
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium(II)-catalyzed γ-selective hydroarylation of alkenyl carbonyl compounds with arylboronic acids†
Rei
Matsuura
a,
Tanner C.
Jankins
a,
David E.
Hill
a,
Kin S.
Yang
a,
Gary M.
Gallego
b,
Shouliang
Yang
b,
Mingying
He
b,
Fen
Wang
b,
Rohan P.
Marsters
a,
Indrawan
McAlpine
b and
Keary M.
Engle
 *a
*a
aDepartment of Chemistry, The Scripps Research Institute, 10550 N Torrey Pines Road, La Jolla, California 92037, USA. E-mail: keary@scripps.edu
bPfizer Oncology Medicinal Chemistry, 10770 Science Center Drive, San Diego, California 92121, USA
First published on 6th September 2018
Abstract
A catalytic γ-selective syn-hydroarylation of alkenyl carbonyl compounds using arylboronic acids has been developed using a substrate directivity approach with a palladium(II) catalyst. This method tolerates a wide range of functionalized (hetero)arylboronic acids and a variety of substitution patterns on the alkene. Preliminary mechanistic studies suggest that transmetalation is rate-limiting.
Introduction
Transition-metal catalyzed 1,4-addition of arylboronic acids to enones and related conjugated alkenes has emerged as a reliable and robust method to introduce an aryl group at the β position of a carbonyl compound or functional group equivalent. In 1997 Miyaura reported a seminal study describing Rh(I)-catalyzed β-selective hydroarylation of enones using arylboronic acids (Scheme 1A).1 In 1998, Miyaura and Hayashi reported an asymmetric version of this reaction.2 Since then, Hayashi has reported an array of β-selective hydroarylation methods for many classes of alkenes in conjugation with electron-withdrawing groups, including esters,3 aldehydes,4 phosphonates,5 and sulfonyls.6 Hayashi has also achieved δ-selective functionalization by utilizing conjugation in α,β,γ,δ-unsaturated systems.7,8 However, expanding this mode of reactivity to C–C π bonds remote from and out of conjugation with a carbonyl group (e.g., the γ-position) remains a challenge.9 Notably, Sigman and co-workers have recently developed a powerful toolkit of remote arylation reactions to access enantioenriched arylated aldehydes from alkenyl alcohols using a redox-relay strategy (Scheme 1B).10,11 The goal of the present study was to develop a redox-neutral method for γ-selective hydroarylation of β,γ-unsaturated carboxylic acid derivatives with arylboronic acids using a substrate directivity approach, a mode of reactivity that would complement existing methods.12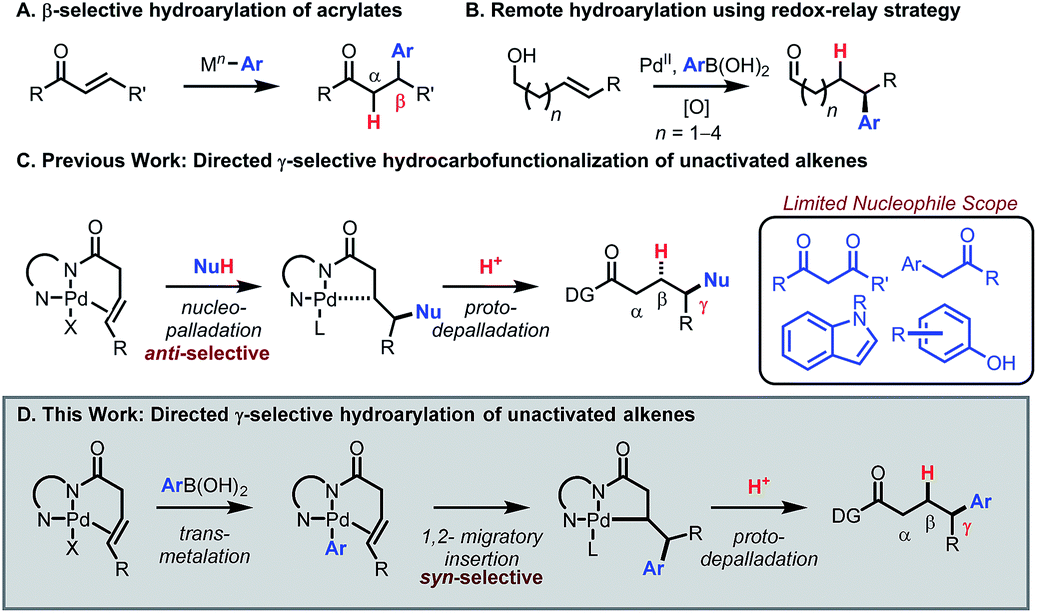 | ||
| Scheme 1 Past and current work in C–C π-bond hydrocarbofunctionalization. | ||
Previously, our group13–18 and others19,20 have developed a suite of chelation-controlled regioselective alkene functionalization reactions using Daugulis's 8-aminoquinoline (AQ)21,22 directing group under palladium(II) catalysis. With 3-butenoic-acid-derived substrates, the AQ directing group forces an incoming nucleophile to attack the γ-position to form the preferred five-membered palladacycle. The intermediate then reacts with an electrophile to yield the hydro- or difunctionalized product. In particular, the hydrocarbofunctionalization of 3-butenoic acid derivatives using various carbon nucleophiles has been reported (Scheme 1C).14,19b Thus far, nucleophile scope has been limited to 1,3-dicarbonyls, aryl carbonyls, indoles, phenols, and related nucleophiles capable of engaging in a Wacker-type anti-nucleopalladation mechanism. The addition of general aryl nucleophiles in this mode of catalysis remains an unmet challenge.20
Given the vast structural diversity of arylboronic acids and their widespread commercial availability, we sought to achieve analogous reactivity with this family of nucleophiles. We envisioned that such a transformation would be useful given that the γ-aryl carboxylic acid motif is found in multiple FDA-approved drugs (e.g., chlorambucil, moexipril and sitagliptin, Fig. 1). The proposed approach would allow such molecules to be synthesized in a modular fashion using synthetic logic that is appealing to medicinal chemists and would allow for facile synthesis of analogues.
