
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe methylation effect in prolonging the pure organic room temperature phosphorescence lifetime†
Zhu
Mao‡
a,
Zhan
Yang‡
a,
Zhenguo
Fan
a,
Eethamukkala
Ubba
a,
Wenlang
Li
a,
Yang
Li
b,
Juan
Zhao
*a,
Zhiyong
Yang
a,
Matthew P.
Aldred
a and
Zhenguo
Chi
 a
a
aPCFM Lab, GDHPPC Lab, Guangdong Engineering Technology, Research Center for High-performance Organic and Polymer Photo-electric, Functional Films, State Key Laboratory of OEMT, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China
bInstrumental Analysis and Research Center, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhaoj95@mail.sysu.edu.cn
First published on 8th October 2018
Abstract
Prolonging the phosphorescence lifetime of pure organic phosphorescent materials by a methyl-substitution strategy is described. We present a chemical strategy for improving the phosphorescence lifetime of triplet excitons under ambient conditions by incorporating methyl groups into the chemical structures. This is observed by a long-lived phosphorescence lifetime of up to 0.83 s detected for methylated 9-(4-(mesitylsulfonyl)phenyl)-9H-carbazole (3M), compared to 0.36 s for 9-(4-(phenylsulfonyl)phenyl)-9H-carbazole (0M) without any methyl groups. Additionally, enhanced phosphorescence efficiency can be obtained at an appropriate methylation degree, because of the smaller ΔEST (singlet and triplet energy gap) and ΔETT* (normal phosphorescence and long-lived phosphorescence energy gap). A comprehensive investigation on the packing mode in the crystalline state reveals that the methyl groups occupy the free volume and result in a suppression of non-radiative decay, accounting for the enhanced phosphorescence lifetime.
Introduction
Materials with long-lived phosphorescence emission exhibit extraordinarily long lifetimes, typically a lifetime of 0.1 s or greater,1 and have a wide range of applications, such as in bio-imaging,2 sensors,3 anti-counterfeiting,4 and displays.5 However, most of these phosphorescent luminogens are limited to inorganic materials with resource-limited transition metals, such as metal sulphides and alkaline earth aluminates/silicates.6 In addition, the preparation processes associate with consuming energy for high temperature sintering and products have high bio-toxicity. Considering these drawbacks of inorganic persistent luminescent materials, one alternative is the utilization of organic long-lived phosphorescence materials. Currently, strategies such as guest–host doping,7 constructing organic frameworks8 and H-type aggregation9 have been proposed to provide a rigid environment for suppressing non-radiative decay of triplet excitons and thus extending the lifetime of organic long-lived phosphorescence. For a higher quantum yield (ΦP) of organic phosphorescence, fast-radiative decay (krT), lower non-radiative decay (knrT) and higher inter-system crossing (ISC) efficiency (ΦISC) are the key parameters (ΦP = krT/(krT + knrT)ΦISC).10 However, there is a contradiction in that fast-radiative decay means a short phosphorescence lifetime (τp). Therefore, the balance between phosphorescence lifetime and ΦP is pivotal to the development of high performance organic long-lived phosphorescence emitters, and some strategies including heavy atom effects,11 dihydrogen bonding,12 and polycrystalline phases13 have been previously reported. In spite of this progress, further detailed studies regarding the prolonging of the phosphorescence lifetime and enhancing the phosphorescence quantum efficiency are still required.Methylation is an important process in bio-systems and disrupted DNA methylation affecting the genes in cells is associated with multiple diseases,14 indicating a shorter life. The methyl group is a weak donor and relatively bulky; therefore, we proposed that the methylation of organic semiconductors could have a significant impact on the exciton behaviours of organic phosphorescence materials, such as lifetime and quantum yield efficiency. To validate this hypothesis, herein we presented an investigation into the effect of methyl groups on the phosphorescence lifetime and efficiency of long-lived phosphorescence materials. Cz-DPS (0M), which emits long-lived phosphorescence with a lifetime τp = 0.36 s under ambient conditions, was reported by our group in 2016.11b Here, we have observed significant changes in the optical and photo-physical properties when the chemical structure of 0M is altered by methylation (Fig. 1). Two compounds 1M and 3M, containing one and three methyl groups, respectively, exhibit long-lived phosphorescence lifetime with 120% (τp = 0.82 s) and 130% (τp = 0.83 s) enhancement compared to 0M (Fig. 1 and 2). Moreover, the ΦP of 1M shows a significant improvement (2.64%) compared to unsubstituted 0M (1.50%).
