
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmart composite films of nanometric thickness based on copper–iodine coordination polymers. Toward sensors†‡
Javier
Conesa-Egea
ab,
Noemí
Nogal
a,
José Ignacio
Martínez
 c,
Vanesa
Fernández-Moreira
d,
Ulises R.
Rodríguez-Mendoza
e,
Javier
González-Platas
e,
Carlos J.
Gómez-García
f,
Salomé
Delgado
ag,
Félix
Zamora
*abg and
Pilar
Amo-Ochoa
*ag
c,
Vanesa
Fernández-Moreira
d,
Ulises R.
Rodríguez-Mendoza
e,
Javier
González-Platas
e,
Carlos J.
Gómez-García
f,
Salomé
Delgado
ag,
Félix
Zamora
*abg and
Pilar
Amo-Ochoa
*ag
aDepartamento de Química Inorgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: felix.zamora@uam.es; pilar.amo@uam.es
bCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
cDepartamento de Nanoestructuras, Superficies, Recubrimientos y Astrofísica Molecular, Instituto de Ciencia de Materiales de Madrid (ICMM-CSIC), 28049 Madrid, Spain
dDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain
eDepartamento de Física and Instituto de Materiales y Nanotecnología (IMN), Universidad de La Laguna, Avda. Astrofísico Fco. Sánchez s/n, La Laguna, Tenerife E-38204, Spain
fInstituto de Ciencia Molecular (ICMol), Universidad de Valencia. C/Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain
gInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 23rd August 2018
Abstract
One-pot reactions between CuI and methyl or methyl 2-amino-isonicotinate give rise to the formation of two coordination polymers (CPs) based on double zig–zag Cu2I2 chains. The presence of a NH2 group in the isonicotinate ligand produces different supramolecular interactions affecting the Cu–Cu distances and symmetry of the Cu2I2 chains. These structural variations significantly modulate their physical properties. Thus, both CPs are semiconductors and also show reversible thermo/mechanoluminescence. X-ray diffraction studies carried out under different temperature and pressure conditions in combination with theoretical calculations have been used to rationalize the multi-stimuli-responsive properties. Importantly, a bottom-up procedure based on fast precipitation leads to nanofibers of both CPs. The dimensions of these nanofibres enable the preparation of thermo/mechanochromic film composites with polyvinylidene difluoride. These films are tens of nanometers in thickness while being centimeters in length, representing smaller thicknesses so far reported for thin-film composites. This nanomaterial integration of CPs could represent a source of alternative nanomaterials for opto-electronic device fabrication.
Introduction
Coordination polymers (CPs) are a family of multifunctional materials showing high structural variety and interesting physico-chemical properties.1 Architectures and properties are directly based on the selection of the building blocks, metal entities and organic/inorganic ligands, required for the construction of their networks. The large number of metal ions and the almost unlimited ligands available to be combined allow obtaining a large number of (multi)functional coordination polymers.2 Some of these compounds also possess dynamic structures, which make them suitable for the preparation of stimuli-response materials, with potential applications as sensors,3 photo-luminescent switches,4 or optical recording devices.5 In some cases, the addition of properties allows the possibility to create new multi-functional stimuli-response materials3,6 whose combination of remarkable physical properties7 has enabled the development of novel devices and potential applications, e.g. thermochromics in paints, plastics and textiles, electrochromics in car mirrors and smart windows, and solvatochromics in biological probes, among others.In this context, CPs based on Cu(I)–I double chains have been shown to be a subfamily with remarkable electrical3,6a,8 and optical properties3,6a,9 but, even more importantly, with very sensitive and flexible structures. Thus, the structures of these materials undergo slight structural changes under physical and/or chemical stimuli, such as vapours, temperature and/or pressure, significantly affecting their physical properties e.g. conductivity and/or emission.
Interestingly, mechanoluminescence in CPs is mostly associated with a phase transition such as a crystal-to-amorphous transition10 and/or an aggregation state change due to mechanical stresses, such as grinding or compressing.11 There are few studies focused on understanding the mechanisms involved in these changes. Very recently Kwon et al. (2017)12 and Conesa-Egea et al. (2017)13 have reported new CPs based on Cu(I)–I double chains undergoing reversible crystal-to-crystal structural changes from ambient to high pressures. These studies allow examining the reversibility of luminescence mechanochromism processes.
These aspects make CPs interesting materials to form novel multi-stimuli-response composites. In this sense, different studies have shown the ability to generate composites in which the integrated CPs retain their original properties.14 The new composites integrating thermoluminescent and/or mechanoluminescent CPs within organic polymers will represent a field which will allow the design of new devices with potential applications in the aerospace,15 transport, packaging,16 and structural failure or impact industry, among others.17 We have to mention that it is a little studied field, especially in the area of mechanoluminescent composite materials where the few published examples are based on the use of microparticles of ZnS with organic matrices.18
Herein we present multifunctional one-dimensional Cu2I2 double-chain CPs anchored with functionalized isonicotinate ligands (MeIN = methyl isonicotinate and NH2–MeIN = methyl 2-aminoisonicotinate). X-ray diffraction studies prove that the presence or absence of an amino group produces a significantly different supramolecular organization affecting the Cu–Cu distances and symmetry of the Cu2I2 chains. Moreover, both CPs are semiconductors and reversibly thermo/mechanoluminescent. Fast precipitation reactions have been successfully used to obtain both CPs as nanofibers. A comparative study of the optical properties of both CPs at micro- and nanoscales has been carried out showing some differences. Finally, the availability of nanomaterials and the deposition method have allowed us to obtain highly homogenous ultra-thin-film composites of the CPs with poly(vinylidene fluoride) (PVDF).19
Results and discussion
Synthesis and structural characterization
The direct reactions between CuI and methyl isonicotinate (MeIN)6a or methyl 2-aminoisonicotinate (NH2–MeIN) in acetonitrile at 25 °C give rise, instantly, to the isolation of nano-crystalline powder materials 1n ([Cu(MeIN)I]n) and 2n ([Cu(NH2–MeIN)I]n), respectively. Micron-size crystals of 1 and 2, named 1m and 2m, can be isolated upon allowing to stand the solutions at 25 °C for 72 h.The structures of 1 and 2 both consist of Cu2II2 double chains anchored with isonicotinate functionalized ligands. The coordination environment around each Cu(I) centre is tetrahedral. The Cu is coordinated to three bridging μ3-iodides and a pyridine nitrogen atom of the terminal ligand (Fig. 1, S1 and S2 and Table S1‡). The isonicotinate ligands are oriented in a different way in both compounds. Thus, the I–Cu–N–C torsion angles are 128.9(11)° and 158.1(8)°, for 1 and 2, respectively.
 | ||
| Fig. 1 Crystal structures of compounds [Cu(MeIN)I]n1 (A) and [Cu(NH2–MeIN)I]n2 (B). Red dashed lines indicate hydrogen bonding interactions; Cu: orange; I: violet; C: grey; N; blue; O: red. | ||
The iodide is tri-coordinated in a distorted pyramidal geometry. The stair-type chain can also be described as composed of planar Cu2I2 squares that share two opposite sides with angles between adjacent squares.
The most significant difference in the packing of the Cu2I2 chains between 1 and 2 arises from the presence of amino groups in 2 which form weak donor–acceptor H-bond interactions with the oxygen of the carboxylic groups of the parallel neighbouring Cu2I2 chain (Fig. 1b). These supramolecular interactions built a rather different 3D network in 2 when compared to the one formed in 1 by only weak van der Waals supramolecular interactions. Interestingly, the Cu–Cu distances along the Cu2I2 chains in 1 and 2 show significant differences that are probably a consequence of the different interchain interactions above mentioned. Thus, while the Cu2I2 chains in 1 are very similar (2.751 and 2.818 Å) leading to an almost symmetric chain, two very different Cu–Cu distances are found in compound 2 (2.682 and 3.514 Å), giving rise to a chain that can be better described as an organization of Cu2I2 dimers connected by weak Cu⋯Cu interactions.
