
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective radical coupling of activated ketones with N-aryl glycines†
Yang
Liu‡
a,
Xiangyuan
Liu‡
a,
Jiangtao
Li
a,
Xiaowei
Zhao
a,
Baokun
Qiao
a and
Zhiyong
Jiang
 *ab
*ab
aKey Laboratory of Natural Medicine and Immuno-Engineering of Henan Province, Henan University, Kaifeng, Henan 475004, P. R. China. E-mail: chmjzy@henu.edu.cn
bHenan Key Laboratory of Organic Functional Molecule and Drug Innovation, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
First published on 27th August 2018
Abstract
Asymmetric H-bonding catalysis as a viable strategy for enantioselective radical coupling of ketones is demonstrated. With a visible-light-mediated dual catalytic system involving a dicyanopyrazine-derived chromophore (DPZ) photosensitizer and a chiral phosphoric acid (CPA), N-aryl glycines with a variety of 1,2-diketones and isatins underwent a redox-neutral radical coupling process and furnished two series of valuable chiral 1,2-amino tertiary alcohols in high yields with good to excellent enantioselectivities (up to 97% ee). In this catalysis platform, the formation of neutral radical intermediates between ketyl and H-bonding catalyst CPA is responsible for presenting stereocontrolling factors. Its success in this work should provide inspiration for expansion to other readily accessible ketones to react with various radical species, thus leading to a productive approach to access chiral tertiary alcohol derivatives.
Introduction
The radical coupling reaction,1 wherein the nearly zero activation energy2 of connecting two distinct odd-electron partners enables a strong tendency to generate a new chemical bond, often furnishes an efficient and direct synthetic pathway to molecules with high functional group tolerance. Therefore, the development of compatible strategies to produce two types of radical species and facilitate the coupling reaction has attracted much attention from chemists over the last few decades.1–3 In 2011, the MacMillan group made a significant breakthrough4 in which such a transformation was realized via visible-light-driven photoredox catalysis.5 This convenient and sustainable platform promptly inspired a number of elegant efforts focusing on radical coupling.6 However, the enantioselective manifold still remains underdeveloped given that few examples have been established.7 This lack of development is mainly due to the high reactivity that results in an elusive precise absolute stereocontrol of the reaction. In 2015, Ooi and co-workers7a reported the first example with excellent enantioselectivity, wherein asymmetric α-coupling of N-arylaminomethanes with aldimines through cooperative catalysis7–10 of an Ir-centered photosensitizer and an ionic Brønsted acid led to chiral 1,2-diamine derivatives featuring a tertiary carbon stereocenter. Almost at the same time, the Meggers group described an asymmetric radical coupling of tertiary amines with 2-trifluoroacetyl imidazoles catalyzed by a chiral iridium complex.7b They also developed a cooperative Ru-centered photosensitizer and Rh-based chiral Lewis acid catalyst for the cross-coupling of 2-acyl imidazoles with α-silylamines.7d In their elegant work, α-aminoalkyl moieties were successfully introduced onto the more challenging quaternary carbon stereogenic center. Inarguably, the judicious use of imidazole as the substituent of ketones is crucial for the enantioselectivity, as its N atom was shown to enhance the ability of stereocontrol by interacting with the chiral Lewis acid. Although the groups of Xiao,6f Shah6g and Yang6h have demonstrated the viability of radical coupling of various readily accessible ketones with different substrates in a racemic manner, an enantioselective variety still constitutes a formidable challenge.In the putative reaction mechanism of the radical-coupling transformations, ketones always experience a single-electron reduction to generate the corresponding ketyl variants.6,7 In 2013, the Knowles group9f revealed the strong basicity of ketyls and their capability of forming neutral ketyl radicals with a chiral Brønsted acid which could provide efficient enantioselective control for the coupling reaction with hydrazones. This prominent H-bonding catalytic strategy recently inspired us to accomplish a highly enantioselective photoreduction of 1,2-diketones.10c Accordingly, we speculated that such an approach would offer the possibility to address the desired radical coupling of undecorated ketones. If so, it should be a promising and general strategy as it would allow diverse radical species to connect with ketones, thus opening a fruitful avenue for the synthesis of the important chiral tertiary alcohols. Here, we report the development of a redox-neutral, enantioselective, radical coupling of N-aryl glycines with activated ketones, including acyclic 1,2-diketones and cyclic isatins (Scheme 1). The association of a dicyanopyrazine-derived chromophore (DPZ) as the photoredox catalyst and a 1,1′-spirobiindane-7,7′-diol (SPINOL)-based chiral phosphoric acid (CPA) as the H-bonding catalyst was demonstrated as being a workable catalytic system. Two series of chiral 1,2-amino tertiary alcohols that are significant structural scaffolds in synthetic and medicinal chemistry11 were prepared in high yields with good to excellent enantioselectivities.