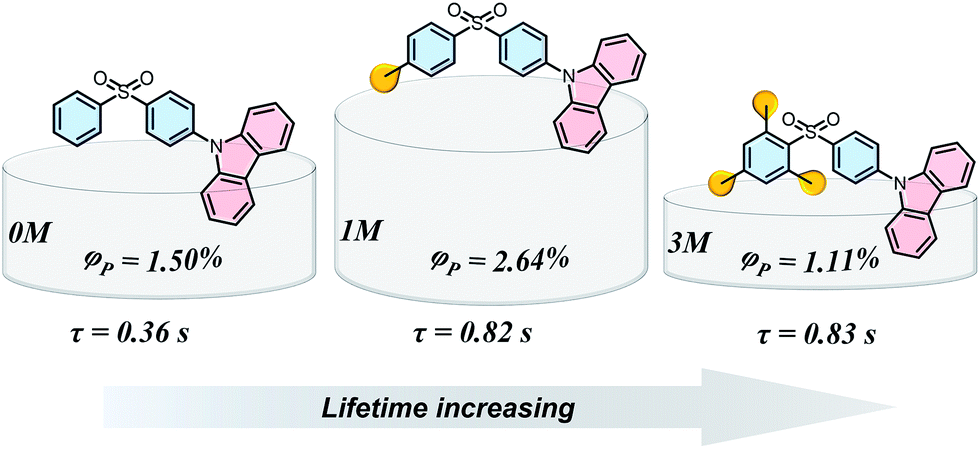 | ||
| Fig. 1 Chemical structures, long-lived phosphorescence lifetime (τ), and phosphorescence efficiency (ΦP) of 0M, 1M, and 3M. | ||
 | ||
| Fig. 2 The long-lived phosphorescence photograph of 0M (upper), 1M (middle), and 3M (bottom) with different delay times. | ||
Results and discussion
Compounds 1M and 3M were both synthesized as shown in Fig. S1† in two-step chemical transformations starting from commercially available sulfonyl chlorides. To explore the changes in the photophysical properties of the derived compounds, we performed systematic investigations in both the solution- and the crystalline-state. In dilute THF solution (1 × 10−5 M), as the methylation degree increases the maximum emission peak is slightly blue-shifted (Fig. 3a). This is attributed to the electron-donating ability of the methyl group(s). The fluorescence lifetimes in THF solutions for 0M, 1M and 3M are 6.84 ns, 6.83 ns and 6.39 ns, respectively (Fig. 3b). Furthermore, the theoretical simulation for the energy level distribution, carried out in a cavity of a tetrahydrofuran (THF) solvent and using the polarizable continuum model, for 0M, 1M and 3M, is as shown in Fig. S2.† These results suggest that the charge distribution is only slightly affected by the number of methyl groups for the isolated molecules.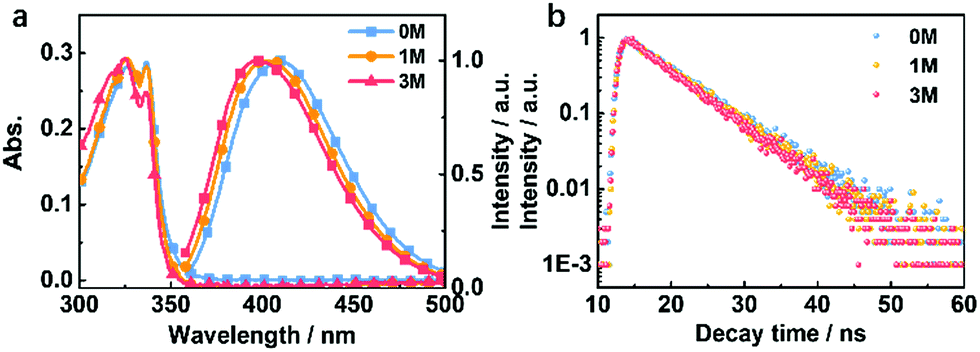 | ||
| Fig. 3 (a) Absorption and fluorescence (300 K) spectra and (b) fluorescence lifetime decay (300 K) curves of 0M, 1M, and 3M in THF solution (10−5 M). | ||
Crystalline powders of the three compounds were obtained by recrystallization from a dichloromethane and ethanol solvent mixture (confirmed by powder X-ray diffraction, Fig. S3†) and display notable differences in optical properties, especially with regard to 1M and 3M. The maximum emission peaks of 1M in the crystalline-state and solution-state are at 423 nm (Fig. 4) and 402 nm, respectively, with a bathochromic shift of 21 nm for the crystalline-state which can be ascribed to π–π stacking.9d For compound 3M, in the crystalline-state the emission peak is located at 376 nm and in the solution-state it is located at 399 nm, with a hypsochromic shift of 23 nm of the crystalline-state that is possibly due to H-aggregation.9a These stacking modes in 1M and 3M can be destroyed by grinding and show blue- and red-shifted emission in 1M and 3M, respectively, as shown in Fig. S4.† Compounds 1M and 3M show fluorescence in the crystalline-state with a lifetime of 9.2 ns and 2.6 ns, respectively (Fig. S5†). The longer fluorescence lifetime of 1M in the crystalline state might be due to the smaller overlap integral of the frontier orbitals of the monomer (Fig. S6†) and π–π stacking of the dimer (Fig. S7†).9d Remarkably, for the three materials under ambient conditions, yellowish long-lived phosphorescence emission can be easily observed by the naked eye when the UV-light excitation source is removed (Fig. 2). For 1M, delayed phosphorescence is observed and the maximum emission peak is located in a longer wavelength region (546 nm) with a lifetime (τ) of 0.82 s, whilst the delayed phosphorescence for 3M is located at 542 nm with a lifetime (τ) of 0.83 s (Fig. 4, right column). All the long-lived phosphorescence decay behaviours have a mono-exponential shape, which indicates a pure triplet emission centre for the long-lived phosphorescence. With respect to 0M and 1M, the delayed emission band located in the fluorescence region (ca. 400–450 nm) is attributed to the triplet–triplet annihilation (TTA) during the decay time of 0M and 1M. These results indicate that the methyl groups do have significant influence over the emission intensity and prolong the existence of the long-lived phosphorescence in the crystalline-states.
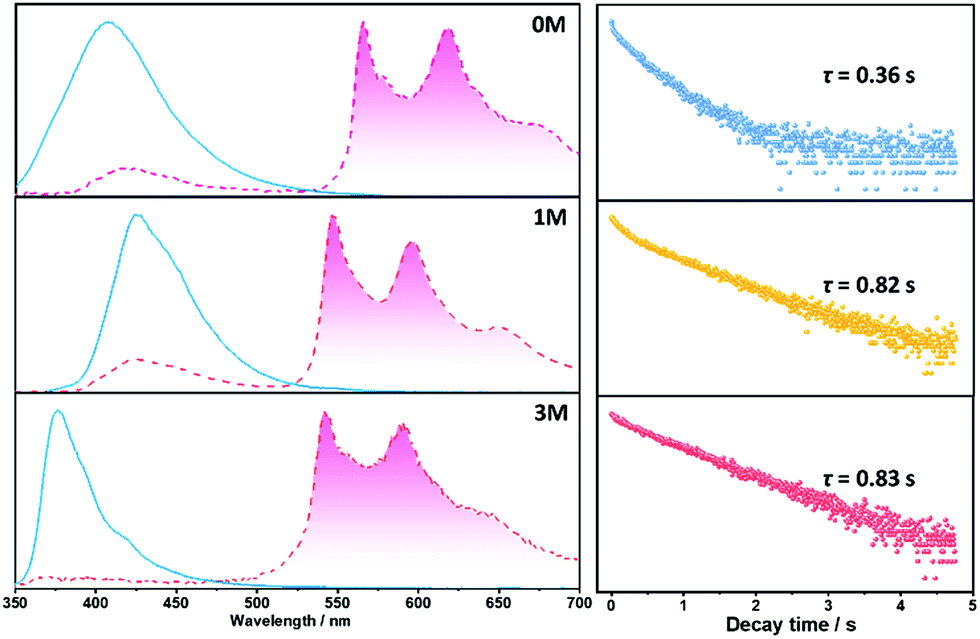 | ||
| Fig. 4 Steady state fluorescence (blue solid line), long-lived phosphorescence (red dashed line filled with red, delay time = 8 ms) spectra of 0M, 1M and 3M, and corresponding maximum emission peak (565 nm, 546 nm and 542 nm for 0M, 1M, and 3M, respectively) of long-lived phosphorescence lifetime recorded under ambient conditions with a 350 nm excitation source. | ||
For further understanding the mechanism of the long-lived phosphorescence of these compounds, the delayed emission was recorded at 77 K (Fig. 5) in the crystalline-state and the clear changes of the emission bands around 450–530 nm at room temperature were seen (Fig. 4, red dashed line). We assume that the phosphorescence emission from the isolated molecule is due to normal phosphorescence. With regard to 1M and 3M, the maximum emission wavelength of the long-lived phosphorescence is 546 nm (2.27 eV) and 542 nm (2.28 eV), respectively, and is much more red-shifted than that of their corresponding normal phosphorescence. The longer wavelength phosphorescence suggests the existence of a lower excited state in the crystalline-state with a much longer lifetime compared to the isolated molecules, which is due to the electronic coupling of adjacent molecules (dimer) in the crystals.9a,11b Single crystals of 1M and 3M were obtained by slow evaporation of saturated solutions from mixed solvents (dichloromethane and ethanol) under the same conditions for the crystalline powders. Single crystal X-ray diffraction was carried out to gain information regarding the packing of the methylated molecules (Fig. S7 and S8†). The intermolecular interactions between adjacent molecular units in the acquired crystals containing C–H⋯π, S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H and π–π stacking cause the construction of the crystalline frameworks. These intermolecular interactions can help suppress the non-radiative pathways of excited molecules. As a consequence, the three crystals emit bright blue fluorescence with relatively high fluorescence quantum yields of 29.1%, 72.5% and 51.1% for 0M, 1M, and 3M, respectively. Compound 1M shows close contact between neighbouring molecules, i.e., co-linear by the head-to-tail style with C–H⋯π (2.719 Å) forming dimer 1, taking the dipole orientation into account, as H-type interaction. The molecular arrangements of the face-to-face interactions reveal that the π–π stacking (3.464 Å) mode aids the construction of dimer 2 and the two adjacent units adopting the anti-parallel packing mode with SO⋯H interaction (2.633 Å) forming dimer 3. These numerous intermolecular interactions contribute to the comparatively high fluorescence quantum yield of 1M in the crystalline state. Likewise, the SO⋯H interactions constructed the framework of the 3M single crystals consisting of the three kinds of dimers (Fig. S8†). Molecules with such plenty of interactions in the crystal lattices improve the potential for phosphorescence emission.
O⋯H and π–π stacking cause the construction of the crystalline frameworks. These intermolecular interactions can help suppress the non-radiative pathways of excited molecules. As a consequence, the three crystals emit bright blue fluorescence with relatively high fluorescence quantum yields of 29.1%, 72.5% and 51.1% for 0M, 1M, and 3M, respectively. Compound 1M shows close contact between neighbouring molecules, i.e., co-linear by the head-to-tail style with C–H⋯π (2.719 Å) forming dimer 1, taking the dipole orientation into account, as H-type interaction. The molecular arrangements of the face-to-face interactions reveal that the π–π stacking (3.464 Å) mode aids the construction of dimer 2 and the two adjacent units adopting the anti-parallel packing mode with SO⋯H interaction (2.633 Å) forming dimer 3. These numerous intermolecular interactions contribute to the comparatively high fluorescence quantum yield of 1M in the crystalline state. Likewise, the SO⋯H interactions constructed the framework of the 3M single crystals consisting of the three kinds of dimers (Fig. S8†). Molecules with such plenty of interactions in the crystal lattices improve the potential for phosphorescence emission.
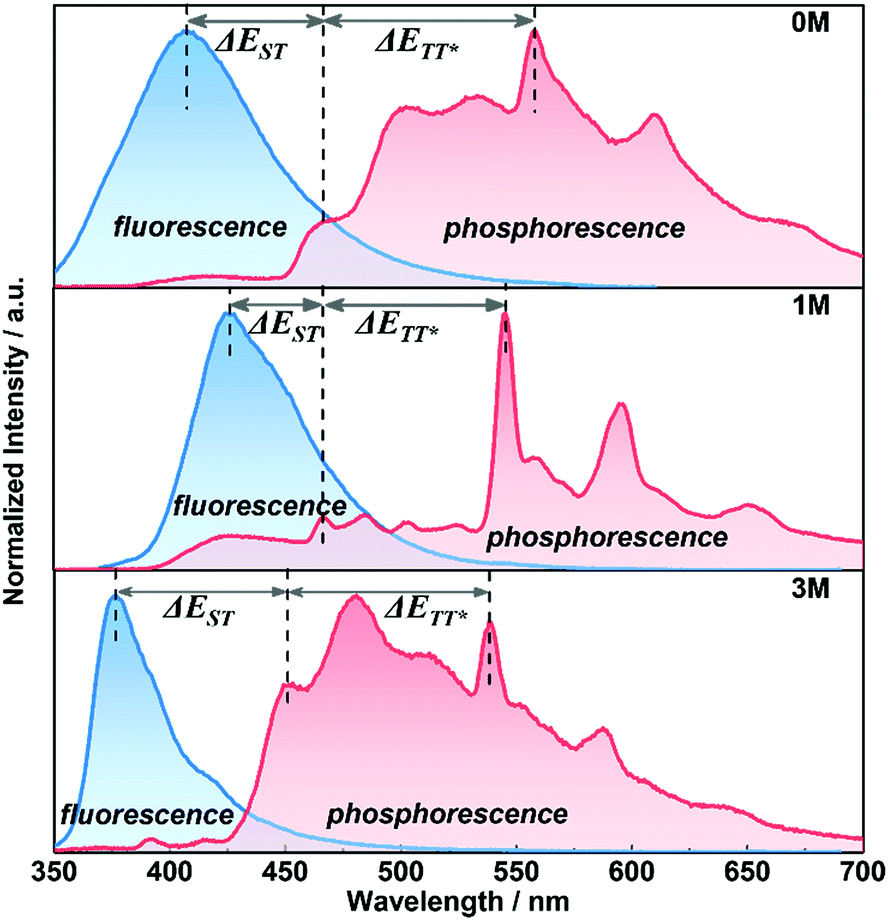 | ||
| Fig. 5 Fluorescence (blue area, 300 K) and phosphorescence spectra (red area, 77 K, and delay time = 8 ms) for 0M, 1M, and 3M from up to bottom. ΔEST = energy gap between the participating S1 and T1 states and ΔETT* = stabilized gap. | ||
Theoretical time-dependent density functional theory (TD-DFT) calculation studies have been carried out to reveal the intermolecular electronic coupling effects in the dimer molecule system.15 As discussed in our previous work,11b the different electronic configurations of the donor (D) and acceptor (A) units make a significant contribution to enhancing the intermolecular electronic coupling (IEC) effects. Hence, we extracted the dimers containing spatially close donor and acceptor units (both dimer 1 for 1M and 3M, Fig. S8†) to evaluate the IEC effects and identify the possible ISC channels in both monomers and dimers (Fig. S9†). The monomers of 0M, 1M, and 3M exhibit an increased energy gap between the lowest excited singlet (S1) and triplet (T1) states, demonstrating that the ability of electron-withdrawing nature of the diphenylsulphone group is reduced by methylation. For instance, the T1, T4, T7, and T9 states of the isolated 1M molecules involve the same orbital composition as the S1 state, indicating good channels for ISC.9a,11b,15a Considering the narrower energy gap between the S1 and Tn states, the ISC channels become more efficient in the dimer models. The energy levels of T7 and S1 states are relatively close in the 1M dimer, and the orbital composition is also well matched, indicating a barrier-less intersystem crossing pathway between S1 and T7. The hot excitons (T7) decay to the lowest triplet state via rapid internal conversion (IC) and then to the ground state, resulting in long-lived phosphorescence at room temperature in 1M with ΦP = 2.64%. Compared to the 1M dimer, compound 3M shows fewer and lower efficiency ISC channels (Fig. S9†), resulting in a relatively low ISC efficiency and contributing to the lowest long-lived phosphorescence efficiency (ΦP = 1.11%) amongst the methylated systems.
By combining the photophysical properties and the packing modes as well as the ISC channels in the single crystals, we have proposed a mechanism for the long-lived phosphorescence in these methylated derivatives (Fig. 6). The short lifetime fluorescence of several nanoseconds results from the radiative decay of the lowest excited singlet state (S1), which can also form triplet excitons through ISC with the help of the electronic configuration according to El-Sayed's rule,16 enabling the occurrence of the normal phosphorescence with typical microseconds to milliseconds lifetime. The long-lived phosphorescence (hundreds of milliseconds) is observed in crystals due to the presence of a new lower-lying triplet energy level 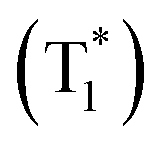 , which is formed by the interaction of adjacent molecules and refers to as IEC.11b In these processes, the splitting of energy between two different excited states shows a great influence on the transition probability. For example, the narrow gap can facilitate the processes of ISC and IC according to the Arrhenius equation.17 As calculated from the fluorescence and cryogenic phosphorescence spectra (Fig. 5), the energy splitting of ΔEST (0.25 eV) and ΔETT* (0.40 eV) of 1M is minimal (Fig. 6), thus facilitating exciton population at the
, which is formed by the interaction of adjacent molecules and refers to as IEC.11b In these processes, the splitting of energy between two different excited states shows a great influence on the transition probability. For example, the narrow gap can facilitate the processes of ISC and IC according to the Arrhenius equation.17 As calculated from the fluorescence and cryogenic phosphorescence spectra (Fig. 5), the energy splitting of ΔEST (0.25 eV) and ΔETT* (0.40 eV) of 1M is minimal (Fig. 6), thus facilitating exciton population at the 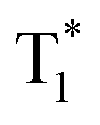 state in crystals. A higher exciton concentration of 1M generates brighter long-lived phosphorescence. Compound 3M exhibits a larger ΔEST (0.53 eV) and ΔETT* (0.47 eV), showing relatively weaker long-lived phosphorescence as depicted in Fig. 2. Interestingly, the emission of normal phosphorescence (T1) in 0M (467 nm, the first phosphorescence peak confirmed at 77 K, Fig. 5) and 3M (449 nm, the first phosphorescence peak at 77 K, Fig. 5) under ambient conditions could be detected but is absent in 1M (Fig. S10 and Table S1†), which is due to the smaller ΔETT* in 1M. Additionally, triplet excitons (T1) generated in 1M can be stabilized quickly to the lower triplet state
state in crystals. A higher exciton concentration of 1M generates brighter long-lived phosphorescence. Compound 3M exhibits a larger ΔEST (0.53 eV) and ΔETT* (0.47 eV), showing relatively weaker long-lived phosphorescence as depicted in Fig. 2. Interestingly, the emission of normal phosphorescence (T1) in 0M (467 nm, the first phosphorescence peak confirmed at 77 K, Fig. 5) and 3M (449 nm, the first phosphorescence peak at 77 K, Fig. 5) under ambient conditions could be detected but is absent in 1M (Fig. S10 and Table S1†), which is due to the smaller ΔETT* in 1M. Additionally, triplet excitons (T1) generated in 1M can be stabilized quickly to the lower triplet state 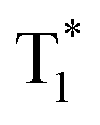 instead of radiative decay, without normal phosphorescence under ambient conditions. The complete transformation of the T1 excitons to the concerned
instead of radiative decay, without normal phosphorescence under ambient conditions. The complete transformation of the T1 excitons to the concerned 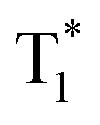 state enhances the brightness of the long-lived phosphorescence; therefore, 1M has the highest long-lived phosphorescence quantum efficiency amongst the three reported compounds. Both theoretical calculations and spectra analysis indicate that the methyl group(s) regulate the energy level distribution within the compounds 1M and 3M, which results in long-lived phosphorescence quantum yield differences.
state enhances the brightness of the long-lived phosphorescence; therefore, 1M has the highest long-lived phosphorescence quantum efficiency amongst the three reported compounds. Both theoretical calculations and spectra analysis indicate that the methyl group(s) regulate the energy level distribution within the compounds 1M and 3M, which results in long-lived phosphorescence quantum yield differences.
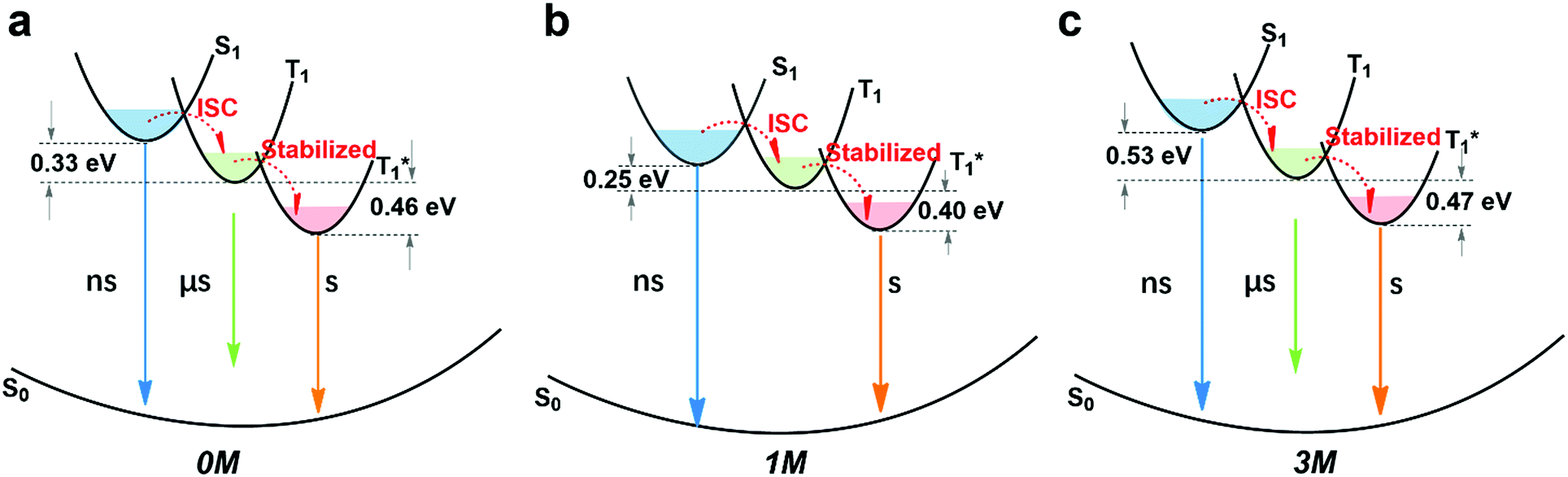 | ||
| Fig. 6 Proposed mechanism of long-lived phosphorescence and energy split (ΔEST and ΔETT*) of (a) 0M, (b) 1M, and (c) 3M. Blue arrows indicate fluorescence, green arrows represent normal phosphorescence, and yellow arrows represent long-lived phosphorescence. | ||
Besides the quantum efficiency, another crucial parameter for organic room temperature long-lived phosphorescence materials is lifetime, which is the reciprocal of the rate of the natural triplet radiative decay. Lifetime strongly depends on the oscillator strength between the lowest excited triplet and ground states for phosphorescence. An appropriate (n, π*) configuration proportion can regulate this process and tune the lifetime of pure organic long-lived phosphorescence materials.16 In our case, the methyl groups have only slight influence on the electronic configuration for isolated molecules, as proven by their similar spectral profiles, such as absorption, fluorescence and phosphorescence emission. Therefore, this indicates that another factor could be the cause of altering the long-lived phosphorescence lifetimes of the methylated compounds. Moreover, the rigid environment means less structural vibration and is essential for stabilizing the triplet excitons for designing pure organic room temperature phosphorescent materials.18 In general, the molecules in a crystal lattice occupy most of the space of a unit cell. However, along with the occupied spaces in crystals, there are plenty of unoccupied spaces, i.e., the free volume, which can afford the structural vibration with nonradiative decay of the excited state. As depicted in Fig. 7, we calculated the free volume distribution of the compounds 0M, 1M and 3M using Materials Studio with a 1.0 Å diameter sensor. Intriguingly, a negative correlation was determined between the long-lived phosphorescence lifetime and fractional free volume (Vf) of the crystals (Fig. 8 and S9a†). As the Vf becomes smaller, longer-lived emission could be observed in the increments of the methylation degree from 0M to 1M and to 3M. A diminished Vf in the crystals of 1M and 3M indicates a more compact packing pattern, accompanied by less space for the structural relaxation of the excited molecules and thus suppresses the non-radiative decay (knrT). Meanwhile, the long-lived lifetime of these three compounds in the crystalline state shows only a slight increase as the temperature decreases (Fig. S11 and Table S2†), which is in contrast to the normal phosphorescence (continuous decreasing with the temperature increasing).19 The near-constant lifetimes at low temperature and room temperature demonstrate the heavy suppression of the non-radiative pathways in these compounds, and the importance of free volume. Consequently, the restricted non-radiative decay leads to long-lived phosphorescence according to the equation τp = 1/(krT + knrT).20 On the other hand, the decreasing energy difference between the maximum excitation and the fluorescence emission (ΔEex–em) of the crystalline solid indicates that there is less energy loss with an increasing number of methyl groups and also the same decreasing trend can be observed in the correlation between Vf and ΔEex–em (Fig. S12b and S13†). Less excited state structural vibrations for 1M and 3M are also evident from the narrower fluorescence full width at half maximum (Fig. 4, blue lines) compared to 0M.21
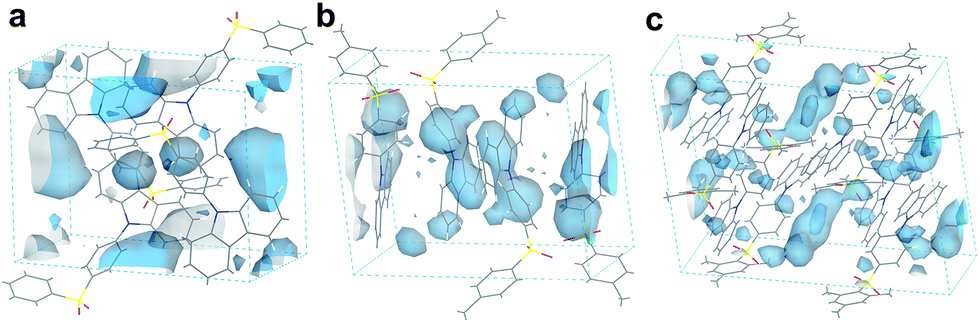 | ||
| Fig. 7 Free volume region in the single crystal cells of (a) 0M, (b) 1M, and (c) 3M. | ||
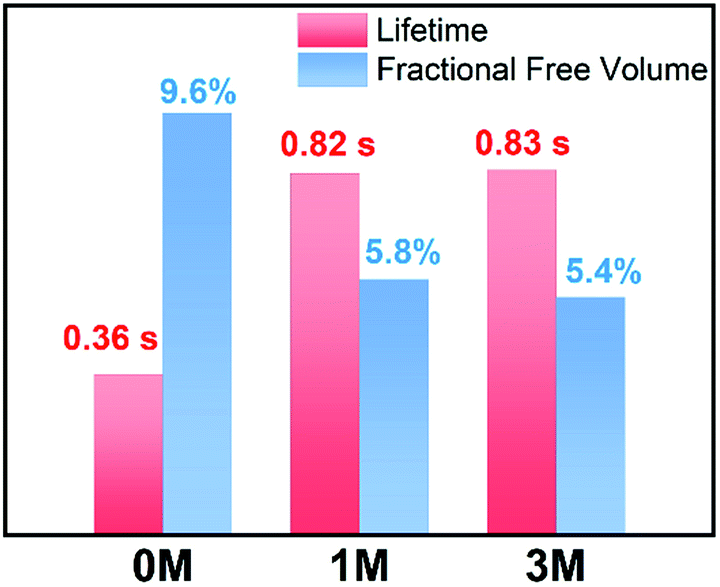 | ||
| Fig. 8 Fractional free volume and long-lived phosphorescence lifetime of 0M, 1M, and 3M. | ||
Furthermore, we compared the Vf and lifetime of several reported typically long-lived phosphorescence compounds (Fig. S14 and Table S3†). A similar noticeable negative trend between τp and Vf was observed (dashed line in Fig. S14†). These results suggest that Vf has a crucial impact on the phosphorescence lifetime. The negative correlation of Vf is very similar to the heavy atom effect in phosphorescent emitters. However, the difference is that the methylation effect accelerates the non-radiative pathways, whilst the heavy atom effect enhances the radiative pathways.10 Inspired by the concept of the life-span based on the methylation in bio-systems, the relationship between Vf and τp proves that the incorporation of methyl groups decreases the free volume in the crystal lattice and confines the excited molecular vibrational relaxations, resulting in the enhancement of the long-lived phosphorescence lifetimes of purely organic phosphorescent emitters. Finally, we have also synthesized additional acridine-type related compounds (DMAC-0M and DMAC-1M) to demonstrate that the methylation effect can also be expanded to other phosphorescence emitters (Fig. S15†). Indeed, for these DMAC-based compounds the phosphorescence lifetime is also enhanced upon methylation, among which DMAC-0M that contains no methyl group exhibits a lifetime of 5.30 μs and DMAC-1M that contains one methyl group exhibits a lifetime of 24.3 μs (Fig. S16†).