Morphological studies
Fig. 2a and 3a show SEM images of 1m and 2m. The length of these crystals is 2000 ± 500 μm and 1500 ± 500 μm, while the width is 150 ± 50 μm and 78 ± 30 μm, respectively. Fast precipitation from the reaction medium allows the isolation of the CPs as nanofibers, 1n and 2n, respectively. Fig. 2c, d and 3c, d show SEM images of nanofibers of 1n and 2n. The fibres are as long as 250 μm in length for both compounds and 811 ± 348 nm for 1n (Fig. 2b–d) and 501 ± 389 nm for 2n (Fig. 3b–d) in width. | ||
| Fig. 2 (A) SEM image of compound [Cu(MeIN)I]n (1m) microcrystals obtained by slow evaporation at 25 °C. (B–D) SEM images of compound [Cu(MeIN)I]n (1n) nanofibers obtained by fast precipitation at 25 °C. | ||
 | ||
| Fig. 3 (A) SEM image of [Cu(NH2–MeIN)I]n microcrystals (2m). (B–D) SEM images of [Cu(NH2–MeIN)I]n nanofibers (2n). | ||
The atomic force microscopy (AFM) study of the fibres obtained from freshly prepared diluted suspensions of 1n and 2n deposited by drop-casting and/or dip-coating on SiO2 shows the formation of bundles, where these fibres displayed heights ranging between 50 and 550 nm for 1n (Fig. 4a and b) and from 5 to 15 nm for 2n (Fig. 4c, d and S10‡). Therefore, fibres of 1n and 2n clearly show a nanometric dimension.
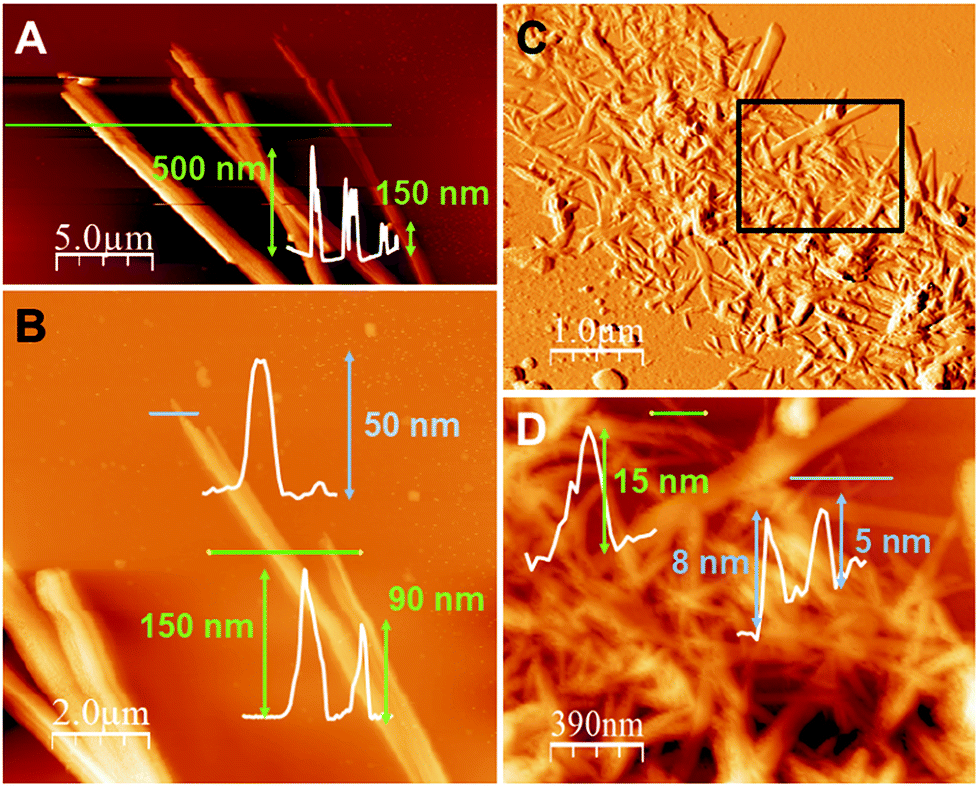 | ||
| Fig. 4 (A) AFM image of [Cu(MeIN)I]n (1n) nano- and sub-microfibers on SiO2 prepared by drop casting, with their height profile across the green line. (B) A zoomed area of (A), with its height profiles across the blue and green lines. (C) AFM image of [Cu(NH2–MeIN)I]n (2n) nanofibers on SiO2 prepared by dip-coating. (D) A zoomed area of (C) the black rectangle, with its height profiles across the green and blue lines. | ||
Switchable luminescence properties
CPs containing d10 metal centres and metallophilic interactions are currently attracting attention due to their excellent photofunctional properties20 which can be switched by environmental changes. For example, emission of Cu(I) complexes can be modulated either by the coordination environment around the metal, by the use of different organic ligands, or by changes in the structure.21 Additionally, studies carried out on some CPs with different nature show that a reduction of the material size can also affect their emission.22 Therefore, the structural analysis of 1 and 2 suggests that they are interesting candidates to carry out emission studies at variable temperature and pressure. Thus, from the qualitative point of view, a naked eye experiment shows that the excitation of 1m/n and 2m/n with an UV lamp (λexc = 365 nm or 312 nm) at room temperature produces a strong orange emission for 1m/n and weak yellow emission for 2m/n (Fig. 5, S13 and S14‡). Upon cooling the materials to 80 K, 1m/n show a slight decrease in their emission intensities, while 2m/n exhibit a significant increase in their emission intensities (Fig. 5b, d, S13 and S14‡). These processes are reversible, and thus warming up the materials from 80 K to 300 K produces a gradual recovery of their initial properties. The comparison between micrometric and nanometric samples shows an influence only in the emission intensity, being more significant for 1n (Fig. 5 and S13‡).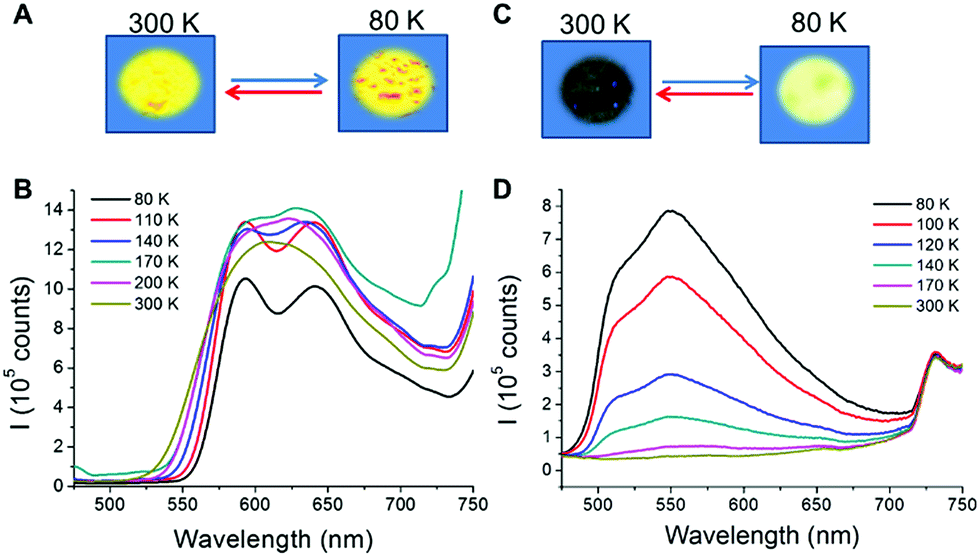 | ||
| Fig. 5 (A) Representation showing the temperature-dependence behaviour of 1n in the solid state under UV lamp excitation (λexc = 365 nm) at room temperature (left) and under liquid nitrogen (right). (B) Temperature-dependent luminescence spectra of 1n. (C) Representation showing the thermochromic behaviour of 2n in the solid state under UV lamp excitation (λexc = 365 nm) at room temperature (left) and under liquid nitrogen (right). (D) Temperature-dependent luminescence spectra of 2n. | ||
To evaluate in further detail these qualitative observations, emission spectra at variable temperature of 1m and 1n as well as 2m and 2n were recorded from 80 to 300 K (Fig. 5 and S13‡). Thus, at 300 K the excitation of the samples with λexc = 440 nm produces a strong emission with an asymmetric band centred at λem = 610 and 614 nm for 1m and 1n, respectively (Fig. 5b and S13a‡), and a weak emission at λemca. 550 nm for 2m and 2n (Fig. 5d and S13b‡). By lowering the temperature from 300 to 80 K, a progressive decrease of the emission intensity (up to 5 times lower at 80 than at 300 K) is observed for 1m, while a progressive increase (up to 50 times higher at 80 than at 300 K) is observed for 2m. In addition, it is remarkable that between 140 and 80 K a structured band with two emission maxima, of similar intensities, centred at 593, 642 nm and a shoulder at ca. 707 nm appeared for 1m and 1n. On the other hand, for 2m and 2n a very asymmetric band centred at ca. 550 nm is observed at 140 and, as temperature decreases, the intensity of this asymmetric band increases. However, in 2n this band is not resolved like a structured band, and a maximum centred at 550 nm together with a shoulder at 516 nm can be observed.