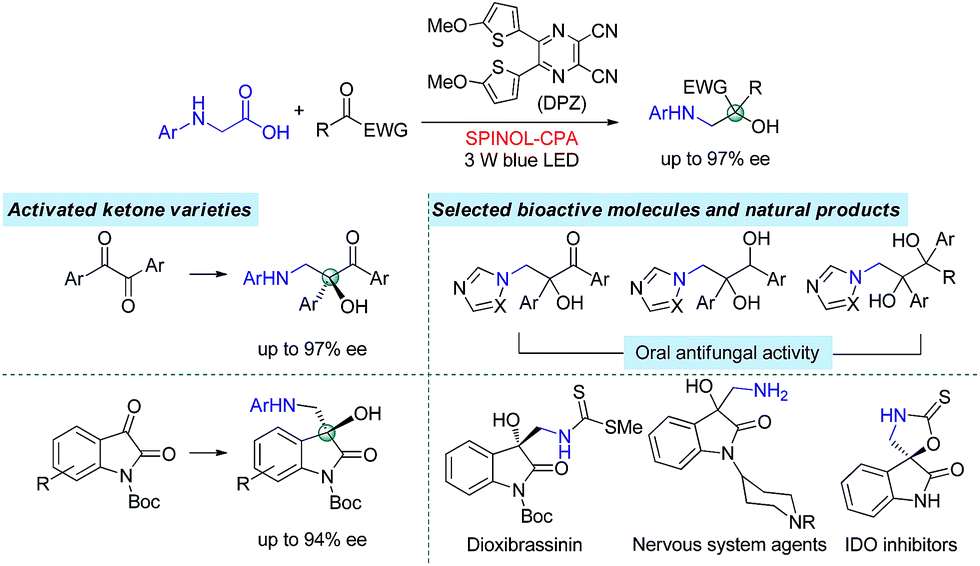 | ||
| Scheme 1 Outline of this work. | ||
Results and discussion
At the onset of our study, we sought to explore the coupling of ketyls derived from 1,2-diketones with α-amino radicals to provide the valuable enantioenriched 1,2-amino tertiary alcohols,11a for which no catalytic asymmetric synthesis has yet been developed. Our developed metal-free DPZ7g,10 was selected as the photosensitizer since 1,2-diketones (e.g. benzil, Ered1/2 = −1.169 V and −1.251 V vs. a saturated calomel electrode (SCE) in CH3CN)10c could be directly reduced by DPZ˙− (Ered1/2 = −1.45 V vs. SCE in CH2Cl2) when the transformation experiences a reductive quenching cycle (Scheme 2). Furthermore, we intend to use N-aryl glycines (e.g. N-phenyl glycine, carboxylate ion, Ep = +0.52 [for the N moiety] and +1.09 V [for the COO− moiety] vs. Ag/AgCl in CH3CN) as the precursors of α-amino radicals given the prominent ability to generate the radical species12via single-electron oxidative decarboxylation by the visible-light-activated DPZ, i.e. *DPZ (Et(S*/S˙−) = +0.91 V vs. in CH2Cl2).7g,10d,e More importantly, the formed α-amino radicals contain a H-bonding donor (N–H) which will be more nucleophilic via a possible interaction with the H-bonding acceptor (X = N, O, etc.) of a chiral bifunctional catalyst, thus facilitating coupling with the electrophilic α-ketone radicals.9s,13 Of note, the use of tertiary amines was susceptible to reduce 1,2-diketones to α-hydroxy ketones under photoredox catalysis.10c,14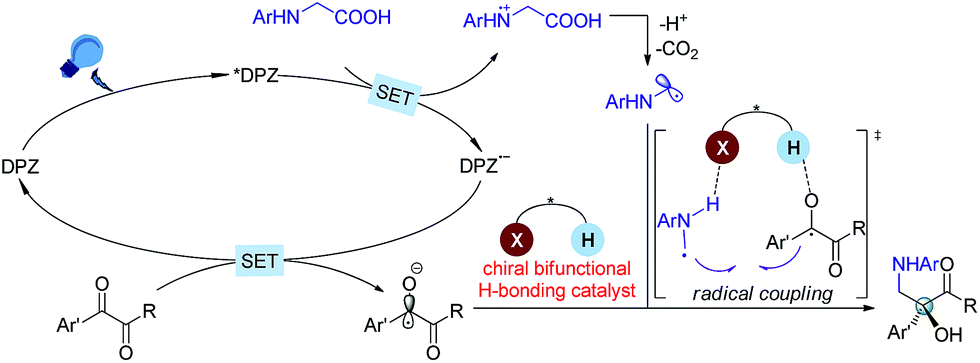 | ||
| Scheme 2 Design plan based on mechanistic considerations. | ||
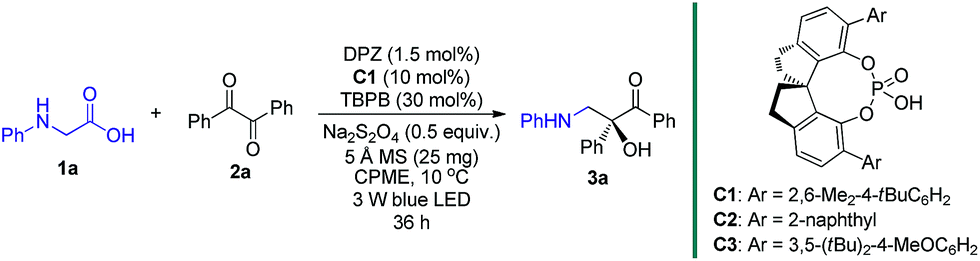
To this end, we began the investigation with N-phenyl glycine 1a and benzil 2a in the presence of DPZ as the photoredox catalyst (Table 1).10c–e The initial investigation showed that the transformation furnished racemic product 3a in 62% yield after 12 h when only using 0.5 mol% DPZ at 25 °C, indicating the feasibility of the reaction and the high reactivity (entry 1). Upon examining a range of chiral H-bonding catalysts and the reaction parameters (see Table S1 in the ESI†), we observed that the reaction conducted in CPME at 10 °C for 36 h in the presence of 1.5 mol% DPZ and 10 mol% chiral SPINOL-CPA15C1 and with the combination of 30 mol% TBPB, 0.5 equiv. Na2S2O4 and 25 mg 5 Å MS as additives affords the desired chiral product 3a in 78% yield with 93% ee (entry 2). Other SPINOL-CPAs (e.g., C2 and C3) with distinct substituents at the 6,6′-positions presented 3a with lower ee values (entries 3–4). [Ru(bpy)3]Cl2 and Rose Bengal as plausible photoredox catalysts were evaluated (entries 5–6), but both the yield and enantioselectivity were decreased. Each of the three additives, which could regulate the strength of H-bonding interaction by exploiting the salt effect (i.e. TBPB10d and Na2S2O4) or diminishing the moisture of the reaction system (i.e. molecular sieves7g), was found to slightly affect the enantioselectivity to the same degree (entries 7–9). The results reveal that all of the additives jointly exerted a positive influence on the stereocontrol of C1. The control experiments confirmed that DPZ, visible light, and an oxygen-free environment are indispensable for the transformation to occur (entries 10–12). Note that the reaction performed under the standard conditions but in the absence of catalyst C1 still produced rac-3a in 53% yield (entry 13), suggesting the existence of a considerable competitive achiral background reaction.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.075 mmol), 2a (0.05 mmol), degassed CPME (1.0 mL), 10 °C, irradiation with blue LED (3 W, 450 nm), 36 h. b Yield of the isolated product. c Determined by HPLC analysis on a chiral stationary phase. d No reaction was detected. e 1a was consumed, but no 3a was obtained. TBPB = tetra-n-butylphosphonium bromide. CPME = cyclopentyl methyl ether. N.A. = not available. | |||
| 1 | 0.5 mol% DPZ in CH2Cl2 at 25 °C and without C1 and any additives, 12 h | 62 | N.A. |
| 2 | None | 78 | 93 |
| 3 | C2 instead of C1 | 74 | 76 |
| 4 | C3 instead of C1 | 73 | 67 |
| 5 | Ru(bpy)3Cl2·6H2O instead of DPZ | 58 | 89 |
| 6 | Rose Bengal instead of DPZ | 69 | 91 |
| 7 | No TBPB | 76 | 89 |
| 8 | No Na2S2O4 | 75 | 90 |
| 9 | No 5 Å MS | 76 | 90 |
| 10 | No DPZ | 35 | 88 |
| 11 | No light | 0d | N.A. |
| 12 | Under air | 0e | N.A. |
| 13 | No C1 | 53 | N.A. |
With the optimal reaction conditions in hand, the scope of this H-bonding catalysis-enabled asymmetric radical coupling strategy was examined (Scheme 3). The reactions of 2a with a variety of N-aryl glycines 1 furnished adducts 3a–g in 78 to 89% yields with 90 to 93% ees within 36 h. Electron-deficient or electron-donating substituents at the para- and meta-positions of the aryl ring presented similar reactivities and enantioselectivities. An attempt at performing the reaction of 1a with 2a in a 1.0 mmol scale presented a similar reactivity and enantioselectivity as 3a, with 87% yield with 93% ee achieved after 48 h (footnote a). With respect to symmetric 1,2-diketones, the reaction tolerated a wide range of aryl substituents regardless of their electronic properties and substitution patterns, and corresponding products 3h–q were obtained in 61 to 89% yields with 84 to 97% ees. Based on the persistent radical effect,13 the slightly lower enantioselectivity for 1,2-diketones (3h–j) with electron-withdrawing substituents is likely due to the stronger racemic background reaction, as the higher stability of these ketyl intermediates would facilitate a coupling with the unstable α-amino radical. For 3-methyl-1-phenyl-1,2-butanedione as a representative of unsymmetrical 1,2-diketones, product 3r was obtained in only 34% ee. However, the modified reaction conditions, that are C2 as the chiral catalyst and 4 Å MS as an additive in CPME at −5 °C, could furnish 3r in 78% ee. The stereochemistry of these adducts was assigned based on the structure of 3q, as solved by single crystal X-ray diffraction.16
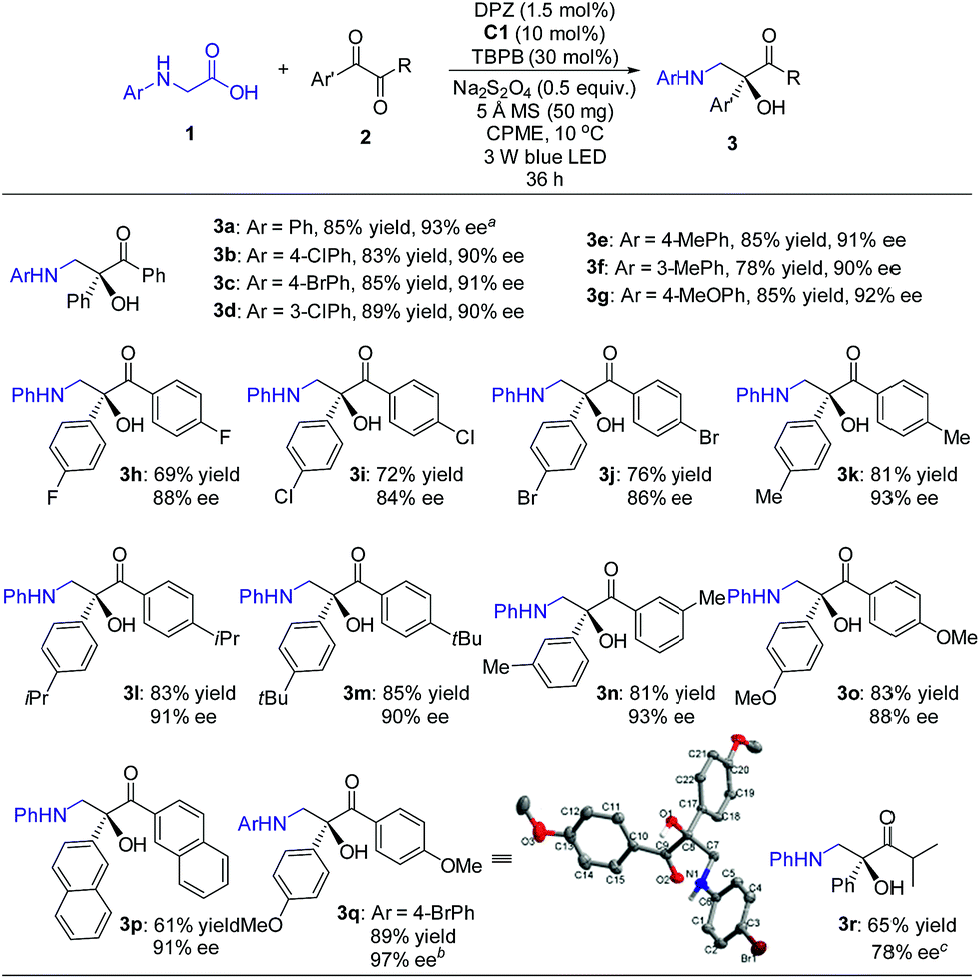 | ||
| Scheme 3 Reactions of N-aryl glycines with 1,2-diketones. Reaction conditions: 1 (0.15 mmol), 2 (0.1 mmol), DPZ (1.5 mol%), C1 (10 mol%), TBPB (30 mol%), Na2S2O4 (0.5 equiv.), 5 Å MS (50 mg), degassed CPME (2.0 mL), 10 °C, irradiation with a blue LED (3 W, 450 nm), 36 h. The yield amount was isolated by flash column chromatography on a silica gel. ee was determined by HPLC analysis on a chiral stationary phase. aOn a 1.0 mmol scale, 48 h, yield of 3a = 87%, ee of 3a = 93%. bThe ee value was obtained after a single recrystallization. Initial data: 85% ee. cReaction conditions: 1 (0.15 mmol), 2 (0.1 mmol), DPZ (1.5 mol%), C2 (10 mol%), 4 Å MS (50 mg), degassed CPME (2.0 mL), −5 °C, 36 h. Under the previous reaction conditions, ee = 34%. | ||
The promising results inspired us to further evaluate this catalytic strategy for isatins, a representative cyclic ketone, to first construct the important chiral 3-hydroxy-3-aminoalkylindolin-2-one derivatives in a direct manner.11b–g As depicted in Scheme 4, under the same catalysis platform but with modified reaction conditions (1.0 mol% DPZ, 20 mol% SPINOL-CPA C3 in THF at 10 °C), the transformations of N-aryl glycine 1h with N-Boc-substituted isatins 4 were complete within 36 h, providing the desired adducts 5a–l with diverse substituents on the aromatic ring of the isatins in 73 to 91% yields with 85 to 94% ees. It was observed that the substituent group at the 4-position (e.g., 5b–c) presented a slightly decreased enantioselectivity, probably owing to the steric hindrance.
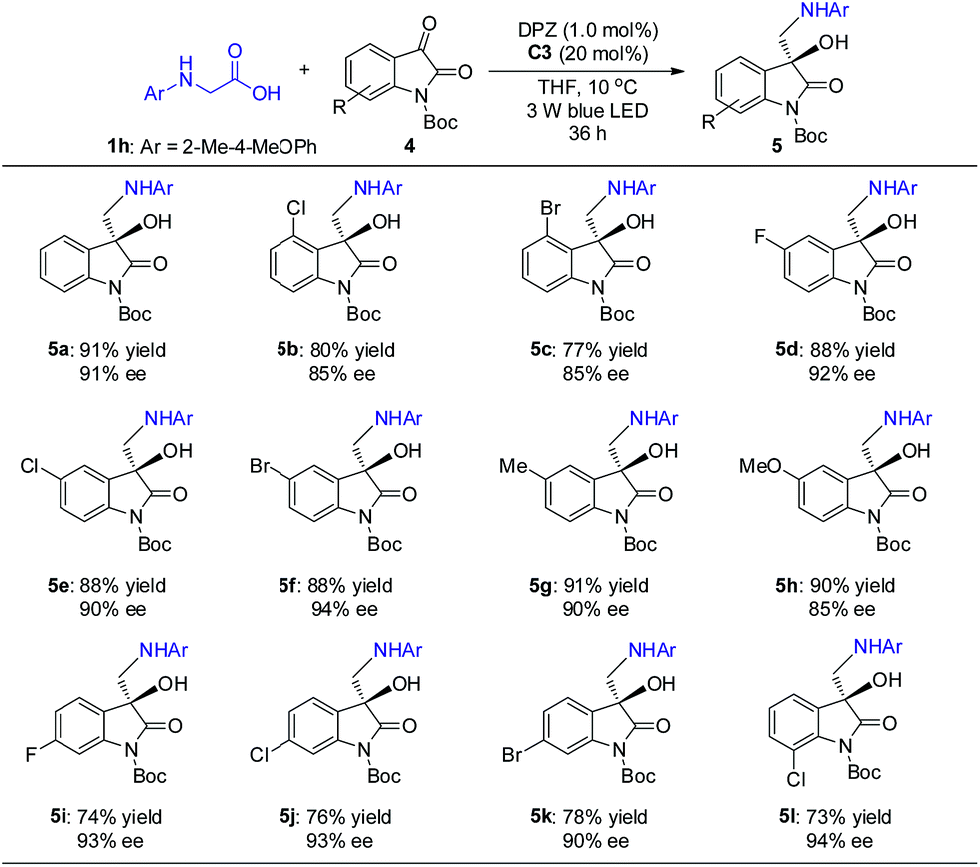 | ||