Conclusions
In summary, we have presented a method for prolonging the lifetime of purely organic room temperature phosphorescence materials. This comprehensive investigation confirms that the incorporation of the methyl group(s) has a crucial effect on the quantum yield and lifetime. Increasing the number of methyl groups prolongs the lifetime at room temperature up to 0.82 s (1M) and 0.83 s (3M) from 0.36 s (0M). The methyl groups effectively decrease the fractional free volumes within the crystal lattice and subsequently suppress the non-radiative decay pathways of the excited structural relaxation. However, excessive methylation of the structures enlarges the energy split (ΔEST and ΔETT*), resulting in a reduction of the phosphorescence quantum yield. Combining the lifetime and quantum yield, we can conclude that an appropriate methylation degree can afford a good balance to improve the integrated performance of long-lived phosphorescence materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC: 51733010, 61605253 and 21672267), the Science and Technology Planning Project of Guangdong (2015B090913003), the China Postdoctoral Science Foundation (2017M620395) and the Fundamental Research Funds for the Central Universities.Notes and references
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920 CrossRef CAS.
- (a) Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CAS; (b) S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160 CrossRef CAS; (c) S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W. W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef; (d) X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872 CrossRef CAS; (e) Q. Miao and K. Pu, Adv. Mater., 2018, 1801778 CrossRef.
- (a) C. Li, X. Tang, L. Zhang, C. Li, Z. Liu, Z. Bo, Y. Q. Dong, Y.-H. Tian, Y. Dong and B. Z. Tang, Adv. Opt. Mater., 2015, 3, 1184 CrossRef CAS; (b) Y. Katsurada, S. Hirata, K. Totani, T. Watanabe and M. Vacha, Adv. Opt. Mater., 2015, 3, 1726 CrossRef CAS.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231 CrossRef CAS.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655 CrossRef CAS PubMed.
- (a) A. Jaina, A. Kumara, S. J. Dhoblea and D. R. Peshwea, Renewable Sustainable Energy Rev., 2016, 65, 135 CrossRef; (b) J. Hölsä, Electrochem. Soc. Interface, 2009, 18, 42 Search PubMed.
- (a) J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589 CrossRef CAS; (b) R. Gao and D. Yan, Chem. Sci., 2017, 8, 590 RSC; (c) S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2016, 7, 1539 CrossRef CAS; (d) N. Notsuka, R. Kabe, K. Goushi and C. Adachi, Adv. Funct. Mater., 2017, 27, 1703902 CrossRef; (e) R. Kabe and C. Adachi, Nature, 2017, 550, 384 CrossRef CAS.
- (a) X. G. Yang and D. P. Yan, Adv. Opt. Mater., 2016, 4, 897 CrossRef CAS; (b) H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Opt. Mater., 2016, 4, 1015 CrossRef CAS; (c) S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 4005 CrossRef CAS.
- (a) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS; (b) J. A. Li, J. Zhou, Z. Mao, Z. Xie, Z. Yang, B. Xu, C. Liu, X. Chen, D. Ren, H. Pan, G. Shi, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 6449 CrossRef CAS; (c) X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef; (d) S. Pan, Z. Chen, X. Zheng, D. Wu, G. Chen, J. Xu, H. Feng and Z. Qian, J. Phys. Chem. Lett., 2018, 9, 3939 CrossRef CAS.
- A. Köhler and H. Bässler, Electronic Processes in Organic Semiconductors, An Introduction, 2015 Search PubMed.
- (a) S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef; (b) Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181 CrossRef CAS; (c) P. Xue, P. Wang, P. Chen, B. Yao, P. Gong, J. Sun, Z. Zhang and R. Lu, Chem. Sci., 2017, 8, 6060 RSC.
- L. Gu, H. Shi, C. Miao, Q. Wu, Z. Cheng, S. Cai, M. Gu, C. Ma, W. Yao, Y. Gao, Z. An and W. Huang, J. Mater. Chem. C, 2018, 6, 226 RSC.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef.
- B. P. Williams and M. Gehring, Nat. Commun., 2017, 8, 2124 CrossRef.
- (a) L. Paul, S. Chakrabarti and K. Ruud, J. Phys. Chem. Lett., 2017, 8, 1253 CrossRef CAS; (b) H. Ma, W. Shi, J. Ren, W. Li, Q. Peng and Z. Shuai, J. Phys. Chem. Lett., 2016, 7, 2893 CrossRef CAS.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592 CAS.
- (a) X. Han, Q. Bai, L. Yao, H. Liu, Y. Gao, J. Li, L. Liu, Y. Liu, X. Li, P. Lu and B. Yang, Adv. Funct. Mater., 2015, 25, 7521 CrossRef; (b) Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326 CrossRef CAS.
- (a) R. Gao, X. Fang and D. Yan, J. Mater. Chem. C, 2018, 6, 4444 RSC; (b) P. Xue, J. Sun, P. Chen, P. Wang, B. Yao, P. Gong, Z. Zhang and R. Lu, Chem. Commun., 2015, 51, 10381 RSC.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y. C. Wu, Y. Zhang, C. Jin, P. Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909 RSC.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1833075 and 1833083. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03019g |
| ‡ Dr Z. Mao and Dr Z. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |