
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regio- and chemoselective Csp3–H arylation of benzylamines by single electron transfer/hydrogen atom transfer synergistic catalysis†
Takafumi
Ide
,
Joshua P.
Barham
 ,
Masashi
Fujita
,
Yuji
Kawato
,
Hiromichi
Egami
and
Yoshitaka
Hamashima
*
,
Masashi
Fujita
,
Yuji
Kawato
,
Hiromichi
Egami
and
Yoshitaka
Hamashima
*
School of Pharmaceutical Sciences, University of Shizuoka, 52-1 Yada, Suruga-ku, Shizuoka 422-8526, Japan. E-mail: hamashima@u-shizuoka-ken.ac.jp
First published on 12th September 2018
Abstract
We present a highly regio- and chemoselective Csp3–H arylation of benzylamines mediated by synergy of single electron transfer (SET) and hydrogen atom transfer (HAT) catalysis. Under well precedented SET catalysis alone, the arylation reaction of N,N-dimethylbenzylamine proceeded via aminium radical cation formation and selectively targeted the N-methyl group. In contrast, addition of PhC(O)SH as a HAT catalyst precursor completely switched the regioselectivity to Csp3–H arylation at the N-benzylic position. Measurement of oxidation potentials indicated that the conjugate base of PhC(O)SH is oxidized in preference to the substrate amine. The discovery of the thiocarboxylate as a novel HAT catalyst allowed for the selective generation of the sulfur-centered radical, so that the N-benzyl selectivity was achieved by overriding the inherent N-methyl and/or N-methylene selectivity under SET catalysis conditions. While visible light-driven α-C–H functionalization of amines has mostly been demonstrated with aniline derivatives and tetrahydroisoquinolines (THIQs), our method is applicable to a variety of primary, secondary and tertiary benzylamines for efficient N-benzylic C–H arylation. Functional group tolerance was high, and various 1,1-diarylmethylamines, including an α,α,α-trisubstituted amine, were obtained in good to excellent yield (up to 98%). Importantly, the reaction is applicable to late-stage functionalization of pharmaceuticals.
Introduction
The amino group is found in many kinds of molecules, including naturally occurring bioactive compounds, pharmaceutical drugs, agrochemicals, and functional materials. Therefore, direct and selective Csp3–H functionalization of amine compounds is extremely useful for rapid derivatization.1,2 In addition, the benzylamine unit is contained in various synthetic intermediates and is a core structure of many pharmaceuticals (Fig. 1A), as exemplified by donepezil (AChE inhibitor) and roxatidine (H2 blocker). To expedite structure–activity-relationship studies, regio- and chemoselective C–H functionalization of benzylamine derivatives is highly attractive. Moreover, 1,1-diarylmethylamine unit is a well-characterized pharmacophore (Fig. 1B).3 Considering the importance of such substructures in medicinal chemistry, mild and efficient methods to construct the 1,1-diarylmethylamine framework are of great interest, and benzylic Csp3–H arylation of benzylamines might be a straightforward and expedient approach.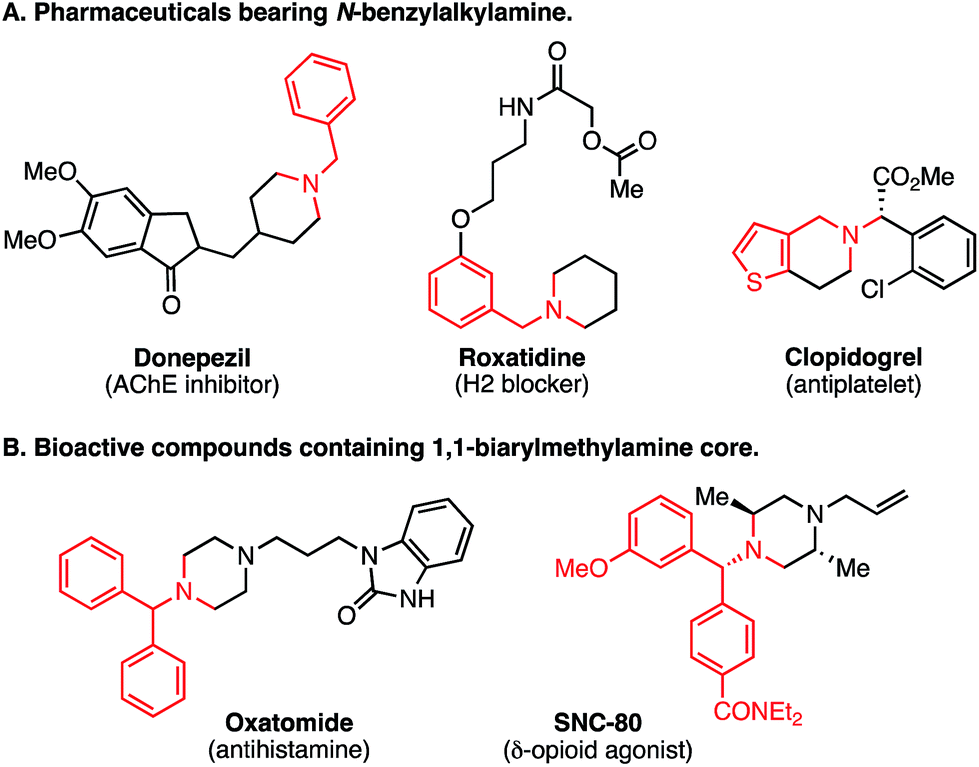 | ||
| Fig. 1 Bioactive compounds containing benzylamines. | ||
Nucleophilic addition of an aryl nucleophile to an imine or an iminium ion is a commonly used strategy to construct the 1,1-diarylmethylamine core.4 On the other hand, recent work has explored direct benzylic Csp3–H arylation of amine derivatives (Scheme 1). Li and co-workers reported an oxidative cross-dehydrogenative coupling reaction of N-aryl tetrahydroisoquinolines (THIQs) via in situ oxidation to iminium ions (Scheme 1A).5 Stephenson's group achieved similar oxidative transformations under photoredox catalysis conditions.6 In addition to the oxidative approach, other researchers have employed a protocol involving deprotonation at the N-benzylic position, followed by transition-metal-catalyzed cross-coupling reaction (Scheme 1B).7 Although a variety of aromatic groups can be incorporated, these approaches normally require harsh reaction conditions, such as strong base and high temperature. Elegant methodologies using transition metal-catalyzed C–H activation have also been disclosed, though in these cases, a directing group is required for the interaction between the transition metal and the benzylic C–H bond (Scheme 1C).8