| Scheme 4 Reactions of N-aryl glycine with isatins. +Reaction conditions: 1h (0.15 mmol), 4 (0.1 mmol), DPZ (1.0 mol%), C1 (20 mol%), degassed THF (2.0 mL), 10 °C, irradiation with a blue LED (3 W, 450 nm), 36 h. The yield amount was isolated by flash column chromatography on silica gel. ee was determined by HPLC analysis on a chiral stationary phase. | ||
The two series of enantiomerically enriched 1,2-amino-alcohols featuring oxo-hetero-quaternary carbon stereocenters are direct precursors to many bioactive natural and non-natural compounds (Scheme 1). For example, the treatment of adduct 3g derived from 1,2-diketone using TCCA readily cleaved the PMP N-protective group (Scheme 5). The resultant amine 6 was then transformed into chiral 1,2-imidazolyl tertiary alcohol 7 that possesses oral antifungal activity11a in 65% yield over two steps with 92% ee. The transformation of adducts from isatins was also carried out. The Boc-protected product 8 was rapidly obtained in 98% yield by treating 5a with (Boc)2O and DMAP. The replacement of 2-methyl-4-methoxyphenyl with Ts as the N-protective group was performed through a sequential process involving the use of TCCA for the deprotection and TsCl for the protection, furnishing product 9 in 72% yield and without diminishing the ee. The results clearly indicate that chiral products 5 are excellent synthetic intermediates for conveniently synthesizing the biologically important 3-hydroxy-3-aminoalkylindolin-2-one variants as shown in Scheme 1.11b–g
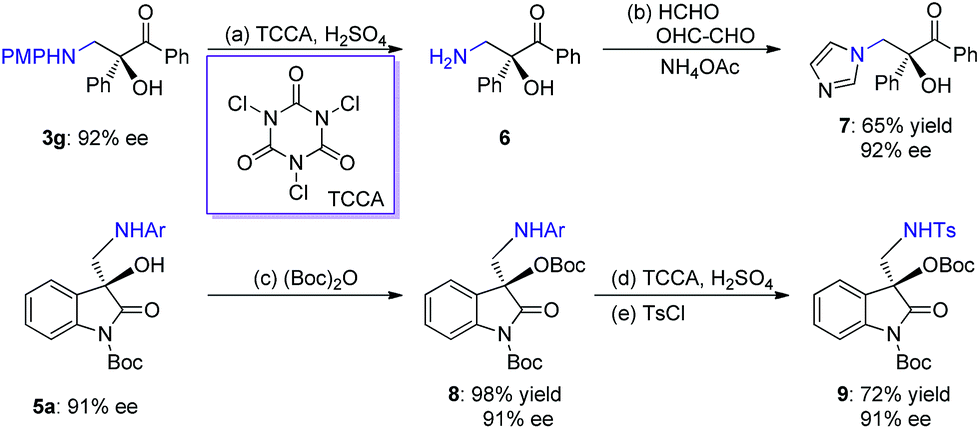 | ||
Scheme 5 Synthetic applications. (a) TCCA (0.5 equiv.), H2SO4 (1 M, aq., 2.0 equiv.), CH3CN/H2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 16 h. (b) HCHO (2.0 equiv.), OHC–CHO (2.0 equiv.), NH4OAc (2.0 equiv.), MeOH, 80 °C, 5 h, 65% yield, 92% ee. (c) (Boc)2O (1.1 equiv.), DMAP (0.2 equiv.), DCM, 0 °C, 0.5 h, 98% yield, 91% ee. (d) TCCA (0.5 equiv.), H2SO4 (1 M, aq., 2.0 equiv.), CH3CN/H2O = 1:1, 16 h. (e) TsCl (2.0 equiv.), Et3N (2.0 equiv.), EtOAc, 0 °C to r.t., 5 h, 72% yield in two steps, 91% ee. PMP = para-methoxyphenyl; TCCA = N,N′,N′′-trichloroisocyanuric acid; (Boc)2O = di-tert-butyl dicarbonate; Boc = tert-butyl carbonate; DMAP = 4-dimethylaminopyridine; TsCl = p-toluenesulfonyl chloride; Ts = tosyl. :1, 16 h. (b) HCHO (2.0 equiv.), OHC–CHO (2.0 equiv.), NH4OAc (2.0 equiv.), MeOH, 80 °C, 5 h, 65% yield, 92% ee. (c) (Boc)2O (1.1 equiv.), DMAP (0.2 equiv.), DCM, 0 °C, 0.5 h, 98% yield, 91% ee. (d) TCCA (0.5 equiv.), H2SO4 (1 M, aq., 2.0 equiv.), CH3CN/H2O = 1:1, 16 h. (e) TsCl (2.0 equiv.), Et3N (2.0 equiv.), EtOAc, 0 °C to r.t., 5 h, 72% yield in two steps, 91% ee. PMP = para-methoxyphenyl; TCCA = N,N′,N′′-trichloroisocyanuric acid; (Boc)2O = di-tert-butyl dicarbonate; Boc = tert-butyl carbonate; DMAP = 4-dimethylaminopyridine; TsCl = p-toluenesulfonyl chloride; Ts = tosyl. | ||