The luminescence emission bands observed in the case of [Cu(L)I]n polymers, where L is an N-donor ligand, are mostly assigned to iodide-to-copper(I) charge transfer (ICCT), metal to ligand charge transfer (MLCT), halogen-to-ligand charge transfer (HLCT), cluster-centred (CC) transition, or the admixture of more than one of these contributions.16 In order to evaluate the emission arising from a ligand-centred (LC) transition, the free ligands methyl isonicotinate and 2-amino methyl isonicotinate were also investigated at 300 K (Fig. S12‡). The emission energy of each Cu(I) polymer is much lower than that of the corresponding free ligands (439 and 410 nm respectively), which rules out the intraligand π–π* transitions.
The stair-type chain structure adopted by 1 and 2 is common for I–Cu–N donor ligand coordination polymers and the emission origin, centred around 520–640 nm, can be most likely due to a mixed iodide-to-ligand and metal-to-ligand charge transfer (IL/MLCT) excited state [3(I + M)LCT].15,23 This assignment is supported when comparing the emission spectra of 1 and 2, in which we can see that the emission is affected by the nature of the substituent on the pyridine ring. Thus, 2 shows emission bands at higher energies than 1, in accordance with a higher HOMO–LUMO gap. Finally, the broad and unstructured bands observed at room temperature are also in accordance with the assignation made as a combination of 3(I + M)LCT states.24
To rationalize the origin of the changes in the emission by lowering the temperature, crystal structures for 1m and 2m were solved at 110 K (Fig. 6). We have previously reported on the unusual structure of 1m,6a where all metallic distances are shorter than 2.80 Å, which is twice the van der Waals radius (only four structures have been reported in the CSD database version 5.36 with similar characteristics25,26). However, 2m adopts the most common disposition of the Cu2I2 units in these stair-step chain structures, showing alternation of long (>2.80 Å) and short (<2.80 Å) distances between Cu and Cu atoms. These structural differences observed along the Cu2I2 chains, symmetric for 1versus asymmetric for 2 (Fig. 1), provoke changes in the emission.
 | ||
| Fig. 6 Cu–I distances (black, Å) and angles (red, degrees) of the 1D chain of compound [Cu(NH2–MeIN)I]n (2m) at 296 K (A) and 110 K (B). | ||
Comparing the thermochromic properties of both compounds, 2m does not display emission at room temperature but, upon cooling, a bright yellow emission is observed increasing the intensity of emission with decreasing temperature. As is described below, the crystal structures of compound 2m at 296 and 110 K show that the space group and the general structural features do not change with the temperature (Tables S1 and S2,‡Fig. 1 and 6). Thus, the Cu–I and Cu⋯Cu distances show the expected decrease with decreasing temperature and the increase observed in the emission intensity upon cooling is in accordance with the increase in structural rigidity, decreasing the non-radiative rate constant27 (Fig. 6). As we have previously described,6a we have also carried out a structural analysis of 1m as a function of temperature (296, 200 and 110 K). Although the space group of 1m is the same at those temperatures, it shows a transition at 125–145 K, confirmed by the study carried out on the thermal dependence of the unit cell parameters. Thus, this structural transition can be attributed to a slight change in the asymmetry of the Cu–Irail bond lengths along the chain, increasing from 0.0171 Å at 296 K to 0.0243 Å at 200 K but decreasing to 0.0208 at 110 K. These changes in the structure will likely induce a variation in the shape and energy of the HOMO and LUMO when changing from room temperature to 80 K.6a
The lifetimes measured for 1n and 1m at 300 and 80 K were 8.1–7.1 and 6.6–8.1 μs, respectively, which fall in the microsecond-order decay lifetime range and are assigned to phosphorescence, arising from a triplet state. The fact that the structured band observed at 80 K, with maxima centred at 592 and 640 nm, has similar lifetime values seems to indicate that both emissions belong to the same structured band emitting from the same excited state and, alike, lifetime measurements for both temperatures suggest that there is only one emission level without any geometrical distortions in the excited state. For 2n, the lifetime measured at 80 K was 1.25 and 1.15 ms for the emissions centred at 516 and 550 nm, respectively. Similar values are also indicative of emissions from the same excited state. On the other hand, 2m has a higher lifetime than 2n, being 1.37 ms for the emission centred at 550 nm. 2m has a higher lifetime than 1m, suggesting a greater rigidity in its structure due to the presence of the amino group attached to the pyridine ring.28
Theoretical calculations of the excitation spectra for the two structures of 1 and 2 resolved by X-ray diffraction at low (110 K) and high (296 K) temperatures have been carried out to rationalize the thermochromic effect evidenced by the experiments. Interestingly, our theoretical approach is able to capture the reduction in the photoluminescence performance observed in compound 2 and very weak changes in the case of compound 1 from the low-temperature to the high-temperature regime. Fig. 7 shows the prominent excitation feature located at around 580 nm for the case of T = 110 K. Nevertheless, the intensity of the peaks decreases by around 35% (2) for the cases of T = 296 K and T = 110 K, with the maximum of the peak located at 600 nm for compound 2 and barely any displacement for compound 1. The decrease in the photoluminescence behaviour may have its origin in the thermal fluctuations that tend to broaden both the VB and CB, subsequently reducing the efficiency of the metal–ligand transition for the case of the high-temperature regime as compared with the T = 110 K case.