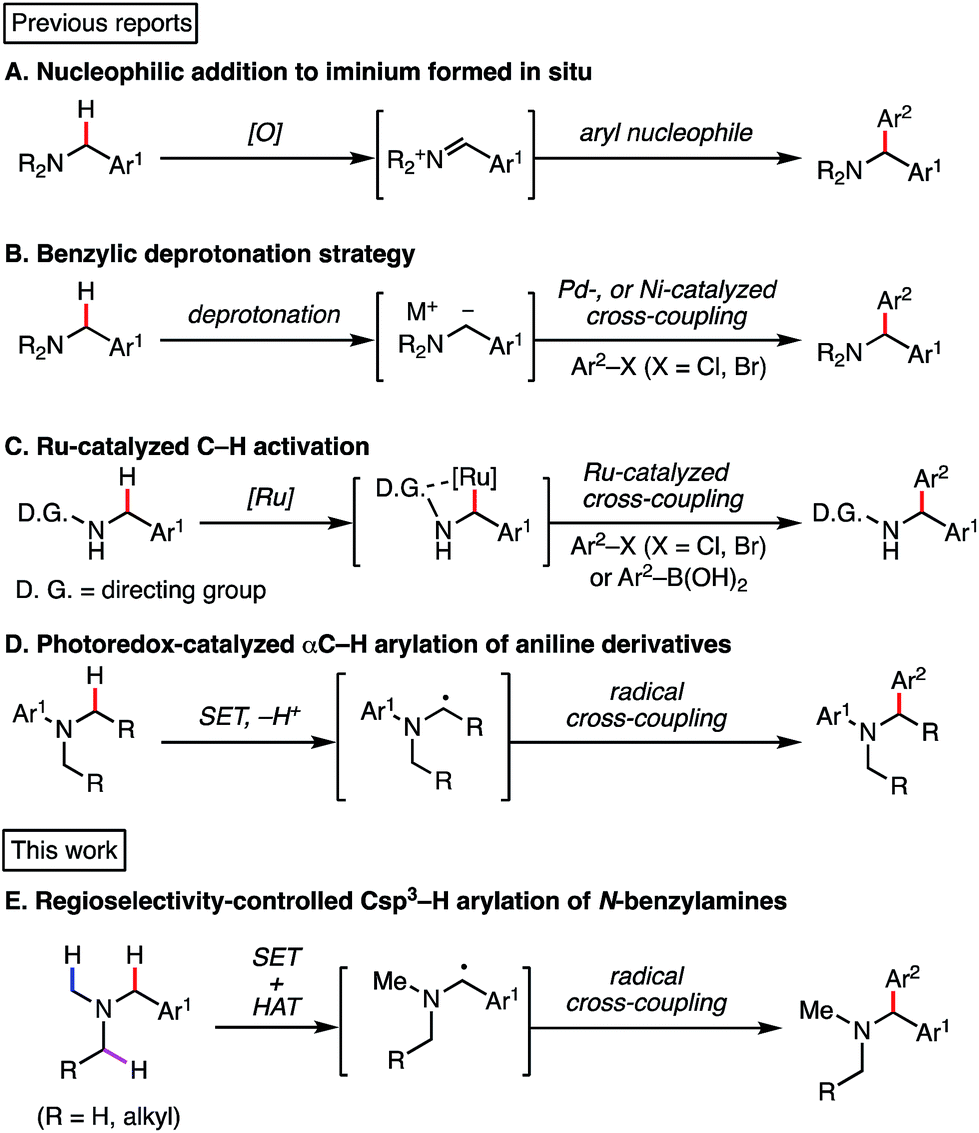 | ||
| Scheme 1 Previous benzylamine arylation strategies and this work. | ||
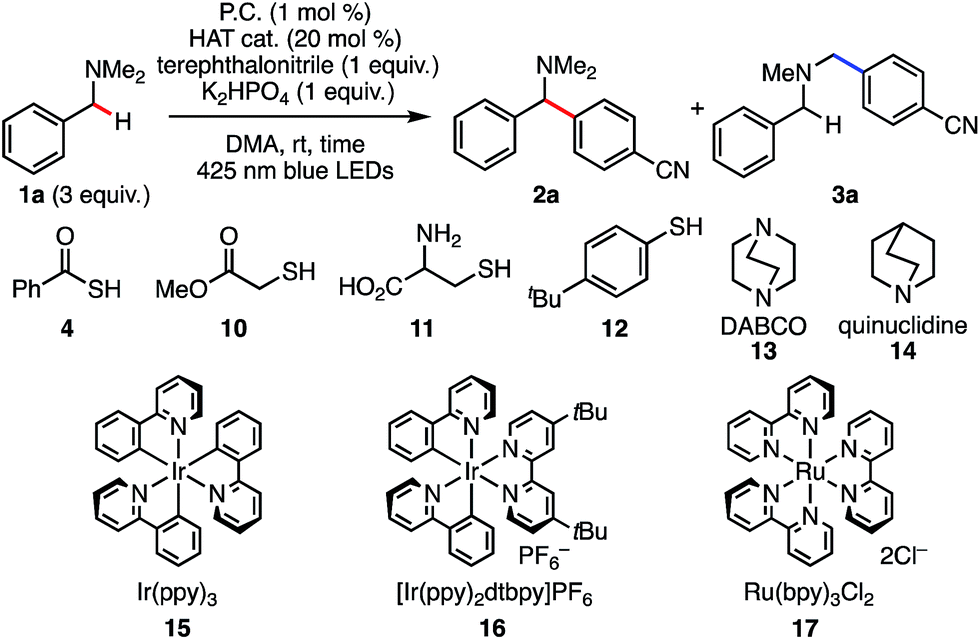
Photoinduced electron transfer has long been examined for C–H functionalization adjacent to a nitrogen atom.9,10 In 2011, MacMillan and co-workers reported a redox-neutral α-C–H arylation of various aniline derivatives using electron-deficient arenes as arylating partners in the presence of a photoredox catalyst (Scheme 1D).11 Following that work, we recently reported a redox-neutral α-C–H alkylation and cyanation of N-aryl-THIQs using activated alkyl halides and tosylcyanide.12 While visible light photoredox functionalization of amines has been well investigated, the substrate scope is generally limited to THIQ and aniline derivatives. On the other hand, there are some reports on photo-mediated SET oxidation of tertiary alkyl amines (e.g., Et3N and iPr2NEt) though they are used as sacrificial reducing agents.13,14 As for benzylic C–H arylation, THIQ derivatives have been investigated actively as benchmark substrates.6e,15,16 It should be noted that THIQs are special compounds in terms of both the oxidation potential and the bond dissociation energy (vide infra), indicating that simple N-alkyl benzylamines should be considered different from standard THIQs. Therefore, selective benzylic transformation of simple alkylamines is still a challenging subject. To our knowledge, there is no general and selective method available for N-benzylic C–H arylation. Herein, we report a regio- and chemoselective C–H functionalization of benzylamines via redox-neutral SET and HAT synergistic catalysis (Scheme 1E). The distinct features of the present reaction are listed below: (1) in addition to well-studied aniline-type compounds, various unprotected N-alkyl benzylamines are available in this reaction. (2) Since PhC(O)SH is easily deprotonated and converted to the corresponding sulfur-centered radical, single electron oxidation of the amine substrates is blocked effectively. SET/HAT synergistic catalysis enables excellent N-benzyl selectivity in preference to inherent N-methyl and N-methylene selectivity observed under SET catalysis conditions. (3) Due to the efficient generation of the HAT catalyst, as little as 1 mol% of PhC(O)SH is sufficient. In addition, an excess amount of starting amine is not required in our reaction. (4) The chemoselectivity is high, and the late-stage functionalization of pharmaceutical drugs was demonstrated successfully.
Results and discussion
According to the reported reaction conditions of photoredox catalysis via an aminium radical cation intermediate,11 we began with the reaction of N,N-dimethylbenzylamine (1a) with terephthalonitrile expecting that the most stable (benzylic and 2°) α-amino radical would be generated (Scheme 2). With Ir(ppy)3 (1 mol%) as a photoredox catalyst and K2HPO4 as base, the reaction afforded two compounds, 2a and 3a, in excellent mass balance (95%), though a long reaction time (12 h) was required for completion. Contrary to our expectation, the major product was not 2a but 3a, which is derived from the thermodynamically less stable non-benzylic and 1° α-amino radical.17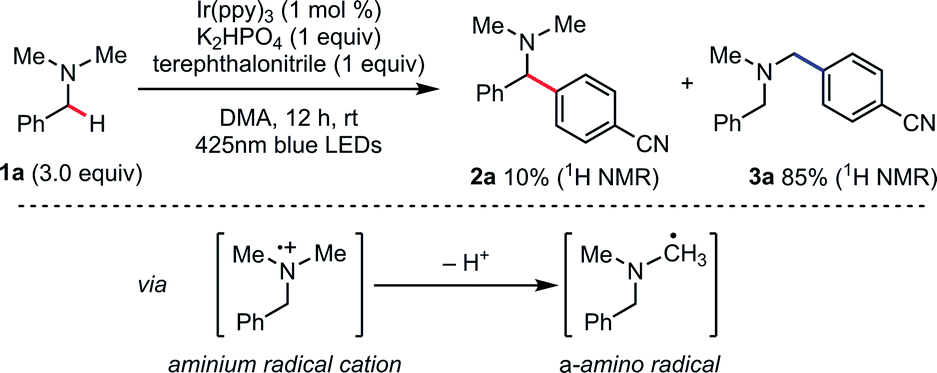 | ||
| Scheme 2 Csp3–H arylation of N,N-dimethylbenzylamine under photoredox catalysis. | ||
While the origin of this regioselectivity is unclear at this point, we think that the kinetic acidity in terms of stereoelectronic and steric factors are crucial. Thus, deprotonation of aminium radical cations to give α-amino radicals might be accelerated by the overlap of the breaking Csp3–H bond orbital with the SOMO orbital on the aminium radical cation.18,19 These results suggested that well-precedented SET catalysis is not applicable and distinct reaction conditions should be devised for achieving the targeted N-benzylic transformation.