Based on the previous examples10,12 and the product structure, the reaction between N-aryl glycines and ketones that underwent a single electron transfer (SET) redox radical coupling process to form the products is reasonable. The Stern–Volmer experiments17 confirmed that the catalytic cycle was triggered from the reductive quenching of *DPZ (Scheme 2). To better understand the role of the chiral CPA in asymmetric induction, the transformation of N-phenyl-N-methyl glycine 10 with benzil 2a under the standard reaction conditions as shown in Table 1 was carried out, and adduct 11 was obtained in 35% yield with 15% ee (Scheme 6). The lower yield than that achieved with the transformation of N-phenyl glycine 1a (entry 2, Table 1) was due to the deteriorated chemoselectivity, as the reduced product of 2a, i.e., benzoin, was obtained in a considerable amount. Note that in the absence of catalyst C1, 3a was also produced in a decreased yield in the reaction of 1a with 2a (entry 13, Table 1). According to the persistent radical effect,13 the results suggest the existence of an interaction between N–H of 1a as a H-bonding donor and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the chiral CPA as a H-bonding acceptor, thus increasing the nucleophilicity of the α-amino radicals. In this context, the chiral CPA should serve as a bifunctional catalyst to activate the reaction and provide stereocontrol for the new C–C bond formation,9s for which a ternary transition state as shown in Scheme 2 is plausible.
O of the chiral CPA as a H-bonding acceptor, thus increasing the nucleophilicity of the α-amino radicals. In this context, the chiral CPA should serve as a bifunctional catalyst to activate the reaction and provide stereocontrol for the new C–C bond formation,9s for which a ternary transition state as shown in Scheme 2 is plausible.
 | ||
| Scheme 6 Transformation of 10 and 2a. | ||
Conclusions