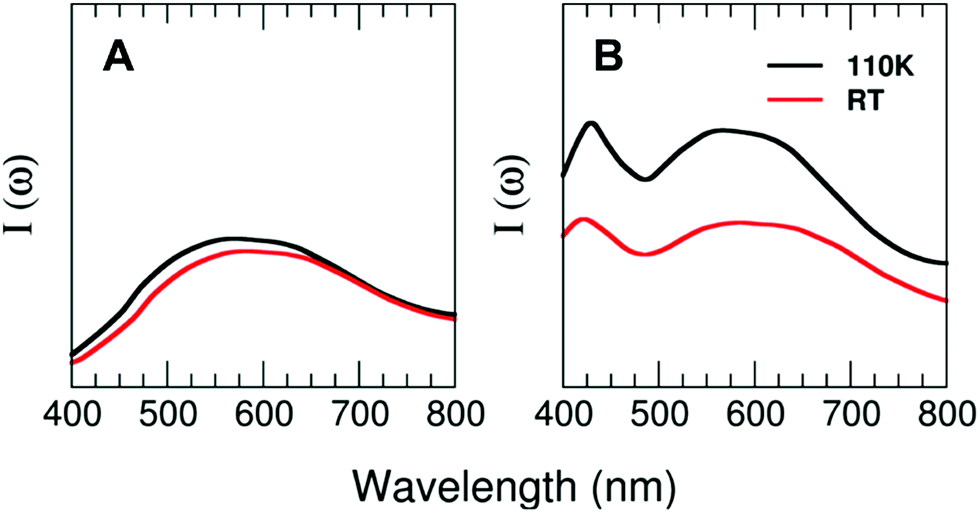 | ||
| Fig. 7 Computed TDDFT photoexcitation spectra (in arb. units) for 1 (A) and 2 (B) as a function of the photon wavelength (in nm) for two different temperatures at standard pressure (T = 110 and 296 K). Ordinate-scale is the same in both graphs for a better comparison. | ||
To study the potential mechanoluminescence behaviour of these compounds, we have prepared pressed pellets at different pressures with the materials in the form of nano- and micro-metric powders for both compounds. The pressed pellets of compound 1m/n show slight changes, their emission going from an intense orange to a weaker reddish orange, which does not change upon decreasing the temperature to 77 K (Fig. S14c‡). However, the pressed pellets of 2n/m (at different pressures from 1 to 5 GPa) show a significant change in their luminescence. Indeed, upon applying pressure their emission is quenched at both 298 and 80 K (Fig. S14f‡), but it is recovered when the pellet is decompressed by grinding, indicating reversibility of the emission with the pressure. To gain knowledge about this behaviour, initial X-ray powder diffraction patterns were collected from the as-synthetized materials (2n/m) in the form of pressed and ground pellets. The results do not show significant differences, suggesting that the structures are the same (Fig. S9‡).
To gain knowledge about the origin of the changes in the emission with the pressure, X-ray diffraction studies on a single crystal of 2m were carried out. We have selected 2 for these studies based on the preliminary results in which 1 did not show significant changes with the pressure. Thus, a crystal of 2m has been introduced into a cell together with a methanol–ethanol–water mixture (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) as a pressure-transmitting medium, which remains hydrostatic in the range of pressure used in our experiment (up to 7.16 GPa).29 The data obtained from this experiment allow calculating the isothermal equations of state (EoS) and the bulk modulus (K0 = 9.7(2 GPa)) (Section S1, Fig. S3–S6, and Table S3‡), explaining the degree of material compressibility. The bulk modulus obtained for 2 is close to the lower limit typical of organometallic compounds (10–20 GPa).30 In general, these lower values are attributable to the deformability of the intermolecular interactions or voids present in the structure. Similar copper iodide Cu–I ladders under hydrostatic pressure have been studied recently, reporting K0 values of 10.2(2) and 14.1(3) GPa and K0′ values of 8.1(3) and 7.4(2) (ref. 9d and 13) demonstrating elastic properties like our title compound.
:3:1) as a pressure-transmitting medium, which remains hydrostatic in the range of pressure used in our experiment (up to 7.16 GPa).29 The data obtained from this experiment allow calculating the isothermal equations of state (EoS) and the bulk modulus (K0 = 9.7(2 GPa)) (Section S1, Fig. S3–S6, and Table S3‡), explaining the degree of material compressibility. The bulk modulus obtained for 2 is close to the lower limit typical of organometallic compounds (10–20 GPa).30 In general, these lower values are attributable to the deformability of the intermolecular interactions or voids present in the structure. Similar copper iodide Cu–I ladders under hydrostatic pressure have been studied recently, reporting K0 values of 10.2(2) and 14.1(3) GPa and K0′ values of 8.1(3) and 7.4(2) (ref. 9d and 13) demonstrating elastic properties like our title compound.
Additionally, the increase of pressure induces a significant variation in some distances and angles of the structure (Table S4 and Fig. S5‡). The evolution of these parameters shows that the main changes produce a slight deformation of the Cu–I ladders. The softest interactions are Cu⋯Cu respect to Cu–I bonds and the stiffest interactions are I⋯I (Fig. S5‡). The layers maintain their planarity and orientation but the small displacement of the ligands over one another appears to be the cause for the changes in angles in the unit-cell parameters with pressure.
Thus, among all the shortenings found in the distances, the most significant variation is found in Cu–Cu distances with a decrease of 10.33% (Cu–CuIII) when the pressure increases to 7.16 GPa (Table S4‡). Other significant distortions with the pressure are reflected in the corresponding Cu–I–Cu and I–Cu–I bond angles along the chain (80.95° for 0 GPa to 74.5° at 7.16 GPa and 104.3° for 0 GPa to 100.7 at 7.16 GPa, respectively) (Fig. S9‡). Finally, the interlayer distances also decrease when the pressure increases (Fig. S6‡).
High pressure emission has also been measured for 2m and 2n. Both spectra (Fig. 8a and b) show a spectral multicomponent structure. Some similarities between them are observed, particularly under ambient conditions (300 K, 0 GPa), where we can see a band located around 500 nm. However, upon applying pressure, differences between them are clearly observed, both in intensity and in spectral shape. Under ambient conditions and with excitation at 375 nm, 2m shows a structured and asymmetric broadband with three components centred approximately at 500 (green), 575 (yellow) and 615 (orange) nm. As pressure increases, changes in the shape and intensity are remarkable; the asymmetry and the structure of the band disappear at 1.3 GPa, retaining only the orange component, and the integrated intensity increases three fold. However, when increasing the pressure, gradual decreases of the intensity and large redshifts of the emission bands of around −440 cm−1 per GPa are also noticeable. Above 6 GPa, intensity is almost negligible (less than 15%) compared to that obtained under ambient conditions. Upon releasing pressure, the system seems to be almost reversible (Fig. 8a).