We considered that a HAT strategy might alter the regioselectivity (Scheme 3). A significant determinant of the selectivity of a HAT process is the C–H bond dissociation energy (BDE). Since BDE is directly associated with the stability of the radical products, a large difference between N-methyl C–H and N-benzyl C–H BDEs should be anticipated. According to the previous studies,20,21 we expected that sulfur-centered radicals would be HAT agents of choice for selective activation of the N-benzyl group. Importantly, however, in order to avoid direct single electron oxidation of N-benzylamine 1a leading to the SET catalysis pathway (Scheme 3, right), the sulfur-centered radical precursor should have a lower anodic peak potential than that of N-benzylamine 1a. For this purpose, we envisaged that the anion of a sulfur atom would undergo more facile oxidation than a typical thiol such as cysteine (E1/2 = +0.85 V vs. SCE for cysteine in CH3CN, pKa = 9.35 for cysteine methyl ester)22 and that a more acidic sulfur compound would be more favorable. Therefore, we selected thiocarboxylic acid 4, whose conjugate base could be generated with a weak base (pKa = 3.2 for thioacetic acid).23 The negatively charged, electron-rich thiocarboxylate species 5 might undergo SET oxidation much faster than the substrate amine, thereby affording the sulfur-centered radical 6 preferentially (Scheme 3, left). This scenario would lead to regioselective C–H activation at the N-benzylic position by overriding the natural N-methyl selectivity observed in Scheme 2.
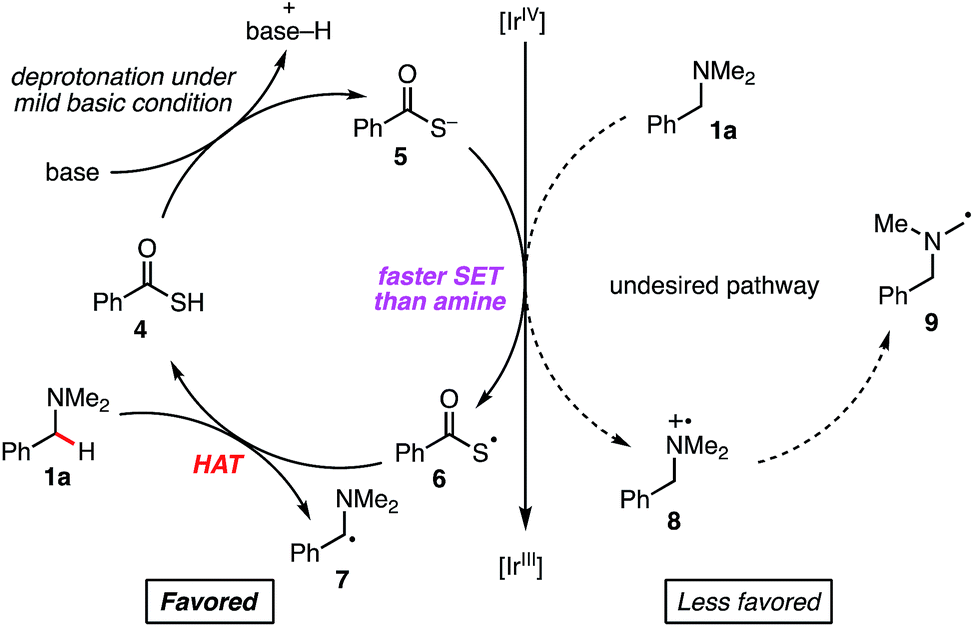 | ||
| Scheme 3 Working hypothesis for regioselective arylation at the N-benzylic position. | ||
To validate our working hypothesis, we measured the oxidation potentials of potassium thiobenzoate (PhC(O)SK) and three N-benzylamines by cyclic voltammetry (CV) in DMA (Fig. 2; see ESI† for potentials vs. SCE and further CV studies). Gratifyingly, PhC(O)SK was found to have a less positive anodic peak potential (Epox (6/5) = +0.80 V vs. Ag/AgCl in DMA) than all the amine compounds examined, even when compared to the aniline-type N-benzyl tetrahydroquinoline (THQ) (Epox = +0.97 V vs. Ag/AgCl in DMA). Therefore, thermodynamics predicted that selective oxidation of the thiobenzoate anion would occur even in the presence of the reducing amine substrates. Furthermore, CV revealed that oxidation of PhC(O)SK by [IrIV(ppy)3]+ (E1/2 (IrIV/IrIII) = +0.96 V vs. Ag/AgCl in DMA) was exergonic, while oxidation of the amines was always endergonic. According to these data, a faster reaction profile is expected compared to the reaction shown in Scheme 2.