In summary, we developed an enantioselective radical coupling of N-aryl glycines with diverse activated ketones, including acyclic 1,2-diketones and cyclic isatins, via visible-light-driven cooperative photoredox and chiral H-bonding catalysis. A range of significant enantioenriched 1,2-amino alcohols that feature an oxo-hetero-quaternary carbon stereocenter were obtained in high yields and ees. This work robustly demonstrates the viability of H-bonding catalysis for the highly reactive radical-coupling of ketones by directly providing a stereocontrolled environment for ketones. We believe that this dual catalytic system can serve as a powerful tool to address a variety of radical coupling reactions of diverse ketones through flexibly selecting a photoredox catalyst and a H-bonding catalyst, thus providing a direct and productive approach to access various chiral tertiary alcohols. We also anticipate that this work will inspire the pursuit of novel enantioselective radical coupling for other oxidative substrates with feasible H-bonding acceptor moieties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants from the NSFC (21672052) and Provincial Innovation Scientists and Technicians Troop Construction Projects are gratefully acknowledged. We also appreciate Mr Yangyang Shen (ICIQ) for the constructive discussions.Notes and references
- Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001 Search PubMed.
- H. Togo, in Advanced Free Radical Reactions for Organic Synthesis, Elsevier Science, Amsterdam, 2004, p. 39 Search PubMed.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef PubMed; (b) H. Yi, G. Zhuang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef PubMed.
- M. Andrew, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef PubMed.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef PubMed; (c) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474 CrossRef PubMed; (d) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef PubMed; (f) J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef.
- For a selected review, see: (a) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193 RSC. For selected examples, see: (b) E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776 CrossRef PubMed; (c) W.-P. Li, Y.-Q. Yuan, M.-L. Zhang, J. Cheng and C.-J. Zhu, Chem. Commun., 2016, 52, 7596 RSC; (d) X.-Q. Yuan, X.-X. Wu, S.-P. Dong, G.-B. Wu and J.-X. Ye, Org. Biomol. Chem., 2016, 14, 7447 RSC; (e) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, J. Am. Chem. Soc., 2016, 138, 12312 CrossRef PubMed; (f) W. Ding, L.-Q. Lu, J. Liu, D. Liu, H.-T. Song and W.-J. Xiao, J. Org. Chem., 2016, 81, 7237 CrossRef PubMed; (g) S. Sharma, S. Sultan, S. Devari and B. A. Shah, Org. Biomol. Chem., 2016, 14, 9645 RSC; (h) C.-M. Wang, D. Song, P.-J. Xia, Z.-P. Ye, J.-A. Xiao, H.-Y. Xiang, X.-Q. Chen and H. Yang, Org. Chem. Front., 2018, 5, 1608 RSC.