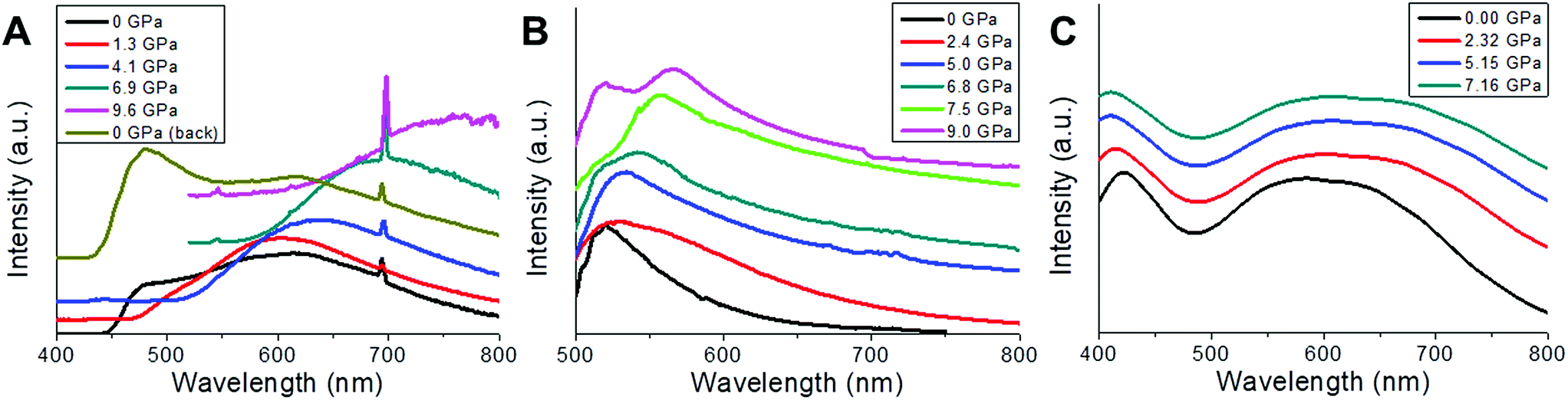 | ||
| Fig. 8 (A) Normalized luminescence spectra of 2m obtained at 298 K under 375 nm laser excitation for different externally applied hydrostatic pressures (P = 0, 1.3, 4.1, 6.9, and 9.6 GPa, and 0 GPa after decompressing) within a range between 400 and 800 nm. (B) Normalized luminescence spectra of 2n obtained at 25 °C under 458 nm laser excitation for different externally applied hydrostatic pressures (P = 0, 2.4, 5.0, 6.8, 7.5, and 9.0 GPa) within a range between 500 and 800 nm. The band at 700 nm corresponds to the ruby emission. (C) Computed TDDFT photoexcitation spectra (in arb. units) for 2 as a function of the photon wavelength (in nm) for different externally applied hydrostatic pressures (P = 0, 2.32, 5.15 and 7.16 GPa). Ordinate-scale is the same in both graphs for a better comparison. | ||
In the case of 2n, no emission was recorded when excited at 375 nm; however upon changing the excitation wavelength to 458 nm an intense emission was observed. Due to the proximity of the excitation wavelength and the starting wavelength of the spectrum, it is impossible to extract valuable information below 500 nm. The emission band under ambient conditions consists of an unstructured asymmetric band with two green components, centred at 520 nm and 555 nm, respectively, the former being larger in amplitude and similar in spectral position to the green component found in 2m. At 0.11 GPa the band broadens around 9 nm and shift to the red by about 5 nm; both tendencies continue until 1.4 GPa, with a red-shift rate of −330 cm−1 per Gpa. From this point, the broadening abruptly changes from 0.6 GPa (74 nm) to 1.4 GPa (116 nm), as a consequence of the increase in the amplitude of the low-energy component, and the band is red-shifted. It seems that the low-energy component, which clearly shifts to the red, dominates up to 7.5 GPa, but beyond this pressure two components are now clearly identified, one remaining at 520 nm (high-energy component) which matches with that observed under ambient conditions, and the low-energy one with red-shifts of −350 cm−1 per GPa, from 7.5 to 9.0 GPa. Regarding intensity, the behaviour is also complex, showing a 4.7 fold increase in the integrated intensity at 2.5 GPa, which after this point gradually decreases by the same amount till 6.8 GPa, just where the splitting of the two components is clear. From that pressure to the last one recorded, an increase of 1.7 times at 9 GPa is observed (Fig. 8b). As it was commented in the previous section, the features of luminescence spectra are closely related to the surrounding environment of the emitter. In this sense a high pressure technique allows gradually modifying and controlling the inter- and intra-molecular interactions so that a correlation between the structure and luminescence can be made. In CuI ladder-type complexes, emission is modulated by some parameters, most of them related to bond angles and distances of the nearby surrounding ions, such as Cu⋯Cu, Cu⋯I, Cu⋯L (ligands), and I–Cu–I, in particular within the Cu2I2 cluster core. The so-called LE emission band usually involves a [Cu2I2] cluster-centred triplet excited state (3CC), which is a combination of iodide-to-metal charge-transfer (3IMCT) and Cu 3d → 4s, 4p transitions (3MCC*), with the latter strongly dependent on Cu–Cu (cuprophilic) interactions, when Cu–Cu distances are lower than the sum of the Van der Waals radii of Cu (2.8 Å), although other contributions can also be considered as 3ILCT and 3MLCT. In order to distinguish among the possible transitions involved, a high-pressure technique can be an interesting tool to check the mechanochromic effects that can be complemented with the thermochromic one, since pressure induced larger changes in bond lengths and angles as well as in interplanar distances than those achieved by changing the temperature. In this kind of complex, different non-excluding transitions can take place as is reflected in the asymmetric and structured ambient condition emission spectrum of 2m and 2n.
Focusing on 2m, the presence of at least three different components in the emission spectrum at 0 GPa is clear, indicating the presence of more than one transition type, and after a pressure of 1.3 GPa was applied, two of them disappear and the one that remains 615 nm undergoes a strong and almost linear red-shift (−440 cm−1 per GPa). This behaviour has been observed in several CuI–L complexes and is attributed to the Cu 3d → 4s, 4p transition (3MCC*) (cuprophilic), where the shortening of the Cu–Cu distances increases the bonding character of the LUMO leading to a red-shift of the band.31 In our sample we have to deal with three different cuprophilic interactions, Cu1–Cu1b with distances of 3.514 Å, Cu1–Cu1c and Cu1–Cu1d (2.682 Å) under ambient conditions, and when pressure is applied both Cu–Cu distances reduce significantly, 3.151 Å (10.3%), Cu1–Cu1d, 2.53 Å (5.6%) at 7.16 GPa (Fig. S5c‡), but only Cu1–Cu1d is less than 2.8 Å, and consequently the cuprophilic transition contributes in an important way to the emission observed for pressures greater than 1.3 GPa. The high-energy component band around 500 nm only appear under ambient conditions and can be tentatively ascribed to a combination of those described above 3IMCT, 3ILCT and 3MLCT. On the other hand, the integrated intensity has its maximum at 1.3 GPa, and after that a gradual quenching is observed until the emission completely disappears at P > 9.0 Gpa. This behaviour can be explained in terms of the pressure-induced electronic degeneracy rupture (explained on the basis of the computed density electronic states and TDDF photoexcitation, see below, Fig. 8c and 9).
 | ||
| Fig. 9 Computed density of electronic states (in arb. units) for 2 as a function of the energy referred to the Fermi level (in eV), for different externally applied hydrostatic pressures (P = 0, 0.72, 1.36, 2.32, 3.08, 4.18, 4.65, 5.15, 5.63, 6.00 and 7.16 GPa). 3D orbital density isosurfaces (with a value of 5 × 10−3 a.u.) are also included for the most representative electronic states and represented over their respective X-ray diffraction structures obtained at 298 K. | ||
In the case of 2n, no orange emission was found but some similarities still remain. The high-energy component (520 nm) of the LE band, which does not change with pressure, resembles that obtained in 2m, and can be ascribed to 3IMCT, 3ILCT and 3MLCT. On the other hand, the low-energy band behaves like the orange component of the 2m sample, with different rate shifts and at shorter wavelengths. It is evident that the surroundings in 2n must suffer from changes compared to 2m due to an increase of the surface-to-volume ratio, modifying the interactions (Fig. 8b).
Fig. 8c shows the computed TDDFT photoexcitation spectra of 2 for different externally applied hydrostatic pressures (P = 0, 2.32, 5.15 and 7.16 GPa) within a range between 400 and 800 nm. Within this wavelength range we can observe a prominent excitation feature centred around 580 nm for the case P = 0, which tends to shift to higher photon wavelengths as the pressure increases. This wide feature has its origin in an electronic transition between the VB and the CB (84%) and the VB−1 and the CB (1%). As usual in this sort of Cu-based polymers, in this case the VB and VB−1 are mostly located in the Cu-based metal skeleton, whilst the CB and CB+1 are located in the organic ligands (Fig. 9). Interestingly, as the pressure starts to increase, the intensity of this broad excitation feature progressively decays, which excellently agrees with the experimental photoluminescence spectra, where the intensity also decays for high pressures. For increasing values of the pressure, theory predicts a slight shift of the feature towards higher wavelengths until being located around 610 nm. Nevertheless, it is well known that this shift should be taken cautiously since these small variations could be framed within the accuracy of this approach. Fig. 8c also shows another excitation feature located at 420 nm for P = 0. This excitation has its origin mostly in an electronic transition between the VB and the CB+1 (96%), which also decays and shifts towards lower wavelengths.