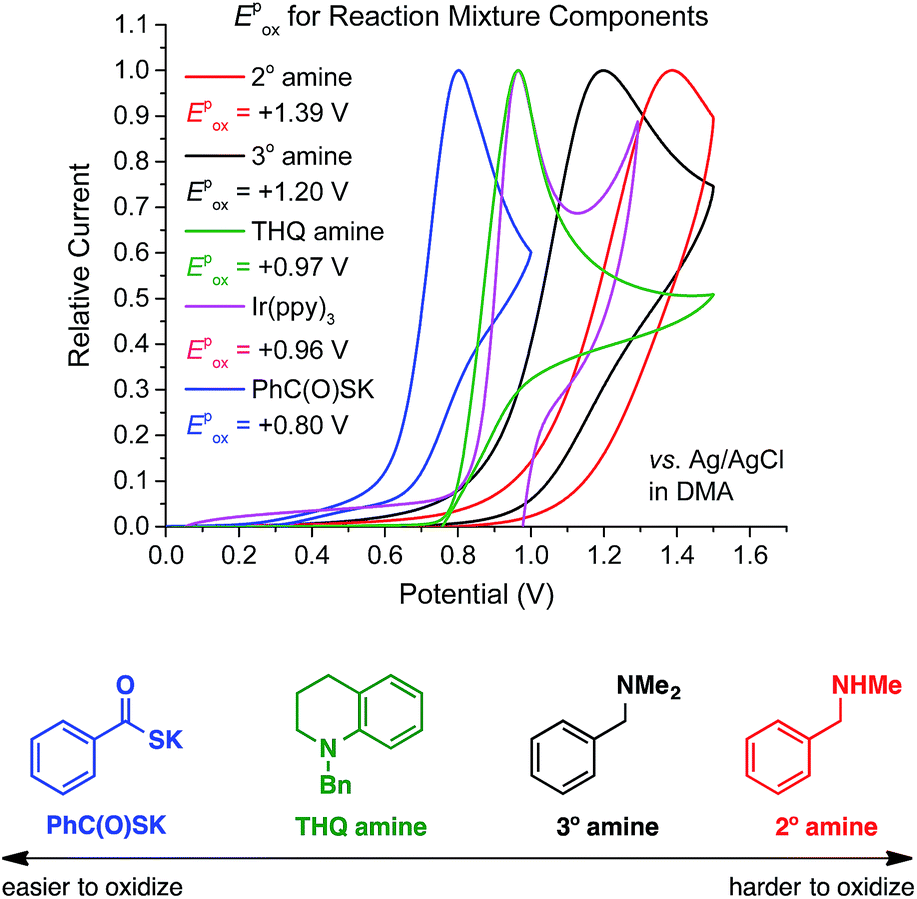 | ||
| Fig. 2 Comparison of oxidation peak potentials measured by cyclic voltammetry. | ||
With the promising CV results in mind, we revisited benzylic C–H arylation of N,N-dimethylbenzylamine (1a) in the presence of PhC(O)SH (4) and other HAT catalyst precursors (Table 1). The arylation proceeded in 91% yield at the benzylic position with excellent regioselectivity (r.r = >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, entry 2), which is consistent with the idea of a mechanistic switchover from SET catalysis to SET/HAT synergistic catalysis. It should be noted that the reaction reached completion within 1 h (entry 3), compared to 12 h required when PhC(O)SH was absent (entry 1). This rate-acceleration might be due to exergonic SET oxidation of the thiocarboxylate and a favorable difference in BDE between PhC(O)S–H (87.4 kcal mol−1)24 and the targeted N-benzylic C–H bond (84.9 kcal mol−1).24 Precatalysts 10 and 11 gave 2a with excellent N-benzyl selectivity. However, the yields were much lower even in the presence of 20 mol% of HAT catalysts (entries 4, 5). We assume that these less acidic thiols are not deprotonated under the reaction conditions, and thus SET oxidation might not be efficient compared to the case of PhC(O)SK (E1/2 = +0.85 V vs. SCE for cysteine in CH3CN,22Epox (6/5) = +0.65 V vs. SCE in CH3CN; see ESI†). Considering the pKa values of the HAT precatalysts, 12 was also examined as a more acidic thiol (entry 6). However, almost no reaction was observed, probably because the generated thiyl radical would be less reactive in the subsequent HAT process (S–H BDE of TolSH = 77–83 kcal mol−1)20 (entry 4). Recently, quinuclidine and DABCO aminium radical cations have been reported to perform HAT with alkylamines at the α position to the nitrogen.25 Given its accessible oxidation potential (Epox = +0.69 V vs. SCE in CH3CN),25b DABCO would undergo SET oxidation selectively over 1a. However, when PhC(O)SK was replaced with DABCO, the reaction did not proceed (entry 7). The reason is not clear at present, but back electron transfer processes such as quenching of the DABCO radical cation by arene radical anion might be sufficiently rapid to interfere with the HAT process.26 When PhC(O)SK was replaced with quinuclidine, the product was formed in only very low yield with low regioselectivity (entry 8), probably because the strong reactivity of quinuclidine radical cation (N+–H BDE of quinuclidine = 100 kcal mol−1)27 cannot effectively differentiate between the C–H bonds at the N-methyl and N-benzyl positions. These results strongly indicate the superiority of PhC(O)SK for the selective N-benzyl arylation over other HAT catalysts.
:1, entry 2), which is consistent with the idea of a mechanistic switchover from SET catalysis to SET/HAT synergistic catalysis. It should be noted that the reaction reached completion within 1 h (entry 3), compared to 12 h required when PhC(O)SH was absent (entry 1). This rate-acceleration might be due to exergonic SET oxidation of the thiocarboxylate and a favorable difference in BDE between PhC(O)S–H (87.4 kcal mol−1)24 and the targeted N-benzylic C–H bond (84.9 kcal mol−1).24 Precatalysts 10 and 11 gave 2a with excellent N-benzyl selectivity. However, the yields were much lower even in the presence of 20 mol% of HAT catalysts (entries 4, 5). We assume that these less acidic thiols are not deprotonated under the reaction conditions, and thus SET oxidation might not be efficient compared to the case of PhC(O)SK (E1/2 = +0.85 V vs. SCE for cysteine in CH3CN,22Epox (6/5) = +0.65 V vs. SCE in CH3CN; see ESI†). Considering the pKa values of the HAT precatalysts, 12 was also examined as a more acidic thiol (entry 6). However, almost no reaction was observed, probably because the generated thiyl radical would be less reactive in the subsequent HAT process (S–H BDE of TolSH = 77–83 kcal mol−1)20 (entry 4). Recently, quinuclidine and DABCO aminium radical cations have been reported to perform HAT with alkylamines at the α position to the nitrogen.25 Given its accessible oxidation potential (Epox = +0.69 V vs. SCE in CH3CN),25b DABCO would undergo SET oxidation selectively over 1a. However, when PhC(O)SK was replaced with DABCO, the reaction did not proceed (entry 7). The reason is not clear at present, but back electron transfer processes such as quenching of the DABCO radical cation by arene radical anion might be sufficiently rapid to interfere with the HAT process.26 When PhC(O)SK was replaced with quinuclidine, the product was formed in only very low yield with low regioselectivity (entry 8), probably because the strong reactivity of quinuclidine radical cation (N+–H BDE of quinuclidine = 100 kcal mol−1)27 cannot effectively differentiate between the C–H bonds at the N-methyl and N-benzyl positions. These results strongly indicate the superiority of PhC(O)SK for the selective N-benzyl arylation over other HAT catalysts.
|
|
|||||
|---|---|---|---|---|---|
| Entry | HAT cat. | P.C. | Time (h) | Yieldb (%) |
2a:3ab |
| a The reactions were run on 0.2 mmol scale. b Yield and regioisomeric ratio were determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. c Data from Scheme 3. d 1 mol% of PhC(O)SH and 0.5 mol% of Ir(ppy)3 were used on a 1 mmol scale. e 2 equiv. of N,N-dimethylbenzylamine was used. f Isolated yield. g 1 equiv. of N,N-dimethylbenzylamine was used. P.C.: photocatalyst. | |||||
| 1c | — | 15 | 12 | 95 | 1:8.5 |
| 2 | 4 | 15 | 12 | 91 | >20:1 |
| 3 | 4 | 15 | 1 | 93 | >20:1 |
| 4 | 10 | 15 | 1 | 23 | >20:1 |
| 5 | 11 | 15 | 1 | 43 | >20:1 |
| 6 | 12 | 15 | 1 | Trace | — |
| 7 | 13 | 15 | 1 | Trace | — |
| 8 | 14 | 15 | 1 | 12 | 1:2 |
| 9 | 4 | 16 | 1 | 42 | >20:1 |
| 10 | 4 | 17 | 1 | No reaction | |
| 11d,e | 4 | 15 | 2 | 90 (87)f | >20:1 |
| 12d,g | 4 | 15 | 2 | 72 | >20:1 |
We also examined other photoredox catalysts. When [Ir(ppy)2dtbbpy]PF6 was used, the product was obtained in a lower yield (entry 9). In this case, reductive quenching of [IrIII]* (E1/2 (*IrIII/IrII) = +0.66 V vs. SCE in CH3CN)28 from the thiocarboxylate (Epox = +0.65 V vs. SCE in CH3CN) is plausible. Since the subsequent SET process between [IrII] (E1/2 (IrIII/IrII) = −1.51 V vs. SCE in CH3CN)28 and terephthalonitrile (Ered1/2 = −1.61 V vs. SCE in CH3CN)29 is thermodynamically disfavored, the reaction might be retarded. On the other hand, the reaction did not proceed with Ru(bpy)3Cl2 (entry 10). Considering the redox potential after the favorable reductive quenching of [RuII]* (E1/2 (*RuII/RuI) = +0.77 V vs. SCE in CH3CN) by PhC(O)SK, SET from [RuI] (E1/2 (RuII/RuI) = −1.33 V vs. SCE in CH3CN)30 to terephthalonitrile (Ered1/2 = −1.61 V vs. SCE in CH3CN)29 is energetically unfavorable. Gratifyingly, we succeeded in decreasing the amount of the catalysts. When we used 0.5 mol% of Ir(ppy)3 and as little as 1 mol% of PhC(O)SH, the reaction proceeded without difficulty to give 2a in 90% yield (entry 11). In contrast, the reaction using 1 mol% of 11 resulted in lower yield and regioselectivity (31% yield, benzyl/Me = 5.2:1). Moreover, a stoichiometric amount of 1a was sufficient to produce 2a in good yield (entry 12), in contrast to most of the preceding examples in which a large excess amount of amine substrate (at least three equivalents of starting amines) was generally needed to achieve good yields. A control experiment confirmed that the reaction does not occur in the dark (see ESI†).