- (a) D. Uraguchi, H. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef PubMed; (b) C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685 CrossRef PubMed; (c) T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953 CrossRef PubMed; (d) J. Ma, K. Harms and E. Meggers, Chem. Commun., 2016, 52, 10183 RSC; (e) Z. Zhou, Y. Li, B. Han, L. Gong and E. Meggers, Chem. Sci., 2017, 8, 5757 RSC; (f) J. Ma, A. R. Rosales, X. Huang, K. Harms, R. Riedel, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 17245 CrossRef PubMed; (g) J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Nat. Commun., 2018, 9, 2445 CrossRef PubMed.
- For selected reviews, see: (a) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (b) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef PubMed; (c) T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307 CrossRef PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef PubMed.
- For selected examples, see: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef PubMed; (b) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef PubMed; (c) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef PubMed; (d) M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062 CrossRef PubMed; (e) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef PubMed; (f) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef PubMed; (g) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef PubMed; (h) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC; (i) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642 CrossRef PubMed; (j) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef PubMed; (k) L. R. Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef PubMed; (l) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef PubMed; (m) A. G. Amador, E. M. Sherbrook and T. P. Yoon, J. Am. Chem. Soc., 2016, 138, 4722 CrossRef PubMed; (n) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636 CrossRef PubMed; (o) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef PubMed; (p) A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017, 9, 1073 CrossRef PubMed; (q) E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322 CrossRef PubMed; (r) E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394 CrossRef PubMed; (s) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef PubMed , and ref. 7.
- (a) G. Wei, C. Zhang, F. Bureš, X. Ye, C.-H. Tan and Z. Jiang, ACS Catal., 2016, 6, 3708 CrossRef; (b) Y. Liu, J. Li, X. Ye, X. Zhao and Z. Jiang, Chem. Commun., 2016, 52, 13955 RSC; (c) L. Lin, X. Bai, X. Ye, X. Zhao, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2017, 56, 13842 CrossRef PubMed; (d) T. Shao, Y. Yin, R. Lee, X. Zhao, G. Chai and Z. Jiang, Adv. Synth. Catal., 2018, 360, 1754 CrossRef; (e) Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083 CrossRef PubMed.
- For the derivatives from 1,2-diketones, see: (a) M. Ogata, H. Matsumoto, K. Takahashi, S. Shimizu, S. Kida, A. Murabayashi, M. Shiro and K. Tawara, J. Med. Chem., 1987, 30, 1054 CrossRef PubMed. For the derivatives from isatins, see: (b) K. Monde, K. Sasaki, A. Shirata and M. Takasugi, Phytochemistry, 1991, 30, 2915 CrossRef; (c) M. Takasugi, K. Monde, N. Katsui and A. Shirata, Chem. Lett., 1987, 1631 CrossRef; (d) M. Mautino, F. Jaipuri, A. Marcinowicz, T. Kesharwani and J. Waldo, PCT Int. Appl., WO 2009073620 A2 20090611, 2009; (e) L. Liu, S. Zhang, F. Xue, G. Lou, H. Zhang, S. Ma, W. Duan and W. Wang, Chem.–Eur. J., 2011, 17, 7791 CrossRef PubMed; (f) Y. Zhang, Z. J. Li, H. S. Xu, Y. Zhang and W. Wang, RSC Adv., 2011, 1, 389 RSC; (g) K. L. Spear and U. Campbell, PCT Int. Appl., WO 2014106238 A1 20140703, 2014.
- (a) C. S. Rajesh, T. L. Thanulingam and S. Das, Tetrahedron, 1997, 53, 16817 CrossRef; (b) L. Chen, C. S. Chao, Y. Pan, S. Dong, Y. C. Teo, J. Wang and C.-H. Tan, Org. Biomol. Chem., 2013, 11, 5922 RSC; (c) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef PubMed; (d) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef PubMed; (e) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11195 Search PubMed; (f) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef PubMed; (g) A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464 CrossRef PubMed.
- A. Studer, Chem.–Eur. J., 2001, 7, 1159 CrossRef.
- I. Willner, T. Tsfania and Y. Eichen, J. Org. Chem., 1990, 55, 2656 CrossRef.
- (a) I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370 CrossRef PubMed; (b) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef PubMed.
- 1836991 (3q) contains the supplementary crystallographic data for this paper.†.
- See the ESI† for the details.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1836991 and 1843336. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02948b |
| ‡ The authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |