio of the formation of 2′ and 3′ (ca. 1:10) was more biased than the statistical ratio of 1:3. The more biased formation of 3′ is possibly attributed to the superiority in the kinetic and/or thermodynamic stability due to its extended conjugated structure. In the next reaction, the resultant imine carbons are subject to cyclization by the attack of the neighboring amino groups, followed by oxidative aromatization to afford benzimidazole moieties (Fig. 7). On the other hand, predominant formation of syn-2 was not observed by changing the amount of Cu(OAc)2·H2O, reaction temperature, or solvent ratio as far as we examined.
In a plausible reaction mechanism shown in Fig. 7, Cu(OAc)2·H2O serves as an oxidant and a Lewis acid to facilitate several steps including oxidation to imine, cyclization, and oxidative aromatization. Because the conversion from 1 to syn-2 or anti-3 is a twelve-electron oxidation reaction, an equimolar amount of Cu(OAc)2·H2O should work as the catalyst in air. Moreover, we found that this reaction proceeded at higher temperature without Cu(OAc)2·H2O. When a solution of 1 in CD3CN was heated at 180 °C under microwave irradiation, syn-2 and anti-3 were formed after 105 min with some impurities (Fig. S39‡). Another oxidant, 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) (12 equiv.), also converted 1 into syn-2 and anti-3, but a substantial amount of byproducts was also formed (Fig. S40‡). These results clearly indicate that O2 in air is the oxidant and that Cu(OAc)2·H2O catalyzes both the oxidation and cyclization steps to cleanly promote this reaction.
Conclusions
In summary, two isomeric benzimidazole[3]arenes syn-2 and anti-3 have been rationally synthesized as a new series of calix[n]arene analogues by the CuII-catalyzed post-macrocyclization transformation reaction of 1via two triimine intermediates 2′ and 3′. The C3- and C1-symmetrical macrocyclic structures and their dynamic inversion behavior in solution were revealed by single-crystal XRD and VT NMR analyses, respectively. The three benzimidazole moieties also led to strong fluorescence and allowed chemical modification at the macrocyclic periphery. As another potential use of benzimidazole[3]arenes, study on metal complexation is in progress, and the resultant supramolecular structures will be reported elsewhere as separate studies. The present rational synthetic method and the potential applications are thus expected to make benzimidazole[3]arenes versatile as with calix[n]arenes and pillar[n]arenes that are being utilized around the world.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the JSPS KAKENHI, Grant Numbers JP16H06509 (Coordination Asymmetry) to M. S. and JP15H05478 (Encouragement of Young Scientists (A)) and JP18H04502 (Soft Crystals) to S. T.Notes and references
-
(a)
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, New York, 2nd edn, 2009, DOI: 10.1002/9780470740880 Search PubMed
; (b) Supramol. Chem., Special Issue of Supramolecular Chemistry for Calix2017, ed. Y. Liu, Taylor & Francis Group, Abingdon, 2018, vol. 30, pp. 7–8 Search PubMed
-
(a) T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef PubMed
-
(a) A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC
-
(a) P. A. Gale, J. L. Sessler, V. Král and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef
- Macrocycles with benzimidazole rings as the backbone:
(a) W. Clegg, J. C. Lockhart and F. H. Musa, J. Chem. Soc., Dalton Trans., 1986, 47–53 RSC
- Benzimidazole-bearing resorcin[4]arene and calix[4]arenes:
(a) L. Trembleau and J. Rebek Jr, Science, 2003, 301, 1219–1220 CrossRef PubMed
-
(a) N. Singh and D. O. Jang, Org. Lett., 2007, 9, 1991–1994 CrossRef PubMed
-
(a) S. Tashiro, R. Kubota and M. Shionoya, J. Am. Chem. Soc., 2012, 134, 2461–2464 CrossRef PubMed
-
(a) W. V. Rossom, W. Maes, L. Kishore, M. Ovaere, L. V. Meervelt and W. Dehaen, Org. Lett., 2008, 10, 585–588 CrossRef PubMed
-
(a) H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795–1804 RSC
- Here the (P)- or (M)-helical isomers of syn-2 and anti-3 are tentatively defined as the clockwise or counterclockwise direction of benzimidaozle moieties (benzimidazole → p-phenylene), respectively, when viewed from the bottom in which more methylene groups are projecting (Fig. S22‡).
-
(a) T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef PubMed
- J. Catalán, E. Mena, F. Fabero and F. Amat-Guerri, J. Chem. Phys., 1992, 96, 2005–2016 CrossRef
Footnotes |
| † Dedicated to the late Mr Akihiro Mizoguchi for his contribution to the development of this study. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and characterization data. CCDC 1585798–1585799 and 1585801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03086c |
| § Current address: Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Kyoto 615-8510, Japan. |
| This journal is © The Royal Society of Chemistry 2018 |