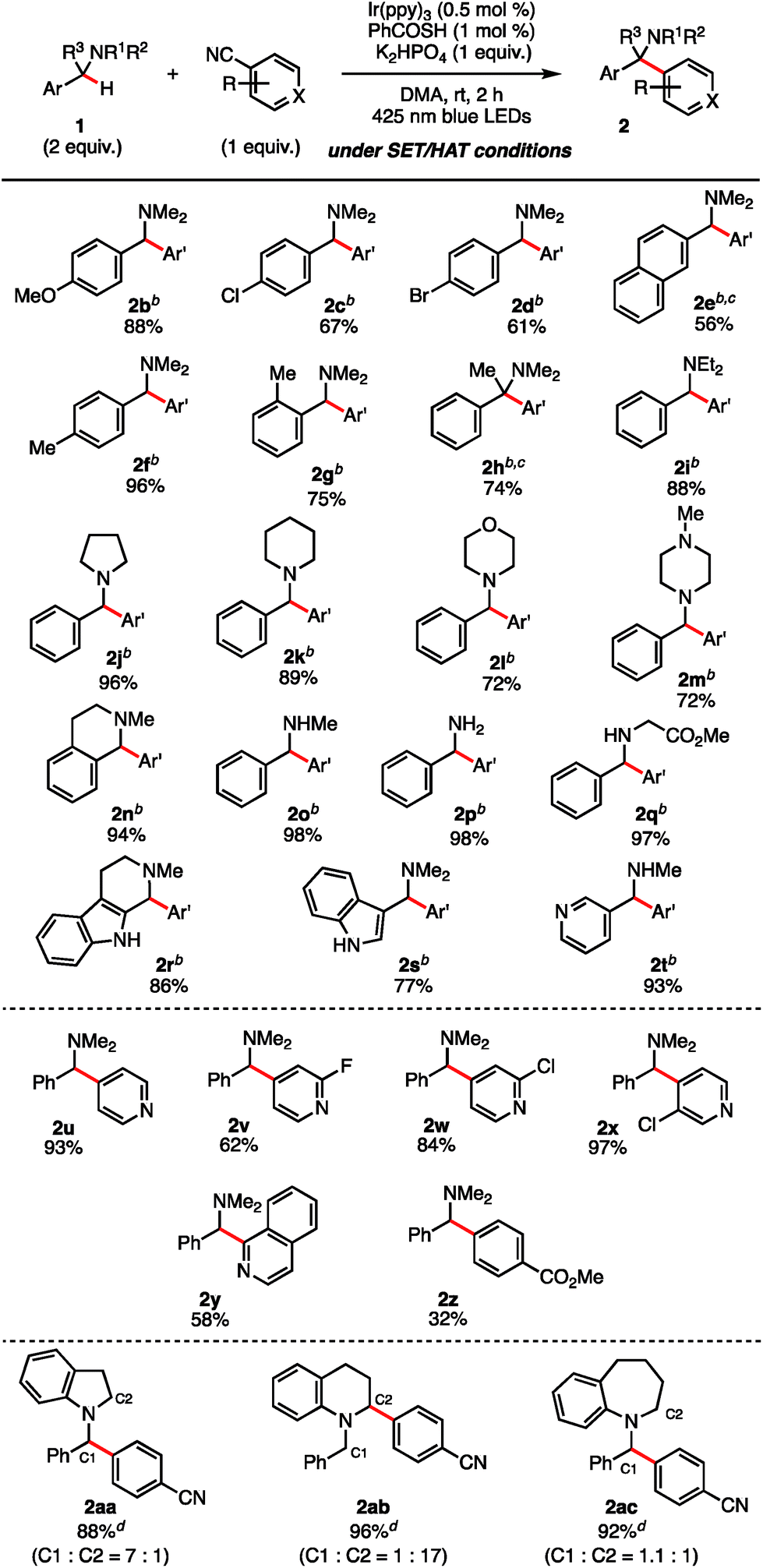
With the optimal reaction conditions identified (Table 1, entry 11), we turned our attention to the substrate scope of the SET/HAT synergistic catalysis for Csp3–H arylation of N-benzylamines (Table 2). N,N-Dimethylbenzylamine derivatives with varying electronic properties were well tolerated, and the desired products (2b–f) were obtained in good yields. Electron-rich derivatives were the highest-yielding, which is consistent with the polarity effects.31 However, para-ester-substituent retarded the reaction and the corresponding arylated product was not obtained, suggesting that strongly electron-withdrawing groups are not suitable for this reaction. Compared to the para-methyl-substituted derivative (2f), the ortho-methyl-substituted derivative gave 2g in a lower yield (75%), probably due to the steric interaction. Similarly, the reaction of the starting benzylamine was much faster than that of the dibenzylamine, so that over-reaction of the products was not observed. Nevertheless, we found that treating sterically congested 1h under the reaction conditions successfully afforded α,α,α-trisubstituted amine 2h in good yield (74%). Different substituted groups on the nitrogen atom did not prohibit the reaction, since ethyl groups (2i), aliphatic cyclic substituents (2j, 2k), and heteroatom-bearing cyclic substituents (2l, 2m) performed equally well under the reaction conditions. N-Methyl THIQ provided 2n in 94% yield as an expected regioisomer.32 Notably, secondary and primary amines having no protective group were also applicable, affording 2o and 2p in 98% and 98% yield, respectively. These are relatively difficult substrates for SET catalysis due to their more positive anodic peak potentials, and so have been less well studied in the literature. Glycine derivative 1q underwent selective N-benzyl functionalization to afford 2q in 97% yield. Moreover, heterocyclic aromatic derivatives were tolerated, providing the corresponding arylated products 2r–2t. Aside from terephthalonitrile, 4-cyanopyridine derivatives and 1-cyanoisoquinoline reacted in a regioselective manner, providing 2u–2y in good to excellent yields. When cyanobenzenes bearing different para-electron-withdrawing groups were examined, the reaction proceeded with excellent regioselectivity, though in less satisfactory yields (2z). For a comparative example, the reaction of 1j without PhC(O)SH was carried out, and a mixture of the C1-arylated 2j (36%) and the C2-arylated 3j (45%) was obtained after 12 h (Scheme 4). These results clearly indicate that the excellent N-benzyl selectivity observed in our reaction is attributed to SET/HAT synergistic catalysis that can outcompete the inherent N-methyl and/or N-methylene selectivity observed under SET catalysis conditions.