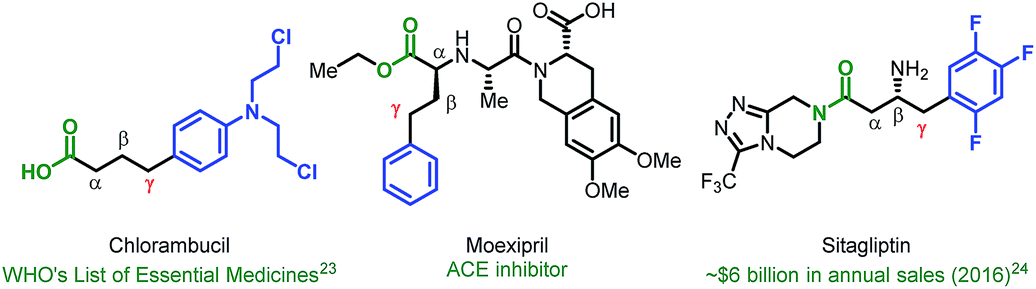 | ||
| Fig. 1 FDA approved drug molecules containing γ-aryl acid motifs. | ||
Moreover, we envisioned that after substrate coordination, the complex would undergo transmetalation with the arylboronic acid, allowing for mechanistically distinct Heck-type syn-selective delivery of the aryl group via 1,2-migratory insertion. Afterwards, the intermediate would undergo protodepalladation to yield the γ-arylated product (Scheme 1D).
Results and discussion
Based on our past work, we initiated our investigation using AQ-masked 3-butenoic acid (1a) as the pilot alkene and phenylboronic acid as the nucleophile. Gratifyingly, we were able to observe the desired hydroarylated product in 27% yield under acidic conditions (Table 1, entry 1). Notably, during preliminary screening we found that AQ out-performed various alternative directing groups (see ESI†). Hence, subsequent optimization focused on AQ-containing alkene 1a. Aromatic solvents were found to be beneficial for the reaction and could be used in combination with common polar solvents (entry 2). Various acids and bases were then screened, leading to the identification of NaF as a comparable alternative to benzoic acid as a promoter (entries 2–6). Interestingly, other fluoride bases did not have a similarly beneficial effect on the reaction (entries 3 and 4). Switching to 100% m-xylene as solvent and lowering the concentration improved the mass balance (entry 7).|
|
|||||
|---|---|---|---|---|---|
| Acid/base | Solvent | Temp | 1a | 3a | |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (10 mol%), acid or base (2 equiv.), water (2.5 equiv.), solvent, temperature, 5 h. b Base (1 equiv.). c 24 h. d 12 h. e 5% catalyst loading. | |||||
| 1 | PhCO2H | MeCN (0.5 M) | 120 °C | 5% | 27% |
| 2 | PhCO2H | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:m-xylene (0.5 M) :1 MeCN:m-xylene (0.5 M) |
120 °C | 3% | 58% |
| 3b | KF | 1:1 MeCN:m-xylene (0.5 M) |
120 °C | 39% | 22% |
| 4b | CsF | 1:1 MeCN:m-xylene (0.5 M) |
120 °C | 51% | 12% |
| 5b | Na2CO3 | 1:1 MeCN:m-xylene (0.5 M) |
120 °C | 52% | 11% |
| 6b | NaF | 1:1 MeCN:m-xylene (0.5 M) |
120 °C | 6% | 56% |
| 7 | NaF | m-xylene (0.1 M) | 120 °C | 41% | 40% |
| 8c | NaF | m-xylene (0.1 M) | 80 °C | 37% | 55% |
| 9c | PhCO2H | m-xylene (0.1 M) | 80 °C | 93% | 3% |
| 10 | NaF | PhCF 3 (0.1 M) | 100 °C | 0% | 91% |
| 11d,e | NaF | PhCF3 (0.1 M) | 100 °C | 39% | 63% |
| 12d | NaF | Toluene (0.1 M) | 100 °C | 33% | 58% |
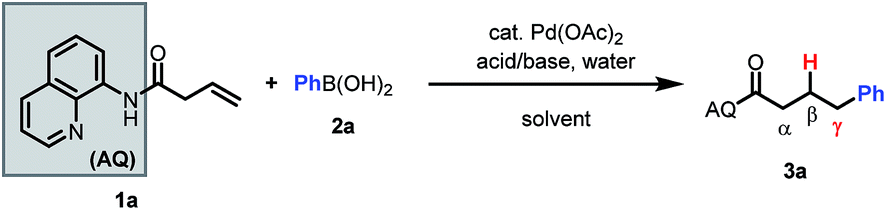
Further optimization of reaction temperature and time, and rescreening of aromatic solvents led to 91% yield of the desired product with NaF in 0.1 M benzotrifluoride (entries 8–10). Decreased catalyst loading lowered the yield and conversion (entry 11). When the reaction was run in toluene, the yield decreased substantially, illustrating the importance of the trifluoromethyl group on benzotrifluoride, perhaps for increased polarity (entry 12). Among several arylboron reaction partners that were attempted (see ESI†), only triphenylboroxine behaved similarly to phenylboronic acid (see ESI†). Boronic esters, on the other hand, did not lead to formation of the desired product.