Fig. 9 shows the computed density of electronic states (in arb. units) for 2 as a function of the energy referred to the Fermi level (in eV), for different externally applied hydrostatic pressures. For the case in which no external pressure is applied (P = 0), theory predicts a canonical narrow-gap p-type semiconducting character, with the Fermi level almost pinning the valence band (VB) of the compound and yielding an electronic gap of around 1.25 eV. Interestingly, as the hydrostatic pressure increases we can clearly observe different emerging effects. The first one consists of a progressive reduction of the band-gap up to a value of 0.8 eV for the case limiting of P = 7.16 GPa. This situation is reached by a totally asymmetrical closure around the Fermi energy with increasing pressure, even flipping the initial p-type semiconducting character at P = 0 GPa into a canonical n-type semiconducting character, with the Fermi level almost pinning the conduction band (CB) of the material at P = 7.16 GPa. The second, and the most interesting, effect is the evident breaking of the electronic degeneracy in most of the electronic states lying within the depicted energy window in the figure. The doubly electronically degenerated CB splits into two electronic states as the external pressure increases. This behaviour is also observed for the, initially at P = 0, doubly degenerated CB+1, as well as for VB−1. The VB does not experience any splitting since it is, even at P = 0, a single occupied electronic state (and no additional electronic impairment effect is observed within our spin-polarized calculations).
Both aforedescribed effects, meaning the sequential asymmetrical band-gap closure as well as the breaking of the electronic degeneracy, have their origin in a pressure-induced electronic degeneracy-rupture, which is a direct consequence of Pauli's exclusion principle, and it has been already reported by our group for other morphologically similar compounds.13 The external hydrostatic pressure effect shortens bond-lengths within the compound (in particular within the metallic Cu chain), which tends to force different electrons to occupy the same electronic state, which is forbidden by Pauli's exclusion principle. Thus, the system electronically reacts by breaking the electronic degeneracy of some populated states to accommodate the pressure-induced forced electrons. Some of these states are directly involved in the visible emission processes of the material. On this basis, this interesting behaviour will have a direct reflection in the optical properties of the system as the external pressure increases.
As previously reported by us,13 the vanishing of the broad excitation feature described in the previous paragraph as the pressure increases can be rationalized in terms of the pressure-induced electronic degeneracy-rupture explained in detail above. The degeneracy-rupture produces a loss of efficiency in the metal–ligand transition, responsible for the photoluminescence behaviour since highly degenerated electronic orbitals contributing to that transition slightly modify their occupancy and symmetry by the effect of the pressure. Increasing pressure produces the compression of bond lengths within the compound, which is the case of the Cu–Cu bond (around a 20% bond-length compression followed by a reduction of around 3° in the dihedral angle from 0 to 7.16 GPa). In particular, this reduction in the Cu–Cu bond, which accommodates occupied electronic states that actively participate in the permitted metal–ligand transition (cuprophilic interaction), has its reflection in a visible vanishing of the photoluminescence. As aforementioned, in the case of the Cu–Cu bonding, electrons belonging to the slightly hybridized occupied Cu dz2 orbitals (within the Cu–Cu bond) are pushed towards each other by the effect of the pressure, forcing them to occupy the same electronic state (Fig. 9). Nevertheless, the system reacts by the electronic degeneracy-rupture slightly modifying the orbital orientation. For the case of P = 0 the slightly hybridized dz2 orbitals located in each Cu atom are oriented facing their lobes to maximize the cuprophilic interaction, and, thus, the photoluminescence is maximum. Nevertheless, for high pressures, with a reduced Cu–Cu bond length, the lobes of the dz2 orbitals spin in such a way to avoid the electronic overlapping. This behaviour produces a reduction in the metal–ligand transition efficiency, which finally leads to the photoluminescence vanishing (see ref. 13 for a detailed explanation).
Composite thin films
The outstanding optical sensing properties of 2n, as well as the mechanical properties of polyvinylidene difluoride (PVDF), prompted us to produce composites of potential interest for the fabrication of new devices. In particular, the production of thin films is of potential interest towards applications. However, the preparation of film composites of nanometric thickness is hampered by the size of the integrated materials. Thus, composites with CPs have been limited to the bulk scale14a,17a,19,32 because of their macroscopic dimensions. Thus, very recently we have demonstrated that the nanoscale production of CP nano-layers enables us to produce composites of sub-micrometric thickness. As 2n consists of nanofibers, it seems to be an excellent candidate for thin-film fabrication. Additionally, 2n shows the ability to reversibly dissolve and recrystallize from DMF solution (Experimental section, ESI section S7‡). This feature has been still little explored but, as we previously demonstrated, it is very useful for the fabrication of CP-based nanomaterials.33 However, it has never been used for composite thin-film preparation.Thus, following the first strategy, a dispersion in DMF of nanofibers of 2n has been prepared at over-saturated concentrations (15 and 30 wt% ESI‡ for details). Alternatively, a bottom-up strategy based on in situ fibres of 2n formed from a DMF solution of 2n with PVDF, in a final concentration of 4% wt, was tested.
For both approaches, the thin-film composites were prepared either by drop-casting (Fig. S17a‡), dip-coating or spin-coating on different substrates (Fig. 10a), and the solvent was eliminated upon soft heating. The so-formed composite-films 2n@PVDF were fully morphologically and structurally characterized (Fig. 10, S17–S26‡). IR and DRX powder diffraction data confirm that the original structure of 2 is retained after the composite formation process in all the cases (Fig. S17e and S18‡).
 | ||
| Fig. 10 (A) Scheme of the spin-coating synthesis of the 2n@PVDF composite. (B) Photography of a 2 × 2 cm 2n@PVDF thin-film. (C and D) SEM images of the 2n@PVDF thin-film with 4% wt of 2n. (E) AFM image of the 2n@PVDF thin film with 4% wt of 2n, and its height profile across the line. | ||
Additionally, AFM, SEM and EDX analyses confirm the formation of highly homogeneous films with thicknesses ranging from 40 μm for the drop-casting method, to thicknesses between 25 and 60 nm for the films prepared by dip-coating or spin-coating (Fig. 10c–e, S17d, S21 and S22‡). The AFM measurements confirmed that, independently from the concentration of 2n in the film composites, those prepared by spin or dip-coating show a roughness of 20–60 nm, likely due to the intrinsic morphology of PVDF, while, for the films prepared by drop-casting, the roughness rises up to 40 μm, without significant differences between concentrations.
Therefore, the most significant effect on the thickness of the film composite depends on the method used for its fabrication. Thus, just spin- and dip-coating methods allow us to form 2n@PVDF films of nanometric thicknesses. It is also worth mentioning that the 2n nano-structuration procedure does not significantly affect the 2n@PVDF thin-film formation. This is understandable because the low dimensions of 2n fibres produced are significantly smaller than the thickness of the 2n@PVDF thin-film formed.
The 2n@PVDF films show both good elasticity and flexibility (Fig. S17a‡). Additionally, the optical bulk properties of compound 2n are also retained in the 2n@PVDF composite showing weak yellow emission (571 nm at 298 K, λexc = 400 nm) that is enhanced upon lowering the temperature (549 and 580 nm at 80 K, λexc = 400 nm) with a similar lifetime, 1.42 ms, to that observed for 2n (Fig. S17b and S25a‡).
Additionally, the emission spectra of 2n@PVDF materials excited at 375 nm have also been measured at ambient and high pressures (Fig. S25b and S26‡). The main difference found between 2n@PVDF and 2n is the presence of a high energy (HE) band at ca. 435 nm. In particular, the emission of the 4% wt 2n@PVDF sample consists of a single asymmetric band that can be de-convoluted into three components, two HE (424 nm and 454 nm) ones with a strong contribution and a weak LE (510 nm) contribution. However, for 15 and 30% wt 2n@PVDF samples the spectra can be clearly divided into two different bands, one HE (435 nm) and one LE (577 nm and 558 nm), respectively. Unfortunately, due to the band overlapping with the diamond emission of the DAC, because of the UV excitation in 375 nm, and the HE emissions in the UV of the samples, it is not possible to resolve these bands for high pressures and, therefore, we can only focus on the pressure evolution of the LE emission bands. Thus, in the 4% wt 2n@PVDF sample regarding the LE component of the band (510 nm), it remains almost constant in wavelength from 0 to 10 GPa, as well as the integrated intensities. On the other hand, the behaviour of 15 and 30% wt 2n@PVDF is similar to that observed for 2m, thereby showing different tendencies. Hence, in both samples the intensity gradually decreases by around 80% with the pressure at 5 GPa compared to their values at 0 GPa, and a red-shift of the LE emission bands is also noticeable. However, in 15% wt 2n@PVDF this red-shift is linear with a slope of around −330 cm−1 per GPa, whereas a more complicated behaviour is observed for 30% wt 2n@PVDF, in which from 2 to 4.7 GPa the red-shift can be considered as linear (−326 cm−1 per GPa). Therefore, a different origin for the transition observed for the 4% wt 2n@PVDF sample with respect to 15 and 30% wt 2n@PVDF samples is expected. Analysing the pressure induced spectral behaviour of the hybrid samples, some conclusions can be extracted; focusing only on the LE band of the 4% wt 2n@PVDF sample, cuprophilic interaction (3MMC) can be disregarded due to the absence of a red shift in the emission band; however, in the other two samples 15 and 30% wt 2n@PVDF, different behaviours are observed showing red-shifts with different rates and quenching of the emission, similar to those considered above (2m and 2n).
Experimental section
All the experimental methods have been moved to the ESI.‡Conclusions
Double copper–iodine chains coordination polymers show structural flexibility allowing the formation of (multi)stimuli response coordination polymer materials. These chains are anchored by terminal ligands coordinated to the copper atoms. In this work we used two different methyl isonicotinate-based organic compounds as terminal ligands to demonstrate that just the presence or absence of a NH2 group produces different supramolecular arrangements affecting their physical properties. Thus, [Cu(MeIN)I]n (1) shows orange emission in a wide range of temperatures enhancing the emission intensity upon lowering the temperature, while [Cu(NH2–MeIN)I]n (2) displays only significant yellow emission at low temperatures. Additionally, 2 shows reversible significant mechanoluminescence properties while 1 only displays a negligible effect. X-ray diffraction studies under different conditions, temperatures and pressures in combination with theoretical calculations help to rationalize the different photoluminescence behaviour as the origin of the thermal fluctuations that tend to broaden both the VB and the CB but with remarkable differences between 1 and 2 as a consequence of their structures.We demonstrate that the ability of 2 to form nanofibers enables the formation of thin-film composites with polyvinylidenedifluoride (PVDF), 2@PVDF. The free-standing films show lengths of several centimetres and thicknesses of tens of nanometres (20–60 nm) which are three orders of magnitude smaller than those previously reported for related thin-film composites of CPs.34 The 2@PVDF thin-film composites show remarkable reversible thermo- and mechanochromic properties even with a low-weight content of 2 (4% wt) while retaining the mechanical properties of PVDF, e.g. elasticity and flexibility.
These results represent the first samples of multi-stimuli-response thin-film composites integrating nanofibers of CPs in their structure. The approach used is simple and innovative and could open new prospects for the fabrication of opto-electronic nano-devices. Our work advances in the field of multifunctional composite nanomaterials based on coordination polymers integrating stimuli-response capabilities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank financial support from the Spanish Ministerio de Economía y Competitividad (MAT2016-77608-C3-1-P, MAT2016-75883-C2-2-P, MAT2010-20843-C02-01, MAT2016-75586-C4-4-P, CTQ2016-75816-C2-1P). J. I. M. acknowledges the financial support by the “Ramón y Cajal” Program of MINECO (Grant RYC-2015-17730) and the EU via the ERC-Synergy Program (Grant ERC-2013-SYG-610256 NANOCOSMOS). J. C. E. acknowledges the financial support by the “FPI-MINECO” Program of MINECO (Grant BES-2015-071534).References
- S. R. Batten, S. M. Neville and D. Turner, Coordination Polymers: Design, Analysis and Applications, RSC Publishing, 2009, vol. 7 Search PubMed.
- V. G. Vegas, R. Lorca, A. Latorre, K. Hassanein, C. J. Gómez-García, O. Castillo, Á. Somoza, F. Zamora and P. Amo-Ochoa, Angew. Chem., Int. Ed., 2017, 56, 987 CrossRef PubMed.
- K. Hassanein, P. Amo-Ochoa, C. J. Gomez-Garcia, S. Delgado, O. Castillo, P. Ocon, J. I. Martinez, J. Perles and F. Zamora, Inorg. Chem., 2015, 54, 10738 CrossRef PubMed.
- J. Tong, Y. Wang, J. Mei, J. Wang, A. Qin, J. Z. Sun and B. Z. Tang, Chem.–Eur. J., 2014, 20, 4661 CrossRef PubMed.
- F. Czerwinski, A. Zielinska-Lipiec and J. A. Szpunar, Acta Mater., 1999, 47, 2553 CrossRef.
- (a) K. Hassanein, J. Conesa-Egea, S. Delgado, O. Castillo, S. Benmansour, J. I. Martinez, G. Abellan, C. J. Gomez-Garcia, F. Zamora and P. Amo-Ochoa, Chem.–Eur. J., 2015, 21, 17282 CrossRef PubMed; (b) P. Amo-Ochoa, K. Hassanein, C. J. Gomez-Garcia, S. Benmansour, J. Perles, O. Castillo, J. I. Martinez, P. Ocon and F. Zamora, Chem. Commun., 2015, 51, 14306 RSC.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC.
- G. Givaja, P. Amo-Ochoa, C. J. Gomez-Garcia and F. Zamora, Chem. Soc. Rev., 2012, 41, 115 RSC.
- (a) N. M. Khatri, M. H. Pablico-Lansigan, W. L. Boncher, J. E. Mertzman, A. C. Labatete, L. M. Grande, D. Wunder, M. J. Prushan, W. G. Zhang, P. S. Halasyamani, J. Monteiro, A. de Bettencourt-Dias and S. L. Stoll, Inorg. Chem., 2016, 55, 11408 CrossRef PubMed; (b) A. N. Dou, Y. C. Du, Q. L. Chen, K. L. Luo, C. Zhang, A. X. Zhu and Q. X. Li, Z. Anorg. Allg. Chem., 2016, 642, 731 CrossRef; (c) S. Demir, H. M. Cepni, N. Bilgin, M. Holynska and F. Yilmaz, Polyhedron, 2016, 115, 236 CrossRef; (d) A. Aguirrechu-Comeron, R. Hernandez-Molina, P. Rodriguez-Hernandez, A. Munoz, U. R. Rodriguez-Mendoza, V. Lavin, R. J. Angel and J. Gonzalez-Platas, Inorg. Chem., 2016, 55, 7476 CrossRef PubMed; (e) W. Ji, J. Qu, S. Jing, D. R. Zhu and W. Huang, Dalton Trans., 2016, 45, 1016 RSC; (f) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566 CrossRef; (g) A. Gallego, O. Castillo, C. J. Gómez-García, F. Zamora and S. Delgado, Inorg. Chem., 2012, 51, 718 CrossRef PubMed.