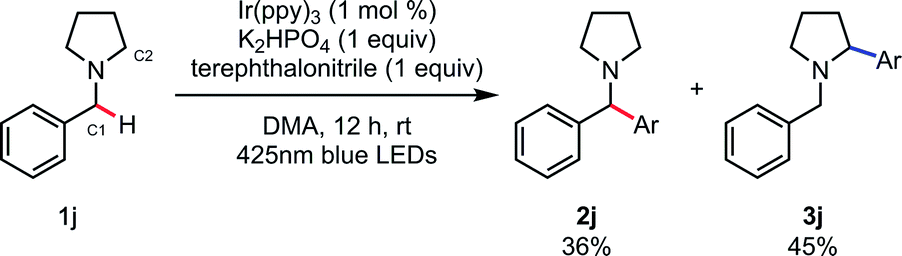 | ||
| Scheme 4 Arylation of 1j under SET conditions. | ||
An interesting phenomenon was observed when comparing cyclic N-benzylaniline-type substrates. Treatment of N-benzyl indoline (1aa) under our reaction conditions resulted mainly in N-benzyl arylation. Nonetheless, under photocatalyst-mediated SET catalysis alone, the regioselectivity was inverted completely from the N-benzyl position to the cyclic N-methylene position (90% yield, C1/C2 = 1:6.7). In contrast, N-benzyltetrahydroquinoline (1ab) reacted under our conditions to give the cyclic N-methylene-arylated product 2ab with high regioselectivity (C1/C2 = 1:17). We postulate that the increase in steric repulsion around the N-benzylic position in moving from the 5- to the 6-membered ring system may be responsible. However, on the other hand, N-benzyltetrahydrobenzoazepine (1ac) gave a 1.1:1 mixture of the C1/C2 regioisomers. The origin of the regioselectivity remains unclear at this point, but we assume that the conformational flexibility within the nitrogen atom-containing ring is important to achieve orbital overlap of the n-orbital on the nitrogen atom with the σ* orbital of the breaking C–H bond, which should have an impact on the BDE.
Next, we evaluated the chemoselectivity of our reaction in more detail. For substrate 1ad, exclusive N-benzyl functionalization occurred in the presence of the benzylic alcohol (Scheme 5A), reflecting the difference in C–H BDEs between the benzylic alcohol side (C–H BDE of benzyl alcohol = 87.5 kcal mol−1)24 and the N-benzyl group (C–H BDE of N,N-dimethylbenzylamine = 84.9 kcal mol−1).24 An intramolecular competitive experiment with 1ae targeted the N-benzylaniline side to produce 2ae in 71% yield (Scheme 5B). We next examined the reaction of a more challenging substrate (Scheme 5C). The C–H arylation reaction of 1af occurred only at the N-benzylic position, even though the BDEs of both N-benzylic C–H bond and N-allylic C–H bond are reported to be close (ca. 85 kcal mol−1 and 83 kcal mol−1, respectively).24 The yield was only moderate due to competitive reactions at the C–C double bond, but no arylation product at the N-allylic position was detected in this reaction. Moreover, we conducted an intermolecular competition experiment using 1b and 1c (1:1). In this case, the reaction mainly proceeded with 1b bearing an electron-donating group, which is indicative of a polarity matching effect (Scheme 5D).31
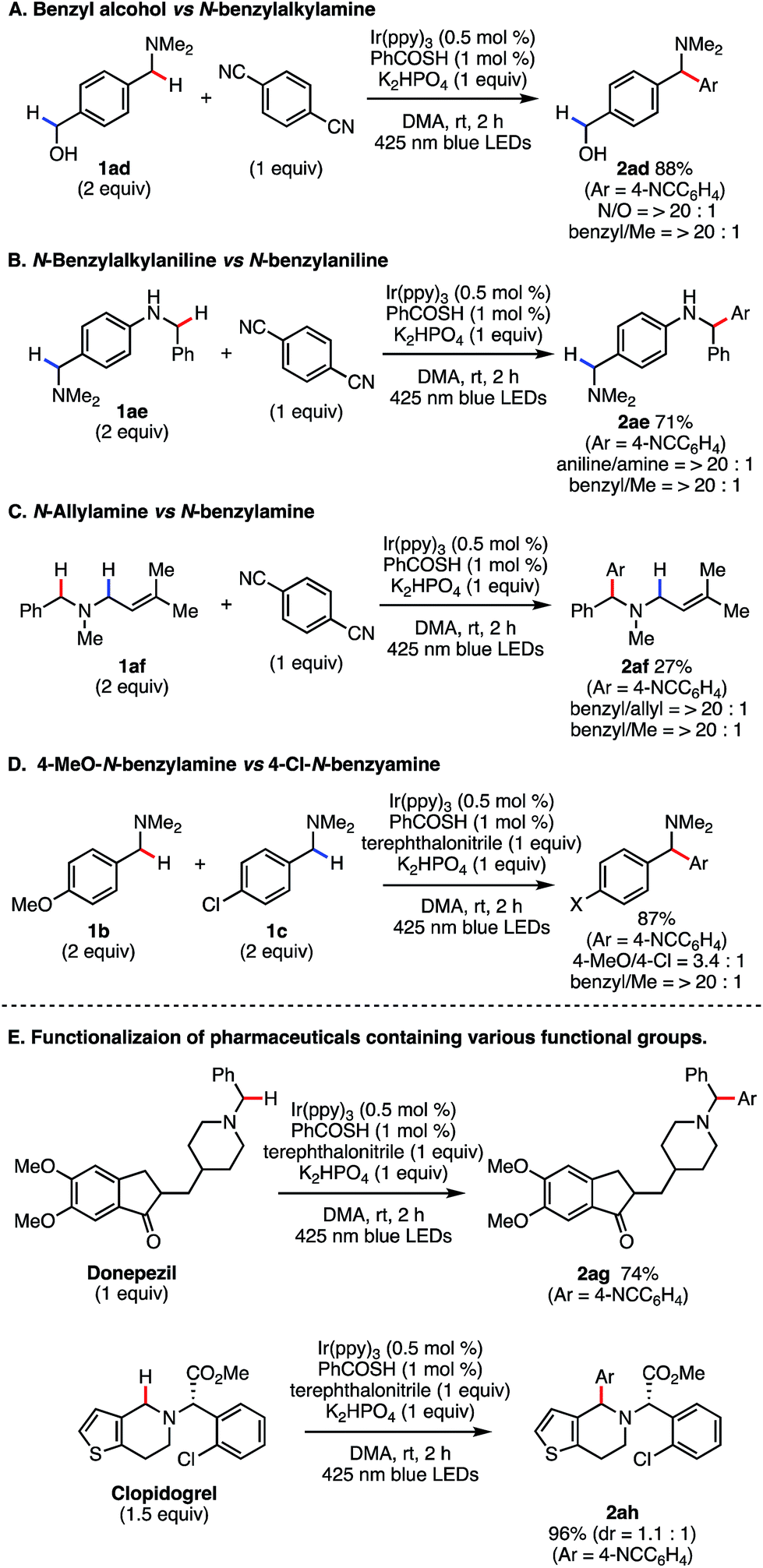 | ||
| Scheme 5 Regio- and chemoselective Csp3–H arylation. | ||
To confirm the synthetic utility of our SET/HAT synergistic catalysis, we evaluated its efficiency in late-stage functionalization. When treated in a 1:1 ratio, donepezil and 7 underwent regio- and chemoselective coupling reaction smoothly to give 1,1-diarylmethylamine 2ag in good yield (74%). Further, even with more complex clopidogrel, C–H arylation at the endocyclic N-benzylic position proceeded regioselectively, furnishing 2ah in 96% yield. These examples clearly demonstrate that the present reaction is applicable to late-stage derivatization of structurally complex bioactive compounds, and thus should be useful to facilitate structure–activity-relationship studies.
All reactions were conducted on 1 mmol scale and isolated yields are shown. Chemo- and regioselectivity were determined by 1H NMR analysis.