Having optimized the reaction, we proceeded to investigate the substrate scope and functional group tolerance.25 As commercial boronic acids can contain varying amounts of water, for each example we screened 0, 1.5, or 2.5 equivalents of water to find the optimal amount (see ESI†). Additionally, some boronic acids with polar functional groups were found to be visibly insoluble in benzotrifluoride. In these cases, a 1:4 mixture of t-BuOH and benzotrifluoride was effective (see ESI†).
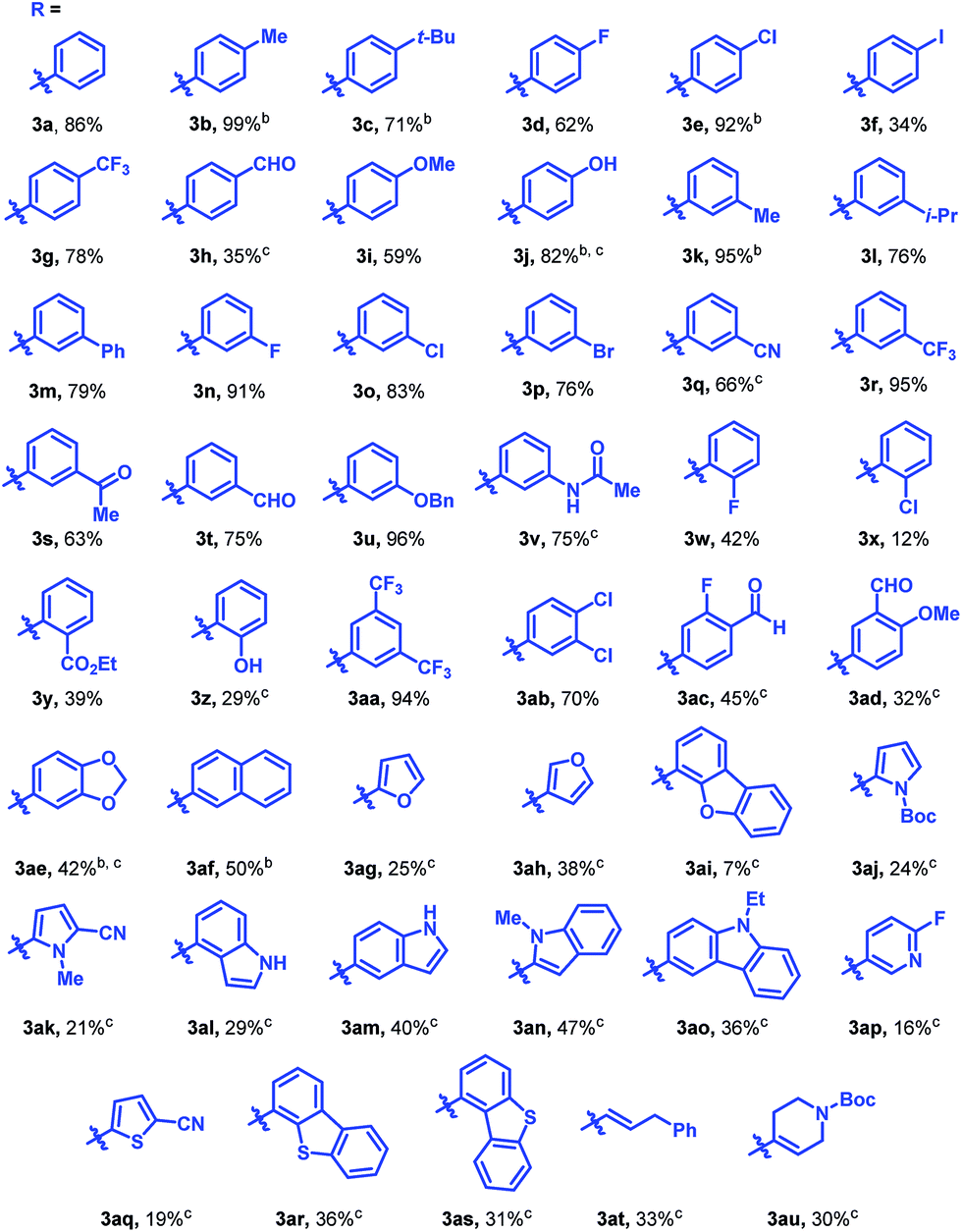
The reaction was found to tolerate an array of functional groups on the aromatic ring of the boronic acid, including both electron-donating and -withdrawing substituents, in both the para and meta positions (Table 2, 3b–3v). Substituents in the ortho position led to diminished yields, presumably due to steric effects, yet in most cases yields were still moderate (3w–z). Our method also tolerated multi-substituted benzenes (3aa–3ad), protected catechols (3ae), naphthalenes (3af), various heterocycles including furans, protected pyrroles, and indoles (3ag–3as), and alkenylboronic acids (3at, 3au). Though pyridine-type heterocycles were not generally compatible, 6-fluoro-3-pyridinylboronic acid was an exception and provided 16% yield (3ap).
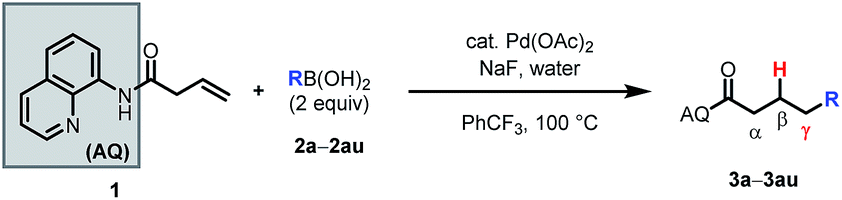
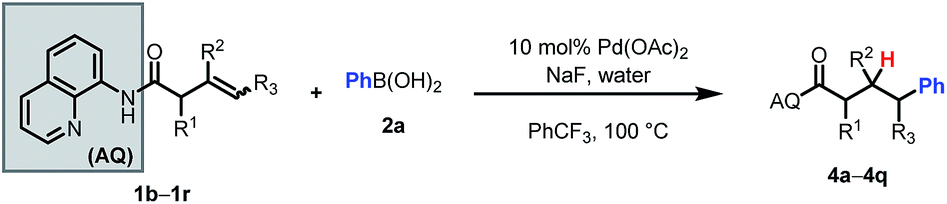
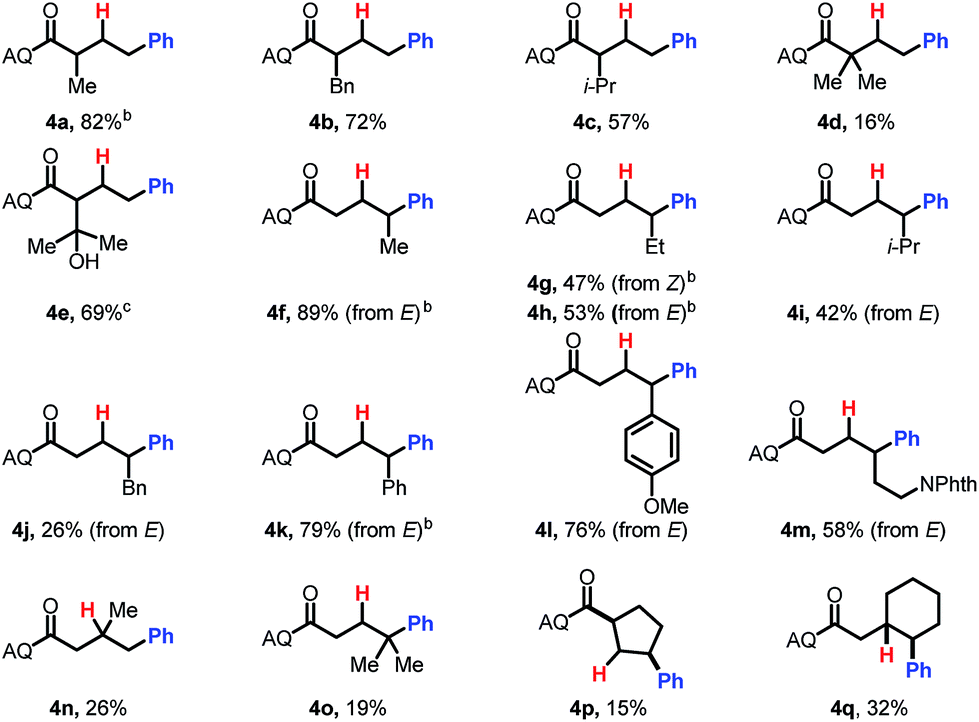
We then probed the alkene scope for our method. Substituents on the α-position were generally well tolerated (Table 3, 4a–e). Internal alkenes were also compatible, providing moderate to good yield (4f–4m). Other sterically hindered alkenes, such as 1,1-disubstituted alkenes (4n) and trisubstituted alkenes (4o) were reactive, albeit in low yields. Alcohols and protected amines on the substrate did not exhibit an inhibitory effect on the transformation (4e, 4m). When cyclic substrates were subjected to the reaction conditions, the corresponding syn-hydroarylated products were exclusively formed (4p, 4q).