- Q. Qi, J. Zhang, B. Xu, B. Li, S. X.-A. Zhang and W. Tian, J. Phys. Chem. C, 2013, 117, 24997 CrossRef.
- C. Y. K. Chan, J. W. Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320 RSC.
- E. Kwon, J. Kim, K. Y. Lee and T. H. Kim, Inorg. Chem., 2017, 56, 943 CrossRef PubMed.
- J. Conesa-Egea, J. Gallardo-Martínez, S. Delgado, J. I. Martínez, J. Gonzalez-Platas, V. Fernández-Moreira, U. R. Rodríguez-Mendoza, P. Ocón, F. Zamora and P. Amo-Ochoa, Small, 2017, 13, 1700965 CrossRef PubMed.
- (a) M. S. Denny and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 9029 CrossRef PubMed; (b) J. Troyano, O. Castillo, J. I. Martinez, V. Fernandez-Moreira, Y. Ballesteros, D. Maspoch, F. Zamora and S. Delgado, Adv. Funct. Mater., 2018, 28, 1704040 CrossRef.
- R. Toivola, P.-N. Lai, J. Yang, S.-H. Jang, A. K. Y. Jen and B. D. Flinn, Compos. Sci. Technol., 2017, 39, 74 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- (a) H.-J. Zhang, R.-Q. Fan, X.-M. Wang, P. Wang, Y.-L. Wang and Y.-L. Yang, Dalton Trans., 2015, 44, 2871 RSC; (b) S. Sivakumar and M. L. P. Reddy, J. Mater. Chem., 2012, 22, 10852 RSC.
- S. M. Jeong, S. Song, S.-K. Lee and N. Y. Ha, Adv. Mater., 2013, 25, 6194 CrossRef PubMed.
- J. B. DeCoste, M. S. Denny Jr, G. W. Peterson, J. J. Mahle and S. M. Cohen, Chem. Sci., 2016, 7, 2711 RSC.
- (a) H. A. Habib, A. Hoffmann, H. A. Höppe, G. Steinfeld and C. Janiak, Inorg. Chem., 2009, 48, 2166 CrossRef PubMed; (b) V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC; (c) E. Cariati, X. Bu and P. C. Ford, Chem. Mater., 2000, 12, 3385 CrossRef; (d) D. M. Ciurtin, N. G. Pschirer, M. D. Smith, U. H. F. Bunz and H.-C. Zur Loye, Chem. Mater., 2001, 13, 2743 CrossRef; (e) F. Wurthner and A. Sautter, Chem. Commun., 2000, 445 RSC.
- S. Sakaida, K. Otsubo, O. Sakata, C. Song, A. Fujiwara, M. Takata and H. Kitagawa, Nat. Chem., 2016, 8, 377 CrossRef PubMed.
- (a) S. Dang, S. Song, J. Feng and H. Zhang, Sci. China Chem., 2015, 58, 973 CrossRef; (b) Y. Yan, J. Chen, N.-N. Zhang, M.-S. Wang, C. Sun, X.-S. Xing, R. Li, J.-G. Xu, F.-K. Zheng and G.-C. Guo, Dalton Trans., 2016, 45, 18074 RSC; (c) Y. Yan, N.-N. Zhang, R. Li, J.-G. Xu, J. Lu, F.-K. Zheng and G.-C. Guo, Eur. J. Inorg. Chem., 2017, 3811 CrossRef.
- (a) P. Guionneau, Dalton Trans., 2014, 43, 382 RSC; (b) R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565 CrossRef PubMed; (c) N. D. Boscher, P. Choquet, D. Duday, N. Kerbellec, J.-C. Lambrechts and R. Maurau, J. Mater. Chem., 2011, 21, 18959 RSC.
- A. Rashid, G. S. Ananthnag, S. Naik, J. T. Mague, D. Panda and M. S. Balakrishna, Dalton Trans., 2014, 43, 11339 RSC.
- (a) Y. Yang, J. Lan and X. Li, Mater. Sci. Eng., A, 2004, 380, 378 CrossRef; (b) F. Baldassarre, M. Cacciola and G. Ciccarella, J. Nanopart. Res., 2015, 17, 1 CrossRef.
- (a) W. Liu, Y. Fang, G. Z. Wei, S. J. Teat, K. Xiong, Z. Hu, W. P. Lustig and J. Li, J. Am. Chem. Soc., 2015, 137, 9400 CrossRef PubMed; (b) E. Bonetti, L. Pasquini and L. Savini, Scale dependent anelasticity and mechanical behavior: The case of nanocrystalline metals, in Nanostructures: Synthesis, Functional Properties and Applications, ed. T. Tsakalakos, I. A. Ovidko and A. K. Vasudevan, Springer Science and Business Media, 2003, vol. 128, pp. 217–237 Search PubMed; (c) C. Huang, H. R. Wang, X. L. Wang, K. A. Gao, J. Wu, H. W. Hou and Y. T. Fan, Chem.–Eur. J., 2016, 22, 6389 CrossRef PubMed.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence. Principles and Applications, Wiley-VCH Verlag GmbH & Co., 2nd edn, 2012 Search PubMed.
- S. A. Corrales, J. M. Cain, K. A. Uhlig, A. M. Mowson, C. Papatriantafyllopoulou, M. K. Peprah, A. Ozarowski, A. J. Tasiopoulos, G. Christou, M. W. Meisel and C. Lampropoulos, Inorg. Chem., 2016, 55, 1367 CrossRef PubMed.
- (a) S. Klotz, J. C. Chervin, P. Munsch and G. L. Marchand, J. Phys. D: Appl. Phys., 2009, 42, 075413 CrossRef; (b) T. H. Kim, Y. W. Shin, J. S. Kim, S. S. Lee and J. Kim, Inorg. Chem. Commun., 2007, 10, 717 CrossRef.
- (a) E. C. Spencer, R. J. Angel, N. L. Ross, B. E. Hanson and J. A. K. Howard, J. Am. Chem. Soc., 2009, 131, 4022 CrossRef PubMed; (b) N. P. Funnell, A. Dawson, D. Francis, A. R. Lennie, W. G. Marshall, S. A. Moggach, J. E. Warren and S. Parsons, CrystEngComm, 2010, 12, 2573 RSC; (c) C. Slebodnick, J. Zhao, R. Angel, B. E. Hanson, Y. Song, Z. Liu and R. J. Hemley, Inorg. Chem., 2004, 43, 5245 CrossRef PubMed.
- T. H. Kim, Y. W. Shin, J. H. Jung, J. S. Kim and J. Kim, Angew. Chem., Int. Ed., 2008, 47, 685 CrossRef PubMed.
- (a) C.-W. Zhao, J.-P. Ma, Q.-K. Liu, X.-R. Wang, Y. Liu, J. Yang, J.-S. Yang and Y.-B. Dong, Chem. Commun., 2016, 52, 5238 RSC; (b) L. Zhai, W.-W. Zhang, J.-L. Zuo and X.-M. Ren, Dalton Trans., 2016, 45, 3372 RSC.
- (a) D. Gentili, G. Givaja, R. Mas-Balleste, M. R. Azani, A. Shehu, F. Leonardi, E. Mateo-Marti, P. Greco, F. Zamora and M. Cavallini, Chem. Sci., 2012, 3, 2047 RSC; (b) R. Mas-Balleste, R. Gonzalez-Prieto, A. Guijarro, M. A. Fernandez-Vindel and F. Zamora, Dalton Trans., 2009, 36, 7341 RSC.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48 CrossRef PubMed.
Footnotes |
| † This paper is dedicated to Ma Pilar Ochoa García. |
| ‡ Electronic supplementary information (ESI) available: Tables of crystallographic data, X-ray powder diffractograms, emission spectra and additional figures. CCDC 1826846–1826858. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03085e |
| This journal is © The Royal Society of Chemistry 2018 |