Finally, on the basis of the observed selectivity and CV measurements, we propose the catalytic cycle shown in Scheme 6. fac-Tris(2-phenylpyridinato)iridium(III) [IrIII(ppy)3] (15) is excited by photo-irradiation (blue LED, 425 nm) and [IrIII(ppy)3]* (18) is generated. This reductant [Ir(ppy)3]* (18) (E1/2 (IrIV/*IrIII) = −1.73 V vs. SCE in CH3CN)33 undergoes SET to electron-deficient arene 20 to afford radical anion intermediate 21 along with [IrIV(ppy)3]+ (19). Subsequently, [IrIV(ppy)3]+ (19) (E1/2 (IrIV/IrIII) = +0.77 V vs. SCE in CH3CN)33 performs SET oxidation of the electron-rich PhC(O)SK (Epox (6/5) = +0.65 V vs. SCE in CH3CN, see ESI†), which outcompetes SET oxidation of the amine substrate leading to the different reaction pathway. The generated sulfur-centered radical 6 regioselectively undergoes HAT regioselectively depending on C–H BDEs. As seen in the previous reports, the resulting N-benzyl radical 22 reacts with the separately formed radical anion intermediate 21 to yield the desired 1,1-diarylmethylamine 2.34
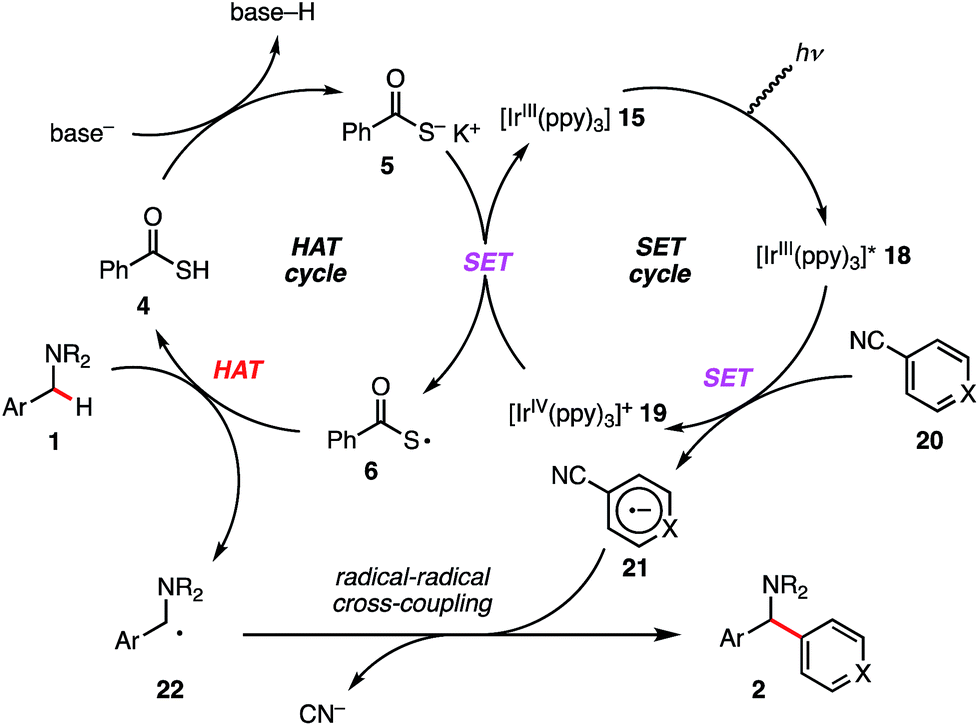 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed a highly regio- and chemoselective Csp3–H arylation of a variety of benzylamine derivatives by employing SET/HAT synergistic catalysis. Under SET catalysis alone, Csp3–H arylation of benzylamines proceeded at the N-methyl and/or cyclic N-methylene positions through an aminium radical cation intermediate. In contrast, synergistic SET and HAT catalysis inverts the regioselectivity. The reaction was completed within 2 h in the presence of as little as 0.5 mol% of the Ir complex and 1 mol% of PhC(O)SH as a HAT catalyst, and various 1,1-diarylmethylamines were obtained in good to excellent yields (56–98%). Importantly, high yields were achieved even when a stoichiometric amount of the benzylamine substrate was employed. From a mechanistic viewpoint, the use of PhC(O)SH as a HAT catalyst precursor is the key to success: SET oxidation of the 3° amine substrates was suppressed effectively due to the favorable oxidation of PhC(O)SK, and the resulting sulfur radical abstracts C–H bonds selectively to give N-benzyl radicals. The excellent regio- and chemoselectivity enables the late-stage N-benzyl arylation of pharmaceuticals with reactive functional groups. Further studies including N-methyl selective functionalization are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Basis for Supporting Innovative Drug Discovering and Life Science Research (BINDS) from AMED, The Research Foundation for Pharmaceutical Sciences, and The FUGAKU Trust for Medicinal Research. T. I. and J. P. B. are grateful for financial support from JSPS.Notes and references
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef PubMed; (c) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef PubMed; (d) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (e) T. Newhuse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef PubMed; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (g) M. C. White, Science, 2012, 335, 807–809 CrossRef PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef PubMed; (i) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef PubMed; (j) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–574 RSC; (k) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef PubMed.
- D. Ameen and T. J. Snape, Med. Chem. Commun., 2013, 4, 893–907 RSC.
- (a) H. Böhme and P. Plappert, Chem. Ber., 1975, 108, 2827–2833 CrossRef; (b) T. Murai and F. Asai, J. Am. Chem. Soc., 2007, 129, 780–781 CrossRef PubMed; (c) S. He, J. Xiao, A. E. Dulcey, B. Lin, A. Rolt, Z. Hu, X. Hu, A. Q. Wang, X. Xu, N. Southall, M. Ferrer, W. Zheng, T. J. Liang and J. J. Marugan, J. Med. Chem., 2016, 59, 841–853 CrossRef PubMed; (d) L.-G. Xie and D. J. Dixon, Chem. Sci., 2017, 8, 7492–7497 RSC; during the preparation of this manuscript, Doyle and co-workers reported an elegant Ni-catalyzed three-component coupling reaction, see: (e) C. Heinz, J. P. Lutz, E. M. Simmons, M. M. Miller, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 2292–2300 CrossRef PubMed.
- For selected examples of N-benzylic C–H arylation via addition of aryl nucleophiles to iminium ions, see: (a) Z. Li and C.-J. Li, J. Am. Chem. Soc., 2005, 127, 6968–6969 CrossRef PubMed; (b) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928–8933 CrossRef PubMed; (c) W. Muramatsu, K. Nakano and C.-J. Li, Org. Lett., 2013, 15, 3650–3653 CrossRef PubMed.
- For selected examples of oxidative N-benzylic C–H functionalization of N-aryl tetrahydroisoquinolines under photoredox catalysis, see: (a) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef PubMed; (b) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94–97 CrossRef PubMed; (c) G. Zhao, C. Yang, L. Guo, H. Sun, C. Chen and W. Xia, Chem. Commun., 2012, 48, 2337–2339 RSC; (d) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem.–Eur. J., 2012, 18, 5170–5174 CrossRef PubMed; (e) J. P. Barham, M. P. John and J. A. Murphy, Beilstein J. Org. Chem., 2014, 10, 2981–2988 CrossRef PubMed; (f) J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280–8283 RSC; (g) H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860–11871 CrossRef PubMed.
- For selected examples of Pd-catalyzed cross-coupling reactions using deprotonation at the benzylic position, see: (a) T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 4689–4691 CrossRef PubMed; (b) T. Niwa, T. Suehiro, H. Yorimitsu and K. Oshima, Tetrahedron, 2009, 65, 5125–5131 CrossRef; (c) G. I. McGrew, J. Temaismithi, P. J. Caroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541–5544 CrossRef PubMed; (d) G. I. McGrew, C. Stanciu, J. Zhang, P. J. Caroll, S. D. Dreher and P. J. Walsh, Angew. Chem., Int. Ed., 2012, 51, 11510–11513 CrossRef PubMed; (e) N. Hussain, B.-S. Kim and P. J. Walsh, Chem.–Eur. J., 2015, 21, 11010–11013 CrossRef PubMed; (f) B.-S. Kim, J. Jiménez, F. Gao and P. J. Walsh, Org. Lett., 2015, 17, 5788–5791 CrossRef PubMed; there are also reported deprotonative cross-coupling reactions under Ni catalysis, see: (g) J. A. Fernández-Salas, E. Marelli and S. P. Nolan, Chem. Sci., 2015, 6, 4973–4977 RSC; (h) X. Cao, S.-C. Sha, M. Li, B.-S. Kim, C. Morgan, R. Huang, X. Yang and P. J. Walsh, Chem. Sci., 2016, 7, 611–618 RSC; (i) G. Gao, Y. Fu, M. Li, B. Wang, B. Zheng, S. Hou and P. J. Walsh, Adv. Synth. Catal., 2017, 359, 2890–2894 CrossRef PubMed.
- (a) N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Org. Lett., 2012, 14, 1930–1933 CrossRef PubMed; (b) N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Org. Lett., 2012, 14, 3792–3795 CrossRef PubMed; (c) N. Dastbaravardeh, K. Kirchner, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2013, 78, 658–672 CrossRef PubMed; (d) N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Eur. J. Org. Chem., 2013, 2878–2890 CrossRef PubMed; (e) N. Y. P. Kumer, R. Jeyachandran and L. Ackermann, J. Org. Chem., 2013, 78, 4145–4152 CrossRef PubMed.
- For seminal reports on PET and its applications, see: (a) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233–240 CrossRef; (b) J. Santamaria, Pure Appl. Chem., 1995, 67, 141–147 Search PubMed; (c) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef PubMed; (d) N. Hoffman, Chem. Rev., 2008, 108, 1052–1103 CrossRef PubMed; (e) N. Hoffman, Synthesis, 2016, 48, 1782–1802 CrossRef; (f) C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef.
- For recent excellent reviews on photoredox catalysis, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed; (c) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed; (d) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (e) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef PubMed; (f) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef PubMed; (g) K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef PubMed . In this report, a pyrrolidine derivative was examined as a single example of an aliphatic amine substrate.
- (a) T. Ide, K. Shimizu, Y. Kawato, H. Egami and Y. Hamashima, Heterocycles, 2017, 95, 738–747 CrossRef; (b) T. Ide, K. Shimizu, H. Egami and Y. Hamashima, Tetrahedron Lett., 2018, 59, 3258–3261 CrossRef.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef PubMed; (b) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef PubMed; (c) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef PubMed; (d) J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef PubMed.
- Arylations of alkylamines, see: (a) A. Singh, A. Arora and J. D. Weaver, Org. Lett., 2013, 15, 5390–5393 CrossRef PubMed; (b) A. Lipp, G. Lahm and T. Opatz, J. Org. Chem., 2016, 81, 4890–4897 CrossRef PubMed.
- (a) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef PubMed; (b) L. R. Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107–4114 CrossRef PubMed; (c) C. Yan, L. Li, Y. Liu and Q. Wang, Org. Lett., 2016, 18, 4686–4689 CrossRef PubMed.
- C–H carboxylation of N-benzylamines under photo-irradiation was reported recently. See: H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef PubMed.
- The N-methyl selectivity has been observed in a few photoredox-catalyzed reactions, see: (a) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC; (b) J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef PubMed; (c) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC; (d) J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef PubMed; for reaction with N-methylmorpholine, see: (e) J. J. Douglas, K. P. Cole and C. R. J. Stephenson, J. Org. Chem., 2014, 79, 11631–11643 CrossRef PubMed; for photooxidative N-demethylation of tertiary amines, see: (f) J. Santamaria, R. Ouchabane and J. Rigaudy, Tetrahedron Lett., 1989, 30, 3977–3980 CrossRef.
- A similar interpretation is given in the ESI† of ref. 16a. For further relevant studies on the kinetic acidity in terms of stereoelectronic and steric factors, see: (a) F. D. Lewis, Acc. Chem. Res., 1986, 19, 401–405 CrossRef; (b) F. D. Lewis, T.-I. Ho and J. T. Simpson, J. Am. Chem. Soc., 1982, 104, 1924–1929 CrossRef; (c) W. Xu and P. S. Mariano, J. Am. Chem. Soc., 1991, 113, 1431–1432 CrossRef; (d) X. Zhang, S.-R. Yeh, S. Hong, M. Freccero, A. Albini, D. E. Falvey and P. S. Mariano, J. Am. Chem. Soc., 1994, 116, 4211–4220 CrossRef; (e) G. W. Dombrowski, J. P. Dinnocenzo, S. Farid, J. L. Goodman and I. R. Gould, J. Org. Chem., 1999, 64, 427–431 CrossRef.
- The detail of the N-methyl selective arylation under SET catalysis conditions will be reported separately in due course.
- S. Escoubet, S. Gastaldi, N. Vanthuyne, G. Gil, D. Siri and M. P. Bertrand, Eur. J. Org. Chem., 2006, 3242–3250 CrossRef.
- For recent selected examples of thiyl radical HAT catalysis, see: (a) K. Ovortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef PubMed; (b) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef PubMed; (c) W. Li, Y. Duan, M. Zhang, J. Cheng and C. Zhu, Chem. Commun., 2016, 52, 7596–7599 RSC; (d) S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204–2207 CrossRef PubMed; see also ref. 14c. For a general review on thiyl radicals, see: (e) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- L. G. Shaidarova, S. A. Ziganshina and G. K. Budnikov, J. Anal. Chem., 2003, 58, 577–582 CrossRef.
- W. P. Jencks and K. Salvesen, J. Am. Chem. Soc., 1971, 93, 4433–4436 CrossRef.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press LLC, Boca Raton, FL, 2003 Search PubMed.
- (a) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef PubMed; (b) J. B. Barham, M. P. John and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 15482–15487 CrossRef PubMed.
- N. Kitamura, R. Obata, H.-B. Kim and S. Tazuke, J. Phys. Chem., 1987, 91, 2033–2035 CrossRef.
- W.-Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778–4783 CrossRef PubMed.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef PubMed.
- Y. Mori, Y. Sakaguchi and H. Hayashi, J. Phys. Chem. A, 2000, 104, 4896–4905 CrossRef.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef.
- (a) L. Feray, N. Kuznetsov and P. Renaud, in Radicals in Organic Synthesis, ed. P. Renaund and M. P. Sibi, WILEY-VCH: Verlag, 2001, pp. 246–278 Search PubMed; (b) J. M. Tedder, Angew. Chem., Int. Ed., 1982, 21, 401–410 CrossRef; (c) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC; (d) V. Paul and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1987, 1322–1324 RSC; (e) P. Kaushal, P. L. H. Mok and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1990, 1663–1670 RSC; (f) K. D. Beare and M. L. Coote, J. Phys. Chem. A, 2004, 108, 7211–7221 CrossRef; (g) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef PubMed; (h) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef PubMed.
- The product 2n was not obtained without PhC(O)SH in our reaction, suggesting that HAT process is necessary to functionalize the benzylic position of N-Me THIQ.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef.
- (a) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef PubMed; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef PubMed; (c) A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, CV data, characterization data of products. See DOI: 10.1039/c8sc02965b |
| This journal is © The Royal Society of Chemistry 2018 |