To demonstrate the practicality and utility of this method, two large-scale reactions were performed. When run on 1 mmol scale or 7.1 mmol scale (1.5 g), the reaction gave comparable yields to the small-scale reaction (Scheme 2A). After hydroarylation, the AQ directing group was readily cleaved to yield the free acid 6a in 94% yield (Scheme 2B). Deprotection using Maulide's milder ozonolysis conditions was also viable, providing free acid 6b, albeit in lower yield (Scheme 2B).26
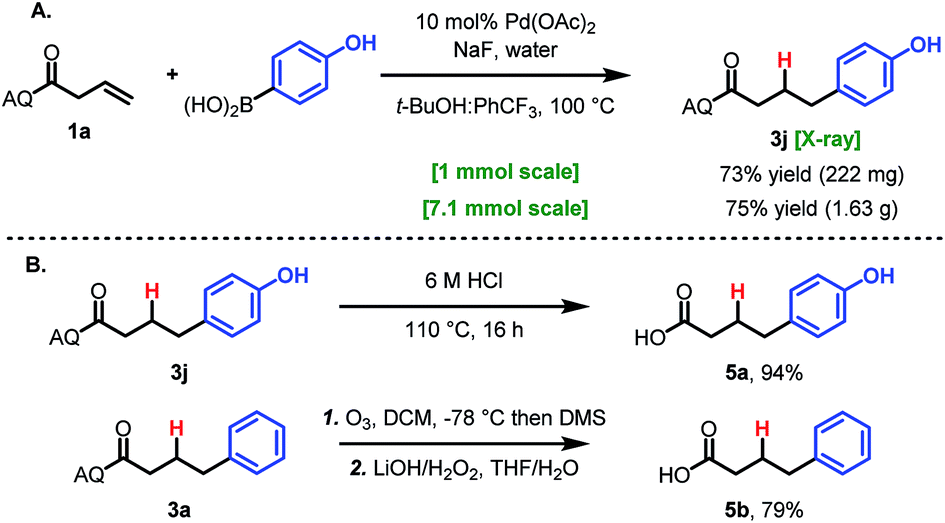 | ||
| Scheme 2 Large-scale synthesis and deprotection. | ||
Finally, a preliminary result indicates that this approach can also be extended to alkyl coupling partners. Using slightly modified conditions with methylboronic acid as the nucleophile, we were able to observe formation of the desired hydromethylation product in 18% yield (Scheme 3). This is a promising finding for future expansion of the reaction scope.
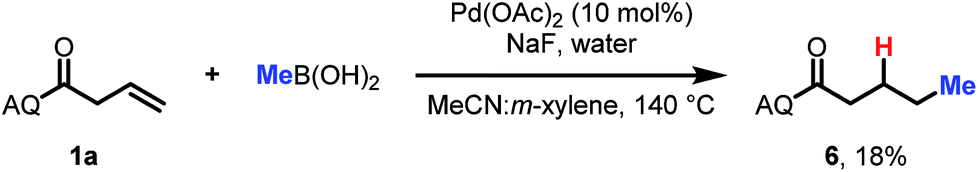 | ||
| Scheme 3 Preliminary result for alkyl boronic acids. | ||
Mechanistic studies
The broad scope of this transformation combined with the critical role of water stoichiometry in catalytic efficiency prompted us to perform several experiments to shed light on key aspects of the reaction mechanism.To identify the origin of the hydrogen atom in the hydroarylated product, deuterium incorporation studies were performed using deuterated reaction components. First, we sought to probe potential H/D exchange reactivity of the starting material and product. When starting material 1a was subjected to reaction conditions using D2O in place of H2O, H/D exchange was observed exclusively at the alkenyl position (Scheme 4A).27 In contrast, H/D exchange with the hydroarylated product 3a was not observed (Scheme 4B). When alkene 1a was then subjected to the standard reaction conditions with D2O, a mixture of bis-protio, mono-protio-mono-deutero, and bis-deutero products was obtained (Scheme 4C). The formation of bis-deutero product combined with the observation that the product does not undergo H/D exchange establishes that water is a competent source of protons/deuterons, consistent with a protodepalladation mechanism.
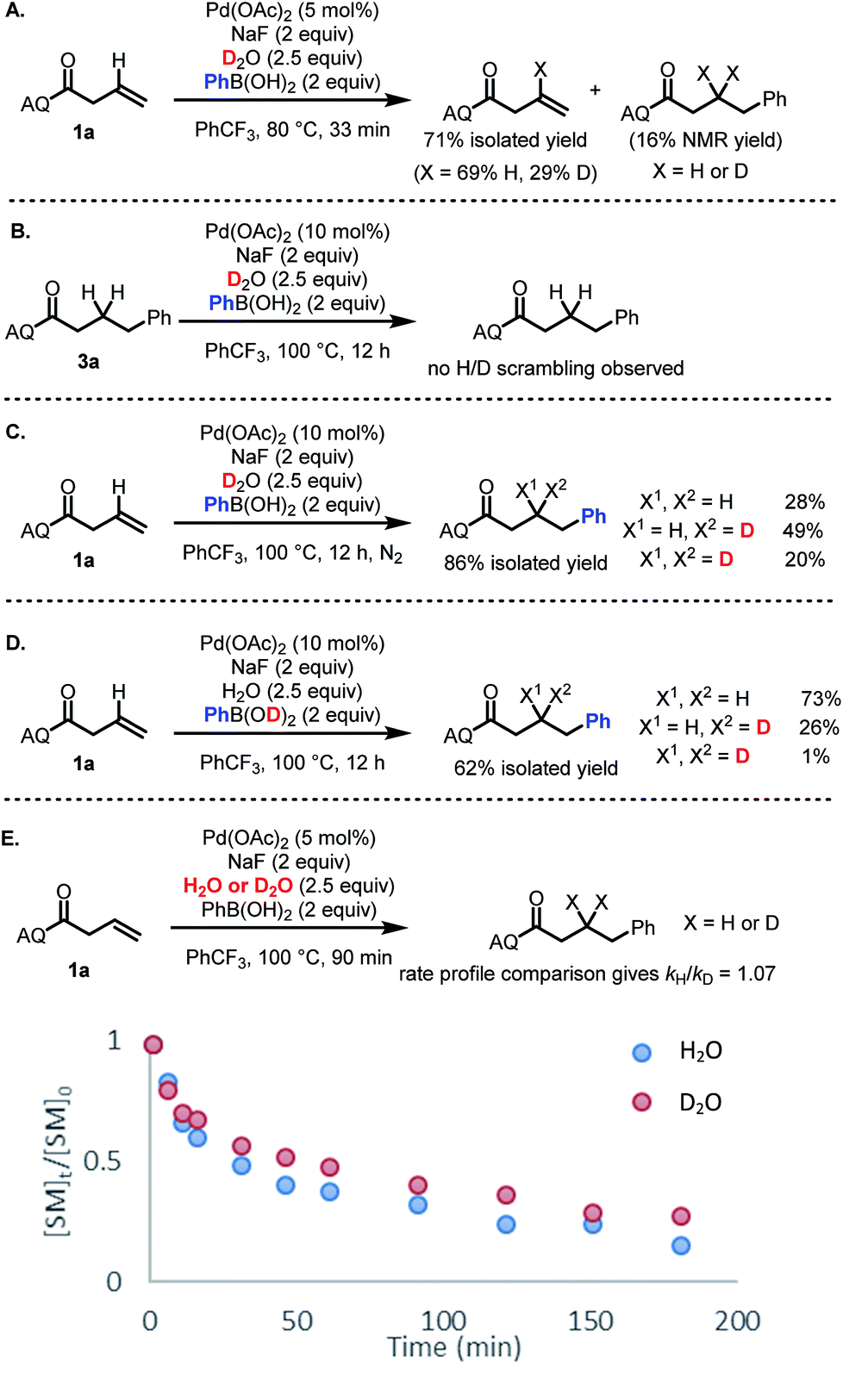 | ||
| Scheme 4 Deuterium-labeling studies. | ||
Interestingly, upon using PhB(OD)2 as the source of deuterons, the reaction gave a dramatically different product distribution, suggesting that while there is some H/D exchange between the boronic acid and reaction medium, not all protons/deuterons in the reaction system are in equilibrium (Scheme 4D). Finally, a comparison of global rate profile between reactions using H2O versus D2O gave kH/kD = 1.07 (Scheme 4E). If protodepalladation were the rate-determining step, we would expect to see a larger KIE (even factoring in the possibility of exchangeable protons/deuterons),28 suggesting that protodepalladation is not the rate-determining step in the present catalytic cycle.
We then performed reaction progress kinetic analysis (RPKA), which is a powerful method to determine the driving forces of a reaction from a minimal number of experiments.29 Same-excess experiments indicated that catalyst deactivation takes place during the reaction and that there is some degree of product inhibition (see ESI†). The general form of the kinetic profiles (as can be seen in the representative cases of the H versus D rate profiles, Scheme 4E), shows a sharp transition between a faster initial rate and a slower rate at higher conversions, which often indicates catalyst deactivation. A plausible pathway for catalyst deactivation is arylboronic acid homocoupling to generate catalytically inactive palladium(0). Indeed, biphenyl was observed by GC-FID during kinetic experiments. A series of different-excess experiments were also performed to determine the orders of the various reactants (summarized in Table 4, see ESI† for complete rate profiles). These experiments indicated positive order in [2a] and negative order in [1a] (see ESI†). Burés's graphical method30 was used to compare rate profiles from experiments with different catalyst amounts, and the results suggested that the reaction is first-order in palladium (see ESI†).
| a Standard conditions: [1a]0 = 0.1 M; [2a]0 = 0.2 M; [Pd(OAc)2] = 0.01 M; [NaF] = 0.2 M; [H2O] = 0.25 M; solvent = PhCF3; 100 °C. |
|---|
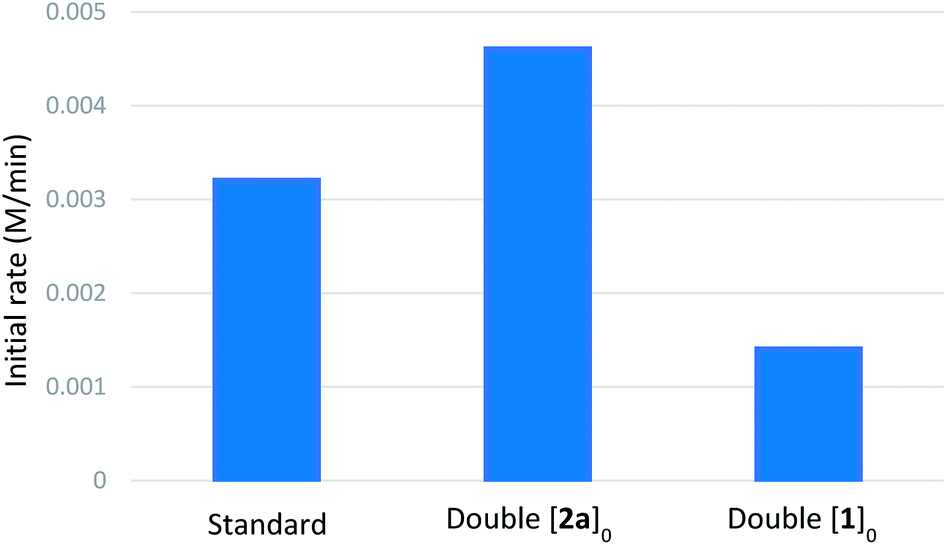
|
Collectively the above experimental observations are consistent with the catalytic cycle depicted in Scheme 5. The Pd(II) catalyst coordinates to the AQ directing group to generate A. The observed H/D scrambling in 1a (Scheme 4A) and the negative order in [1a] are consistent with the formation of an off-cycle complex A′via C(alkenyl)–H activation. Intermediate A reacts via base-promoted, rate-controlling transmetalation to form the aryl palladium complex B which then undergoes syn-selective 1,2-migratory insertion to form Pd(II) complex C. An irreversible protodepalladation step involving water gives product-bound palladium complex D, which dissociates to release the product and regenerate the catalyst.
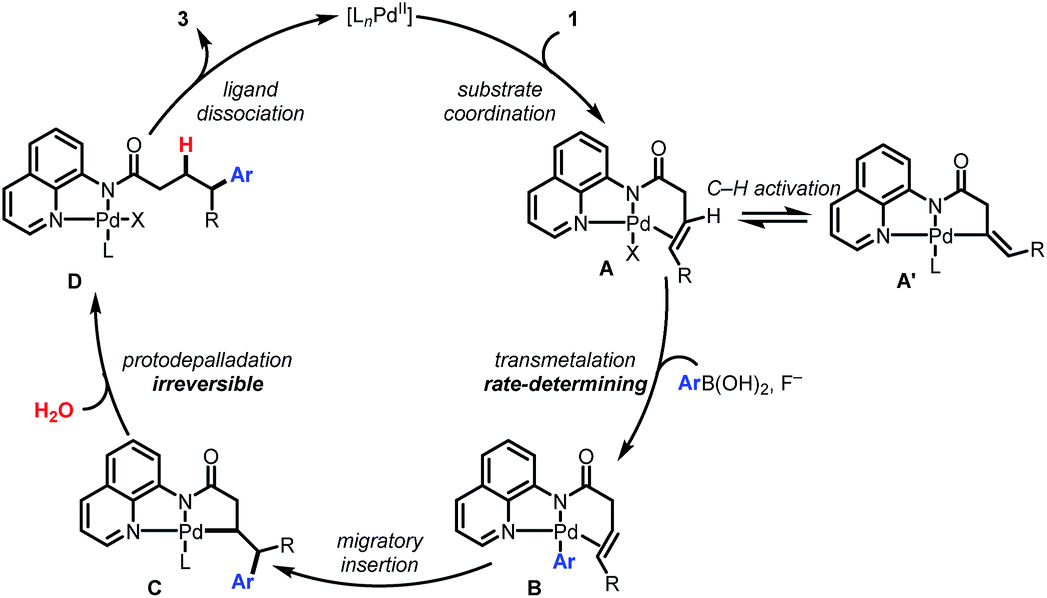 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed a regioselective hydroarylation of 3-butenoic acid derivatives using readily available arylboronic acids and a removable 8-aminoquinoline directing group. Unlike previous methods that utilize anti-nucleopalladation, this reaction proceeds via transmetalation and syn-insertion. This reactivity paradigm dramatically broadens the range of nucleophiles that can be employed and allows for the preparation of diastereoisomeric products that are distinct from previous methods. The reaction was found to tolerate a wide range of substituents and functional groups on the boronic acid and was completely regio- and stereoselective. This method can also be run on larger scales without significant decrease in yield, and facile removal of the directing group was also demonstrated. Future investigation will focus on expanding the scope to include more alkenyl and alkyl boronic acids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by The Scripps Research Institute (TSRI), Pfizer, Inc., Bristol-Myers Squibb (Unrestricted Grant) and the National Institutes of Health (1R35GM125052). We thank Prof. Arnold L. Rheingold and Dr Milan Gembicky (UCSD) for X-ray crystallographic analysis, Dr Dee-Hua Huang and Dr Laura Pasternack for assistance with NMR spectroscopy, Dr Jason S. Chen and Brittany Sanchez for help with compound purification, Prof. Donna G. Blackmond with guidance with RPKA, and Prof. Will R. Gutekunst (Georgia Tech) and Prof. Nuno Maulide (University of Vienna) for helpful discussion.Notes and references
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229 CrossRef.
- Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579 CrossRef.
- Y. Takaya, T. Senda, H. Kurushima, M. Ogasawara and T. Hayashi, Tetrahedron: Asymmetry, 1999, 10, 4047 CrossRef.
- T. Hayashi, N. Tokunaga, K. Okamoto and R. Shintani, Chem. Lett., 2005, 34, 1480 CrossRef.
- T. Hayashi, T. Senda, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 1999, 121, 11591 CrossRef.
- T. Nishimura, Y. Takiguchi and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 9086 CrossRef PubMed.
- T. Hayashi, S. Yamamoto and N. Tokunaga, Angew. Chem., Int. Ed., 2005, 44, 4224 CrossRef PubMed.
- T. Nishimura, Y. Yasuhara and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 5164 CrossRef PubMed.
- For representative reports on Pd(II)-catalyzed β-selective hydroarylation using arylboronic acids, see: (a) T. Nishikata, Y. Yamamoto, I. D. Gridnev and N. Miyaura, Organometallics, 2005, 24, 5025 CrossRef; (b) T. Nishikata, Y. Yamamoto and N. Miyaura, Adv. Synth. Catal., 2007, 349, 1759 CrossRef; (c) T. Nishikata, Y. Yamamoto and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 2768 CrossRef PubMed; (d) X. Lu and S. Lin, J. Org. Chem., 2005, 70, 9651 CrossRef PubMed; (e) B. Zhao and X. Lu, Org. Lett., 2006, 8, 5987 CrossRef PubMed; (f) F. Gini, B. Hessen and A. J. Minnaard, Org. Lett., 2005, 7, 5309 CrossRef PubMed; (g) K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902 CrossRef PubMed; (h) A. L. Gottumukkala, K. Matcha, M. Lutz, J. G. de Vries and A. J. Minnaard, Chem.–Eur. J., 2012, 18, 6907 CrossRef PubMed; (i) F. Wang, F. Chen, M. Qu, T. Li, Y. Liu and M. Shi, Chem. Commun., 2013, 49, 3360 RSC; (j) Q. He, F. Xie, G. Fu, M. Quan, C. Shen, G. Yang, I. D. Gridnev and W. Zhang, Org. Lett., 2015, 17, 2250 CrossRef PubMed; (k) S. Chen, L. Wu, Q. Shao, G. Yang and W. Zhang, Chem. Commun., 2018, 54, 2522 RSC.
- E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef PubMed.
- T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef PubMed.
- For representative recent reports on alternative catalytic approaches to alkene hydroarylation, see: (a) S. M. Podhajsky, Y. Iwai, A. Cook-Sneathen and M. S. Sigman, Tetrahedron, 2011, 67, 4435 CrossRef PubMed; (b) Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215 CrossRef PubMed; (c) K. Semba, K. Ariyama, H. Zheng, R. Kameyama, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2016, 55, 6275 CrossRef PubMed; (d) S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779 CrossRef PubMed; (e) S. D. Friis, M. T. Pirnot, L. N. Dupuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 7242 CrossRef PubMed; (f) L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461 CrossRef PubMed.
- J. A. Gurak Jr, K. S. Yang, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 5805 CrossRef PubMed.
- K. S. Yang, J. A. Gurak Jr, Z. Liu and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 14705 CrossRef PubMed.
- Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122 CrossRef PubMed.
- J. A. Gurak Jr, V. T. Tran, M. M. Sroda and K. M. Engle, Tetrahedron, 2017, 73, 3636 CrossRef.
- Z. Liu, Y. Wang, Z. Wang, T. Zeng, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 11261 CrossRef PubMed.
- Z. Liu, H.-Q. Ni, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 3223 CrossRef PubMed.
- (a) E. P. A. Talbot, T. d. A. Fernandes, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 4101 CrossRef PubMed; (b) H. Wang, Z. Bai, T. Jiao, Z. Deng, H. Tong, G. He, Q. Peng and G. Chen, J. Am. Chem. Soc., 2018, 140, 3542 CrossRef PubMed.
- Two related studies were disclosed while this manuscript was in revision. For a report using nickel(0) catalysis, see: (a) H. Lv, L.-J. Xiao, D. Zhao and Q.-L. Zhou, Chem. Sci., 2018, 9, 6839 RSC. For a reductive Heck approach, see: (b) C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T.-P. Loh, J. Am. Chem. Soc., 2018, 140, 9332 CrossRef PubMed.
- For a representative review, see: O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef PubMed.
- For representative reports on Pd(II)-catalyzed directed γ-arylation of carbonyl compounds via C(sp3)–H activation, see: (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef PubMed; (b) G. He, S.-Y. Zhang, W. A. Nack, R. Pearson, J. Rabb-Lynch and G. Chen, Org. Lett., 2014, 16, 6488 CrossRef PubMed; (c) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301 CrossRef.
- The 2017 WHO Essential Medicines List can be found at http://www.who.int/medicines/publications/essentialmedicines/en/.
- The annual sales for Januvia® (Sitagliptin) can be found at http://investors.merck.com/news/press-release-details/2017/Merck-Announces-Fourth-Quarter-and-Full-Year-2016-Financial-Results.
- For low-yielding examples, the reactions typically only reached low conversion. In these cases, roughly 90% of the material could be accounted for as starting alkene (1) or product (3); the remaining material (approximately 10%) was consumed via alkene reduction..
- M. Berger, R. Chauhan, C. A. B. Rodrigues and N. Maulide, Chem.–Eur. J., 2016, 22, 16805 CrossRef PubMed.
- For an example of similar reactivity involving a six-membered palladacycle, see: M. Liu, P. Yang, M. K. Karunananda, Y. Wang, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 5805 CrossRef PubMed.
- J. Derosa, A. L. Cantu, M. N. Boulous, M. L. O'Duill, J. L. Turnbull, Z. Liu, D. M. De La Torre and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 5183 CrossRef PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1835959. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03081b |
| This journal is © The Royal Society of Chemistry 2018 |