
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZ-scheme solar water splitting via self-assembly of photosystem I-catalyst hybrids in thylakoid membranes†
Lisa M.
Utschig
 *,
Sarah R.
Soltau‡
,
Karen L.
Mulfort
,
Jens
Niklas
and
Oleg G.
Poluektov
*,
Sarah R.
Soltau‡
,
Karen L.
Mulfort
,
Jens
Niklas
and
Oleg G.
Poluektov
Chemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA. E-mail: utschig@anl.gov
First published on 29th October 2018
Abstract
Nature's solar energy converters, the Photosystem I (PSI) and Photosystem II (PSII) reaction center proteins, flawlessly manage photon capture and conversion processes in plants, algae, and cyanobacteria to drive oxygenic water-splitting and carbon fixation. Herein, we utilize the native photosynthetic Z-scheme electron transport chain to drive hydrogen production from thylakoid membranes by directional electron transport to abiotic catalysts bound at the stromal end of PSI. Pt-nanoparticles readily self-assemble with PSI in spinach and cyanobacterial membranes as evidenced by light-driven H2 production in the presence of a mediating electron shuttle protein and the sacrificial electron donor sodium ascorbate. EPR characterization confirms placement of the Pt-nanoparticles on the acceptor end of PSI. In the absence of sacrificial reductant, H2 production at PSI occurs via coupling to light-induced PSII O2 evolution as confirmed by correlation of catalytic activity to the presence or absence of the PSII inhibitor DCMU. To create a more sustainable system, first-row transition metal molecular cobaloxime and nickel diphosphine catalysts were found to perform photocatalysis when bound in situ to cyanobacterial thylakoid membranes. Thus, the self-assembly of abiotic catalysts with photosynthetic membranes demonstrates a tenable method for accomplishing solar overall water splitting to generate H2, a renewable and clean fuel. This work benchmarks a significant advance toward improving photosynthetic efficiency for solar fuel production.
Introduction
Sunlight-driven water splitting provides a pathway to store available solar energy in energy-dense chemical bonds of molecules. Of particular interest is the solar-powered production of H2, a clean and renewable energy source that can replace carbon-based fossil fuels and help provide for ever-increasing global energy demands.1,2 Many current strategies to achieve these so-called “solar fuels” are inspired by Nature's photosynthetic machinery that converts light energy to chemical energy. In plants, algae, and cyanobacteria, two large integral membrane reaction center (RC) proteins work together in a coupled electron transfer Z-scheme: light-driven oxidation of water is carried out by Photosystem II (PSII) whereas Photosystem I (PSI) catalyzes the light-driven transmembrane transfer of an electron from reduced plastocyanin or cytochrome c6 to oxidized ferredoxin or flavodoxin.3 These electrons are then used to produce NADPH, an electron source for Calvin cycle CO2 fixation. Herein, we explore redirecting the electrons normally used for NADP+ reduction toward abiotic catalysts for light-driven H2 production from thylakoid membranes. A precedent of sorts for this work includes bioelectrodes developed using thylakoid membranes, but these focus exclusively on producing electricity and are limited by the diurnal solar cycle.4–7 Therefore, we investigate the possibility of enhancing photosynthetic efficiency for direct production of storable and transportable solar fuels by creating alternative pathways that utilize the excess reducing equivalents produced by photosynthetic electron transfer under high light conditions.8Several inherent features make PSI a powerful photochemical module poised for H2 generation. These include: a quantum yield that approaches 1, a long-lived charge-separated state of ∼60 ms, and an electrochemical potential of −580 mV (vs. NHE) for the terminal electron acceptor FB (a [4Fe–4S] cluster) that provides sufficient driving force to reduce protons to H2 at neutral pH.9 PSI's photogenerated electrons have been successfully coupled to hydrogenase enzymes,10–12 platinum systems,13–15 and molecular catalysts.16,17 These studies focus on the reductive half-reaction of water-splitting, using purified PSI isolated from thylakoid membranes and, thus, require sacrificial electron donors to reduce the oxidized donor of PSI, P700+, so that two successive photon-induced electrons are available at the catalyst site for H2 reduction.18
In this study, we examine the feasibility of achieving complete water-splitting by utilizing the electron originating from PSII photoexcitation to reduce P700+, thereby removing the need for sacrificial redox reagents. To accomplish this, we examine binding abiotic H2 catalysts to the acceptor side of PSI in situ and coupling the electron transfer between PSII and PSI to the bound catalyst via the natural Z-scheme provided by the membrane environment (Fig. 1). For non-membrane systems, highly efficient photocatalysis of H2 can be obtained for an electrostatically-directed self-assembled stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) complex of PSI and Pt nanoparticle.15 The nanoparticle used in our previous study has a similar charge and size as the electron acceptor proteins to PSI,19 and, thus readily binds to a basic patch provided by the stromal side of PSI for protein docking.20 This PSI-Pt nanoparticle hybrid remains one of the most effective photocatalytic Pt-based PSI systems to date.18 Likewise, molecular catalysts self-assemble with native PSI by tucking themselves into hydrophobic pockets provided by the large protein matrix.16,17
:1) complex of PSI and Pt nanoparticle.15 The nanoparticle used in our previous study has a similar charge and size as the electron acceptor proteins to PSI,19 and, thus readily binds to a basic patch provided by the stromal side of PSI for protein docking.20 This PSI-Pt nanoparticle hybrid remains one of the most effective photocatalytic Pt-based PSI systems to date.18 Likewise, molecular catalysts self-assemble with native PSI by tucking themselves into hydrophobic pockets provided by the large protein matrix.16,17
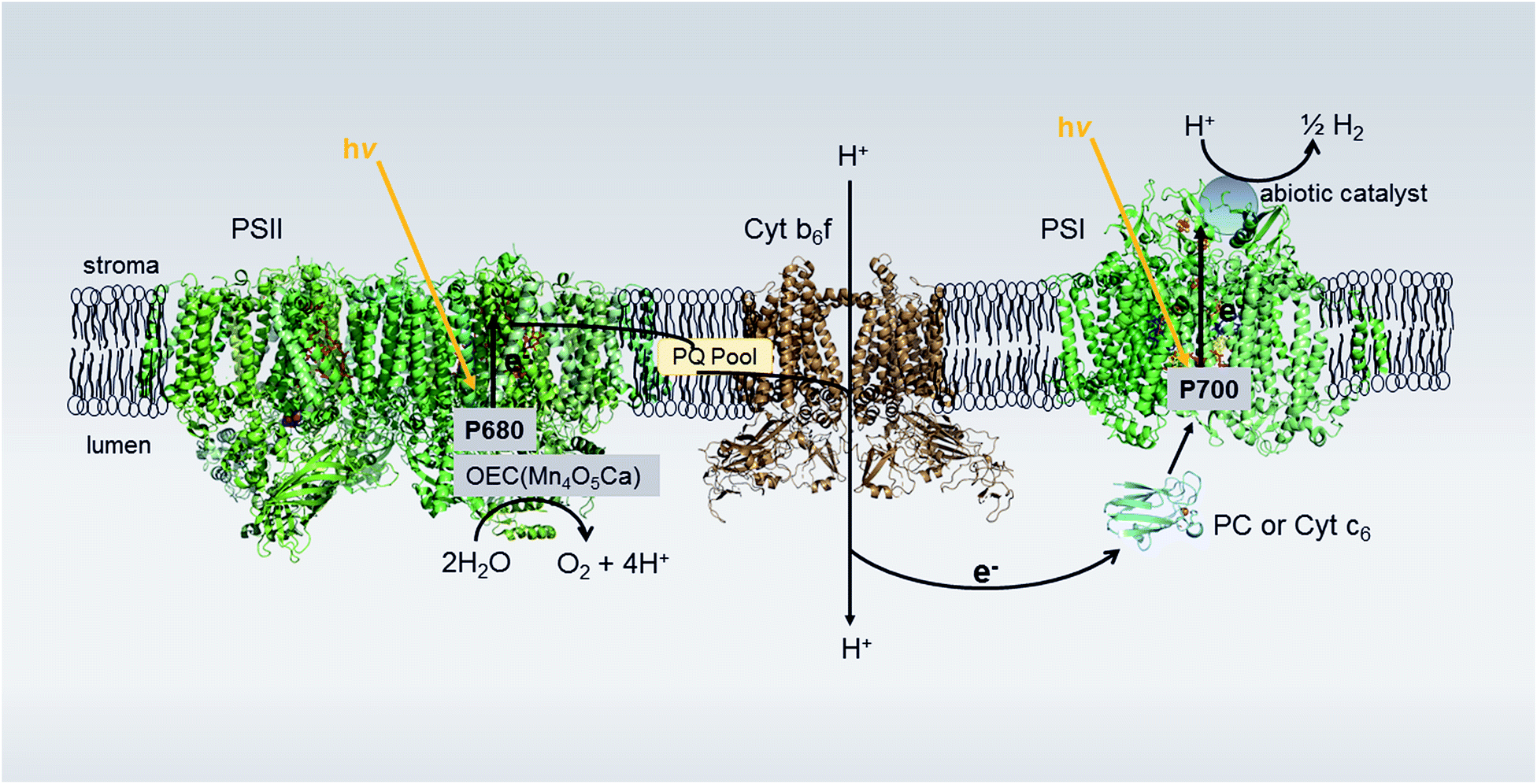 | ||
| Fig. 1 Proposed photosynthetic Z-scheme electron transport in thylakoid membranes for H2 production. Photons absorbed by Photosystem II (PSII) are used to oxidize water in the oxygen-evolving complex (OEC) connected to PSII. The extracted electrons are passed on to Photosystem I (PSI) via the plastoquinone (PQ) pool, the cytochrome b6f complex (Cyt b6f) and the luminal electron transfer proteins plastocyanin (PC) or cytochrome c6 (Cyt c6). Upon light excitation, PSI transfers electrons from the luminal side to the stromal side of the membrane where, in the native system, the soluble electron transfer protein ferredoxin (Fd) is reduced, transporting electrons to ferredoxin-NADP+ oxidoreductase (FNR) for the reduction of NADP+ to NADPH. In our system, bound catalyst at the acceptor end of PSI hijacks the light-generated electrons meant for Fd, utilizing these electrons for H2 production. Protein structures: PSII (2axt), Cyt b6f (2D2C), PC (1bxu), PSI (1JB0). | ||
In 1985, Greenbaum reported photocatalytic H2 production from platinized chloroplasts.21 Metallic Pt was photoprecipitated onto the acceptor end of PSI using a mixture of hexachloroplatinate (IV) and spinach chloroplasts.22 Yet even after 33 years, photoprecipitation remains the only reported method for abiotic catalyst assembly with thylakoids.22 Inspired by this work, we target an advanced strategy that uses the self-assembly of well-resolved and characterized electrostatically charged Pt-nanoparticles and synthetic molecular catalysts with thylakoids in the dark and extend the previous studies to show the Z-scheme nature of the membrane electron transport. Importantly, self-assembled systems have rates of H2 production 103 to 104 times faster15,16 than those of photoprecipitated colloidal Pt PSI hybrids using the isolated protein.14,23 The stromal end of PSI extends beyond the membrane plane, and therefore, should be solvent accessible for catalyst self-assembly to the membrane. We now explore translating our self-assembling hybrid methodologies from isolated PSI systems to PSI embedded within thylakoid membranes and quantify H2 production via the photosynthetic Z-scheme.
Results and discussion
Self-assembly of Pt nanoparticles with spinach thylakoid membranes for light-driven H2 production
Spinach thylakoids24 and mercaptosuccinic-acid-stabilized Pt nanoparticles (∼3.0 nm)25 were prepared according to previously published methods. Pt nanoparticles were added to freshly purified thylakoid membranes from spinach to yield final concentrations of 0.7 mg ml−1 [Chl] and 1.2 μM Pt nanoparticles. The mixture was tumbled overnight in the dark at 4 °C. The membranes were pelleted at 10 K rpm for 10 m and the supernatant was decanted. The pellets were then resuspended and pelleted three times using 20 mM MES (pH 6.3) as a wash buffer to remove unbound Pt-nanoparticles from the membrane surfaces.In the presence of a sacrificial electron donor (SED), these washed spinach membrane/Pt nanoparticle complexes readily produce H2 upon illumination with visible light. Hydrogen measurements were performed in a sealed and N2-purged 5.3 ml spectrophotometer cell with a path length of 1.0 cm. Membrane/Pt nanoparticle complexes were added to a final concentration of 0.04 mg ml−1 Chl in 10 mM MES, pH 6.2. The final reaction mixture contained 100 mM sodium ascorbate as SED and 1 mM 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) as an inhibitor of PSII (Fig. 2A). Plastocyanin purifies along with the thylakoid membrane, and thus, no additional mediator protein was added to reduce P700+. The sample was illuminated with a 300 W xenon lamp using a 500 nm long-pass filter, a heat absorbing filter (KG-2, Schott) and a 29 cm water filter and the intensity of light measured behind the sample was 1000 μE m−2 s−1. Samples of the headspace were removed at 30–60 m intervals and analysed for H2 content by gas chromatography. For this system, the rate of H2 production was determined to be 4 μmol H2 (mg Chl)−1 h−1 (Fig. 3A). The number of PSI molecules per total chlorophyll in the spinach membranes was estimated by quantitation of P700+ with EPR spectroscopy (Fig. S1†). For our spinach membranes, we determined >600 chlorophyll molecules per PSI RC which is consistent with early reports of ∼500 chlorophyll molecules per PSI RC in higher plant chloroplasts.26 Based on this number, the rate for H2 generation is, in more conventional units, 2100 mol H2 (mol PSI)−1 h−1, a TOF which compares well with other photocatalytic PSI hybrid systems.18 The amount of H2 generated increased linearly for 1.5 hours under these experimental conditions, followed by a decrease over time due to depletion of the added electron donor ascorbate. The system completely stopped generating H2 after approximately 3.5 hours. Spinach membranes without Pt nanoparticles added produced negligible H2 under the same experimental conditions (Fig. S2†).
 | ||
| Fig. 2 Schematic representation of experimental set-ups for (A) H2 production using a sacrificial electron donor (SED) and DCMU to block electron transfer from PSII (B) H2 production using Z-scheme electron transport via PSII and (C) EPR spectroscopic experiments to examine light-induced electron transport in PSI and to the acceptor flavodoxin protein (Fld). | ||
 | ||
| Fig. 3 Time profiles of H2 and O2 production following illumination of thylakoid/Pt nanoparticle complexes. (A) H2 evolution from spinach thylakoid/Pt nanoparticle (NP) complex with 100 mM sodium ascorbate and 1 mM DCMU; (B) H2 evolution from spinach thylakoid/Pt NP complex with 20 μM cyt c6 (dark squares) or without cyt c6 (open circles), no sodium ascorbate; (C) O2 evolution from spinach thylakoid/Pt NP complex (open triangles) plotted with H2 data in (C); (D) H2 evolution from S. leopoliensis thylakoid/Pt NP complexes in 100 mM sodium ascorbate with 4 μM cyt c6 and 1 mM DCMU; (E) H2 evolution from S. leopoliensis thylakoid/Pt NP complexes with 12 μM cyt c6, no sodium ascorbate; (F) O2 evolution (open triangle) and H2 evolution (dark squares) from S. leopoliensis thylakoid/Pt NP complexes with 12 μM cyt c6. All samples were in 10 mM MES pH 6.2. | ||
To test for Z-scheme H2 production from the membrane/Pt nanoparticle system, we monitored H2 photocatalysis in the absence of the SED, ascorbate, and opened up electron flow from PSII by removing the PSII inhibitor DCMU (Fig. 2B). Simultaneous light-driven H2 and O2 production were observed at a rates of 0.1 μmol H2 (mg Chl)−1 h−1 and 2 μmol O2 (mg Chl)−1 h−1 (Fig. 3B and C). To support our hypothesis that the H2 observed is via PSII water oxidation, light-induced electron transfer from PSII was blocked by the addition of 1 mM DCMU. Under these conditions, H2 production was not observed in the absence of SED. Although other systems have employed MES as a SED in reductive half-reactions, we have confirmed that it does not act as a SED for PSI as no measurable H2 is observed for the MES buffer system, without sodium ascorbate in solution for both the thylakoid system (Fig. S3†) as well as the isolated PSI-Pt nanoparticle hybrid (Fig. S4†).
A 2:1 ratio of H2:O2 for the full water-splitting reaction is expected, yet we observe a 1:20 ratio. We know from our previous work on aqueous H2 photocatalysis using PSI-Pt nanoparticle biohybrids15 that the quantum efficiency of the reductive half of reaction is near 100%. Likewise, we observe a 40-fold higher rate of H2 production in the membrane system with SED present than without (Table 1), indicating that electron delivery through PSI to the catalyst is not the limiting factor. Rather, we think that the discrepancy in the H2:O2 ratio reflects a low efficiency of electron flow from PSII to PSI due to the complexity of the multi-component electron transfer chain in thylakoids (Fig. 1). In higher plant chloroplasts, PSI and PSII are located in different structural regions: PSI is located in the unstacked stroma membranes and the edges of the stacked grana membranes, whereas PSII is found only in the stacked grana membranes.3 The cytochrome b6f (Cyt b6f) complex is uniformly distributed between both membrane environments3 with a limited plastoquinone (PQ) pool (6.7 PQ/PSII) shuttling reducing equivalents between PSII and Cyt b6f.27 The delivery of electrons from PSII via Cyt b6f to PSI is a diffusion-controlled process. To test this, we added excess mediator protein, cyt c6, which, like plastocyanin, is a shuttle protein that donates an electron to P700+. In the presence of 20 μM cyt c6 the rate of H2 production increased threefold with H2 production proceeding for 2 hours (Fig. 3B).
| System | Catalyst | SEDa | TOF [μmol H2 (mg Chl)−1 h−1] | TOFb [mol H2 (mol PSI)−1 h−1] |
|---|---|---|---|---|
| a Sacrificial electron donor: 100 mM sodium ascorbate. b Estimated from P700+ signal ratios determined by EPR (see ESI Fig. S1). c Ref. 28. d Ref. 29. e No measurable H2 detected in GC traces (see ESI Fig. S2). | ||||
| Spinachc | Pt, photoprecipitated | No | 0.032 | — |
| Spinachd | Pt, photoprecipitated | No | 0.08 | — |
| Spinach | None | Yes | 0e | 0 |
| Spinach | Pt-NP | Yes | 4 | >2100 |
| Spinach | Pt-NP | No | 0.1 | >50 |
| S. leopoliensis | None | Yes | 0e | 0 |
| S. leopoliensis | Pt-NP | Yes | 14 | 1500 |
| S. leopoliensis | Pt-NP | No | 0.4 | 40 |
| T. lividus | Pt-NP | Yes | 0.2 | — |
| S. leopoliensis | Cobaloxime | Yes | 1 | 110 |
| S. leopoliensis | Ni diphosphine | Yes | 3 | 320 |
| S. leopoliensis | Ni diphosphine | No | 0.03 | 3 |
We compare these measurements to those reported for photoprecipitated Pt colloidal spinach systems (Table 1). Spinach chloroplast preparations (later referred to as thylakoids), yielded rates of 32 and 80 nmol H2 (mg Chl)−1 h−1.28,29 These values are consistent with the 100 nmol H2 (mg Chl)−1 h−1 rate observed for our self-assembled spinach thylakoid/Pt nanoparticle preparation. Hexachloroplatinate (IV) acts as a Hill acceptor, an artificial acceptor of electrons from PSII, resulting in photocatalytic O2 evolution.30 Without an artificial acceptor, the reported O2 level was observed to be very low due to the small oxidized plastoquinone pool,28 as we have observed in our current study.
Cyanobacterial-based membrane systems for photocatalysis
Toward future biochemical engineering possibilities, we extend these studies to cyanobacterial systems. To the best of our knowledge, these type of experiments have never been performed with cyanobacterial membranes. The membranes were isolated from Synechococcus leopoliensis and Thermosynechococcus lividus.31 Pt nanoparticles were added to thawed cyanobacterial thylakoid membranes at a final concentration of 0.14 mg ml−1 Chl and 0.6 μM Pt nanoparticle and tumbled overnight at 4 °C. Multiple pellet/wash cycles were performed with 20 mM Tris–Cl, pH 8.0 buffer, to remove unbound nanoparticles from the membranes.Light-induced H2 production was measured from the resultant cyanobacterial thylakoid complexes. The membrane/Pt nanoparticle pellets were resuspended using 10 mM MES buffer, pH 6.2, in spectrophotometer cells purged with N2 to a final concentration of 0.02–0.03 mg ml−1 Chl. The final reaction mixture contained 100 mM sodium ascorbate as the SED and 1 mM DCMU as an inhibitor of PSII (Fig. 2A). Unlike spinach thylakoids, cyanobacterial membrane preparations do not contain plastocyanin. Therefore, 4 μM cyt c6 was added to each reaction mixture to help mediate reduction of P700+. Samples were illuminated as described above for spinach samples. The intensity of light as measured behind the sample ranged from 700 to 900 μE m−2 s−1. For S. leopoliensis, the mesophilic species, H2 production was observed at a rate of 14 μmol H2 (mg Chl)−1 h−1 for 4 hours (Fig. 3D) or 1500 mol H2 (mol PSI)−1 h−1 based on EPR quantification of PSI content in the membrane (Fig. S1†). For the thermophilic species T. lividus, however, very minimal levels of H2 production were observed, 0.2 μmol H2 (mg Chl)−1 h−1 (Fig. S5†). PSI isolated from both S. leopoliensis and T. lividus readily form highly active photocatalysts with Pt nanoparticles, with rates up to 244 μmol H2 (mg Chl)−1 h−1.15 Thus, the low amount of H2 indicates that binding of Pt nanoparticles to PSI in the T. lividus membrane system is restricted and we postulate that phycobilisomes32 may block the self-assembly of Pt nanoparticles. In control experiments, cyanobacterial membranes alone (no Pt nanoparticle added) produced no measurable H2 under the photocatalysis experimental conditions (Fig. S2†).
Z-scheme H2 and O2 production from our S. leopoliensis membrane/Pt nanoparticle complexes in the absence of sodium ascorbate and DCMU was measured (Fig. 2B). Impressively, this system steadily generated H2 and O2 for over 8 hours. The fastest rate, 0.4 μmol H2 (mg Chl)−1 h−1, was observed over the first 2 hours of illumination using 12 μM cyt c6 as a mediator (Fig. 3E). The O2 rate matched the rate of H2 production, 0.4 μmol O2 (mg Chl)−1 h−1 (Fig. 3F). Though not the optimal 2:1 ratio of H2:O2 expected for the full water-splitting reaction, a 1:1 ratio observed for the cyanobacterial membrane reflects a much better efficiency for electron transport from PSII to PSI than that observed for the spinach membranes (1:20). The number of PSI molecules per total chlorophyll in the S. leopoliensis membranes (estimated by quantitation of P700+ with EPR spectroscopy, Fig. S1†) is >5-fold higher than in spinach membranes, which, in part could explain the higher efficiency. In the presence of 1 mM DCMU, H2 evolution was completely inhibited, consistent with the electron for reducing P700+ originating from PSII. Control experiments show that MES buffer does not act as a SED in this system (Fig. S3 and S4†).
Spectroscopically probing the catalyst location and electron transfer pathway
Photocatalysis provides evidence that Pt nanoparticles bind to the stromal end of PSI in spinach and S. leopoliensis membranes. To provide further insight into protein–catalyst interactions in the membrane system, EPR spectroscopy was used to explore light-induced electron transfer reactions. Similar to isolated PSI, we believe that Pt nanoparticles readily self-assemble by electrostatically associating with a basic patch provided by the stromal subunits of PSI that extend beyond the membrane surface.19,20 In this manner, nanoparticles mimic acceptor protein docking with PSI.15 To test this, we examined interprotein electron transfer between PSI and one of its acceptor proteins, flavodoxin (Fld), in membranes with or without Pt nanoparticle bound. Fld replaces ferredoxin as an electron acceptor under iron deficiency in most cyanobacteria and is capable of substituting for ferredoxin in several ferredoxin-driven redox reactions, including reduction of ferredoxin-NADP+ reductase (FNR).33 Whereas ferredoxin contains a Fe–S cluster which spectroscopically overlaps with the three terminal [4Fe–4S] clusters of PSI, Fld contains a flavin mononucleotide (FMN) cofactor which is spectroscopically distinct. In addition, we have access to fully deuterated Fld that enables the signals of reduced flavin acceptor and oxidized primary donor, P700+, to be well-resolved and distinguished at X-band (9.5 GHz) EPR.34S. leopoliensis membranes at 1.6 mg ml−1 Chl were incubated with 100 μM deuterated flavodoxin in the presence of 0.3 mM dichloro(phenol)indophenol (DCPIP) and 10 mM sodium ascorbate as donors to P700+. Fig. 4A shows the X-band EPR spectra of samples that were illuminated for 10 s at room temperature with visible light, prior to freezing in liquid N2. A Fld− signal for the semiquinone FMN radical34 is observed for native membranes, however, the amplitude of this signal is significantly reduced for the membrane/Pt-nanoparticle sample. These results are consistent with the Pt nanoparticle either prohibiting docking of the Fld protein to PSI, or if Fld can dock to PSI-Pt, electron transfer to the Pt nanoparticle is preferential to that of Fld (Fig. 4B).15 This competition measurement supports the hypothesis that the Pt nanoparticle mimics acceptor protein binding to PSI.
 | ||
| Fig. 4 EPR spectroscopy of light-induced electron transfer in photosystem I-Pt nanoparticle thylakoid membranes. (A) cw X-band EPR spectra at 10 K were acquired after 10 s room temperature illumination of S. leopoliensis membranes in the presence of excess 2H-Fld, with (red) or without (black) Pt nanoparticles (NP) bound to the membrane. (B) Schematic explanation of the EPR results; Pt NP blocks binding of Fld to the stromal end of PSI or both Pt NP and Fld bind to PSI, but electron transfer to Pt NP is preferred. (C) Low temperature (10 K) light-induced reduction of the terminal Fe–S clusters of PSI in the membrane environment as observed with X-band EPR spectroscopy, with (red) or without (black) Pt NP bound to the membrane. (D) Schematic explanation of the EPR results; when Pt NP is bound to PSI in the membrane environment, charge separation occurs to the Pt NP and not the terminal Fe–S clusters FA and FB, which act only as an intermediate electron transfer cofactors. | ||
In another set of experiments, we looked at the inherent light-induced low-temperature reduction of the terminal Fe–S clusters FA and FB of PSI. When PSI is frozen in the dark and then illuminated at cryogenic temperatures, only a single electron from P700 can be transferred to either of the terminal electron acceptors FA or FB, but not both, in a given RC, yielding either P700+FA− RCs or P700+FB− RCs.35 The resultant EPR spectrum represents a superposition of the individual broad rhombic EPR signals of FA− (g = 2.05, 1.95, 1.85) and FB− (g = 2.07, 1.93, 1.88).35Fig. 4C shows the low temperature, light-induced cw X-band EPR spectra of [4Fe–4S] clusters FA and FB in S. leopoliensis membranes and membrane/Pt-nanoparticle complexes. Both samples contained 1.6 mg ml−1 Chl, 0.3 mM DCPIP and 10 mM sodium ascorbate. In the Pt sample, the signal intensity for FA− and FB− is greatly reduced compared to the native membrane. Thus, we can estimate there is a small amount of electrons residing on either of the terminal Fe–S clusters following low-temperature (10 K) illumination, but the majority of the electrons transferred at low temperature are located on the Pt-nanoparticle (which is EPR silent). This assertion is supported by the light-induced increase in signal intensity of P700+ at low temperature, indicating that charge separation has occurred (Fig. S6†). These results support that low-temperature electron transfer occurs between PSI and the nanoparticle, and provide spectroscopic evidence of Pt-nanoparticle binding to the stromal end of PSI, near FA and FB (Fig. 4D).15
Toward sustainability: earth-abundant catalytic thylakoid systems
Photosynthetic systems must be both efficient and scalable to be a practical component of a solar energy future. To that end, we explore the incorporation of sustainable first-row transition metal molecular catalysts with thylakoid membranes.Hydrogenase enzymes are highly active and earth-abundant H2 catalysts.36,37 However, efforts for re-directing electron transfer from ferredoxin (reduced by PSI) to hydrogenase are limited by the acute O2 sensitivity38 of hydrogenase and require decoupling PSII O2 generation from hydrogenase activity in reengineered organisms.39–41 Thus, hydrogenase-based systems face challenges if coupled to the native Z-scheme which requires electrons from PSII; although one can envision future systems where the O2-sensitive [FeFe]-hydrogenases are replaced by the more O2-tolerant [NiFe]-hydrogenases.42,43 Cobaloxime44 and nickel diphosphine catalysts45 are more tolerant to O2 in their ground states and provide relatively low cost alternatives to rare and expensive metals, such as platinum. Furthermore, molecular catalysts provide a higher metal-atom efficiency than nanoparticle catalysts which have very few active sites among many spectator atoms. We have previously shown that the Co(dmgH)2pyCl and [Ni(PPh2NPh2)2](BF4)2 molecular catalysts (where dmgH = dimethylglyoximate, py = pyridine, and Ph = phenyl) (Fig. 5) can be successfully linked to PSI photochemistry for H2 production in isolated PSI hybrids.16,17 Both catalysts were found to self-assemble with native PSI in aqueous solution at pH 7 and that binding is dominated by hydrophobic interactions, with each molecular catalyst tucking itself into the hydrophobic pockets provided by the large PSI protein matrix (∼350 kDa).46
 | ||
| Fig. 5 Molecular first row transition metal catalysts used in the current study. (A) Ni diphosphine catalyst, [Ni(PPh2NPh2)2](BF4)2, and (B) cobaloxime catalyst, Co(dmgH)2pyCl. | ||
To test the self-assembly of molecular catalysts to PSI in its membrane environment, S. leopoliensis thylakoid membranes at a final concentration of 0.2 mg ml−1 Chl were incubated with 200 μM [Co(dmgH)2pyCl] or [Ni(PPh2NPh2)2](BF4)2. The samples were tumbled overnight in the dark at 4 °C. Multiple pellet/resuspension cycles were performed with fresh 20 mM Tris–Cl, pH 8.0 buffer to wash away unassociated molecular catalysts from the membrane surfaces. Light-induced H2 production was measured from the molecular catalyst-membrane complexes. The membrane/molecular catalyst pellets were resuspended in spectrophotometer cells purged with N2 to a final concentration of 0.02–0.03 mg ml−1 Chl with 10 mM MES, pH 6.1, 100 mM sodium ascorbate, 1 mM DCMU, and 12 μM cyt c6. Samples were illuminated as described above for spinach samples. The intensity of light as measured behind the sample was 1000 μE m−2 s−1. H2 production was observed at a rate of 1 μmol H2 (mg Chl)−1 h−1 for 1 hour for the cobaloxime membranes and 3 μmol H2 (mg Chl)−1 h−1 for 6 hours for the nickel diphosphine membranes (Fig. 6A). Z-scheme H2 and O2 production were measured for the nickel diphosphine system and found to be 0.03 μmol H2 (mg Chl)−1 h−1 and 0.5 μmol O2 (mg Chl)−1 h−1 (Fig. 6B). Although catalytic activity of Ni diphosphines have been reported to be sensitive to O2,47 we anticipate that the very low O2 levels arising from the native PQ pool are most likely not an issue in the thylakoid system. Table 1 compares the rates for light-induced H2 production from each of the membrane-catalyst systems reported herein.
 | ||
| Fig. 6 Time profiles of H2 and O2 production following illumination of thylakoid-Ni diphosphine complexes. S. leopoliensis thylakoid/Ni diphosphine complex with (A) 100 mM sodium ascorbate, 12 μM cyt c6, and 1 mM DCMU; (B) with 40 μM cyt c6, no sodium ascorbate (H2, black squares; O2, open triangles). Inset: H2 production from panel (B). Both samples were in 10 mM MES pH 6.1. | ||
The observance of H2 production suggests that at least some of the Co(dmgH)2pyCl and [Ni(PPh2NPh2)2](BF4)2 molecular catalysts bind near the protein surface on the acceptor end of PSI. It is expected, however, that catalyst binding may not be specific to PSI, with additional arbitrary binding sites provided by hydrophobic pockets found throughout the large membrane structure. A promising feature of molecular catalysts, however, is that they are synthetically tuneable, with ligand structures that can be synthesized for covalent binding to targeted protein sites, such as histidine or cysteine residues.48 This could provide a method for achieving directed binding of molecular catalysts by covalent linkage to engineered surface residues on the stromal subunits of PSI. Another method is to use inherent protein–protein electrostatic interactions between PSI and its acceptor proteins, utilizing the small proteins to carry the catalyst to the docking site provided by PSI.17
Conclusions
Sunlight-driven production of hydrogen from water provides a sustainable approach to achieve a clean, renewable alternative fuel to fossil fuels. Herein, we demonstrate unique systems that link PSII water oxidation to the reductive proton-coupled chemistry of self-assembled PSI-catalyst constructs in photosynthetic membranes. Both Pt-nanoparticles and synthetic molecular catalysts readily self-assemble with thylakoids via electrostatic or hydrophobic interactions, generating viable complexes that use light to rapidly produce hydrogen directly from water. We show that it is feasible to bind synthetic molecule catalysts to thylakoid membranes and make a functional, inexpensive solar fuel producing system, addressing a key challenge of scalability for making solar fuels a viable energy source. This work provides the basis for future studies that use synthetic catalysts, tuned through known chemical modifications, for in vivo delivery systems that target PSI. Interfacing abiotic catalysts with photosynthetic membranes provides a method to utilize Nature's optimized light-driven Z-scheme chemistry and points to a possible means to enhance photosynthetic efficiency toward solar fuel production by creating an alternative electron transfer pathway during downregulation of photosynthesis under high light intensities.8 These benchmark studies are a positive step toward the implementation of in vivo approaches to generate living photosynthetic systems as a sustainable energy solution.Experimental
Thylakoid preparation
Baby spinach (10 ounces, store bought) was washed and dark adapted overnight at 4 °C. The following steps were performed in a dark laboratory with a green headlamp as the only light source. The spinach leaves were destemmed and placed in a pre-chilled blender (Sunbeam, 1.5 L) with ice cold 20 mM Hepes buffer, pH 7.4, containing 0.4 M NaCl, 4 mM MgCl2, 5 mM EDTA, and 1 mg ml−1 BSA. 6–8 high speed pulses were used to grind the leaves. The ground spinach was then rapidly transferred to a pre-chilled Hamilton Beach Big Mouth juice extractor. Spinach juice was collected in a beaker on ice, then transferred to cold centrifuge bottles and spun at 6500 rpm for 6 m in a Beckman Coulter Avanti J-26 XP with a JLA-16.250 rotor at 4 °C. The resultant pellets were resuspended in 20 mM Hepes buffer, pH 7.5, containing 0.15 M NaCl, 4 mM MgCl2, 1 mM EDTA, and 1 mg ml−1 BSA. The suspension was spun for 6 m at 7000 rpm. The pellet was resuspended in 20 mM MES, pH 6.0, buffer with 15 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mg ml−1 BSA, and 30% ethylene glycol. The sample was pelleted at 10000 rpm for 8 m, resuspended a second time in the same buffer, and pelleted at 11500 rpm for 8 m. The resultant thylakoid pellet was resuspended to a final concentration of ∼4 mg ml−1 Chl. Chl content was measured in 80% cold acetone.49 O2 evolution was measured in the presence of external electron acceptors (Fig. S7†). Aliquots of the membranes were flash frozen with liquid N2, and stored in −80 °C freezer until use.
For cyanobacterial thylakoid isolation, freezer stocks of Synechococcus leopoliensis (UTEX625) or Thermosynechococcus lividus (PCC6717) cells (12–14 g) grown in AC medium at 40 °C or 45 °C, respectively, were resuspended and homogenized with 50 mM Tris–Cl, pH 8.0, and 3 mM EDTA. Following 20 m stirring at room temperature, the cell suspension was placed in a pre-chilled Bead-Beater (BioSpec Products, Inc) with 0.1 mm glass beads. The sample was beat for 8 × 15 s spurts, with 4 m rest in between with cooling in a surrounding ice bath. The resultant mixture was spun at 3000 rpm for 5 m in a Beckman Coulter Avanti J-26 XP with a JLA-16.250 rotor at 4 °C. The pellets were discarded and the supernatant was layered in a 10 ml/15 ml ratio with 0.5 M sucrose, 10 mM EDTA, and 50 mM Tris–Cl pH 7.99. The tubes were spun at 20000 rpm for 1 h in a Beckman L-60 ultrafuge with a 60 Ti rotor. The pellets were resupended to a final concentration of 1.0–1.6 mg ml−1 Chl in 50 mM Tris–Cl, pH 8.0, and 3 mM EDTA and stored at −80 °C freezer until use. Chl content was measured in 100% methanol.49
Thylakoid/Pt nanoparticle complex assembly
The synthesis of 3 nm diameter spherical Pt nanoparticles was carried out according to literature procedures,25 and characterized as previously reported.15 The following procedures were performed in a dark laboratory. Pt nanoparticles, 3 μM in MilliQ water, were added to freshly purified thylakoid membranes from spinach to a final concentrations of 0.7 mg ml−1 [Chl] and 1.2 μM Pt nanoparticle. The final buffer contained 8 mM MES pH 6.0, 6 mM NaCl, 2 mM MgCl2, and 0.4 mM EDTA. The mixture was tumbled overnight (Labquake rotisserie) in the dark at 4 °C. The membranes were pelleted at 10 K rpm for 10 m. The supernatant was carefully pipetted off the top of the pellet. The pellets were then resuspended in fresh 400 μl 20 mM MES buffer (pH 6.3) by slowly drawing up/expelling down with a Pipetman (Rainin). The samples were then repelleted. This procedure was repeated 3 times to remove unbound Pt nanoparticles from the membrane surfaces.Cyanobacterial thylakoid/Pt nanoparticle complexes were prepared by a similar process. The following procedures were performed under dim light laboratory conditions. Pt nanoparticles, 3 μM in MilliQ water, were added to thawed S. leopoliensis or T. lividus thylakoid membranes at a final concentration of 0.14 mg ml−1 Chl and 0.6 μM Pt nanoparticle in a buffer containing 6 mM Tris–Cl pH 7.9 and 0.3 mM EDTA. The samples were tumbled overnight in the dark at 4 °C. Multiple pellet/wash cycles were performed with 20 mM Tris–Cl, pH 8.0 buffer. Thylakoid/Pt nanoparticles samples were stored at-80 °C freezer until use in H2 evolution or EPR experiments.
Thylakoid/molecular catalyst complex assembly
Chemicals for the synthesis of the molecular catalysts were purchased from Sigma-Aldrich and used as received. Co(dmgH)2pyCl was prepared as previously described.50 The 1,3,5,7-tetraphenyl-1,5-diaza-3,7-diphosphacyclooctane [PPh2NPh2] ligand and Ni catalyst [Ni(PPh2NPh2)2](BF4)2 were synthesized according to published methods.51,52 200 μM Co(dmgH)2pyCl or [Ni(PPh2NPh2)2](BF4)2 from 6 mM and 3 mM stock solutions in DMSO was added to 0.2 mg ml−1 Chl S. leopoliensis thylakoid membranes in 6 mM Tris–Cl pH 7.9 and 0.3 mM EDTA. Following overnight incubation in the dark at 4 °C, the samples were pelleted/resuspended with fresh 20 mM Tris–Cl, pH 8.0 buffer in 3 subsequent cycles to wash away adventitious molecular catalysts from the membrane surfaces. H2 experiments were run the same day.H2 and O2 measurements
Photocatalytic hydrogen and oxygen production for TOF was determined using a 300 W Xe lamp (PerkinElmer) with a 500 nm long-pass filter, a 29 cm water filter, and a heat absorbing filter (KG-2, Schott). The light intensity was measured behind each sample using a MQ-100 Quantum meter (Apogee Instruments Inc.). Photocatalysis experiments were performed in a N2-purged, sealed 5.3 ml spectrophotometer cell with a path length of 1.0 cm. Samples (100 μl) were taken from the headspace and analysed for H2 and O2 by gas chromatography with a Varian CP-4900A GC equipped with a 10 m 5-angstrom molecular sieves column with a thermal conductivity detector and UHP N2 carrier gas. H2 calibration curves were constructed using injections of 3% H2 in N2 as a known standard. O2 calibration curves were determined under a N2 atmosphere in a glove bag using injections of O2 in N2 as a known standard.EPR measurements
EPR thylakoid samples were prepared as discussed above at a final concentration of 1.6 mg ml−1 Chl S. leopoliensis membranes. The flavodoxin samples contained 100 μM deuterated flavodoxin53 in the presence of 0.3 mM DCPIP and 10 mM sodium ascorbate as donors to P700+. The samples were placed in quartz EPR tubes, degassed in a nitrogen box, capped and illuminated for 10 s at room temperature with visible light, prior to freezing in liquid N2. For the low temperature electron transfer experiments, S. leopoliensis native membrane and membrane/Pt-nanoparticle samples were placed in quartz EPR tubes and dark-adapted for 20 min at room temperature prior to freezing in liquid nitrogen. Both samples contained 1.6 mg ml−1 Chl, 0.3 mM DCPIP and 10 mM sodium ascorbate. cw X-band (9.5 GHz) EPR measurements were carried out with a Bruker ELEXSYS II E500 EPR spectrometer (Bruker Biospin Corp, Rheinstetten, Germany) equipped with a TE102 rectangular EPR resonator (Bruker ER 4102ST) and a helium gas-flow cryostat (ICE Oxford, UK). Temperature control was provided by an ITC (Oxford Instruments, UK). The samples were transferred from liquid nitrogen to the pre-cooled resonator at 10 K. To measure the low temperature light-induced protein activity, X-band EPR spectra were recorded at 10 K shortly after continuous illumination with a white light LED (Thorlabs).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank A. Wagner for growth of the cyanobacteria. This work is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under Contract No. DE-AC02-06CH11357.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. USA, 2007, 104, 20142 CAS.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science Ltd, Malden, USA, 2002 Search PubMed.
- J. O. Calkins, Y. Umasankar, H. O'Neill and R. P. Ramasamy, Energy Environ. Sci., 2013, 6, 1891–1900 RSC.
- H. Hamadi, K. Kasan, S. C. Emek, Y. Dilgin, H.-E. Akerlund, P.-A. Albertsson, D. Leech and L. Gorton, ChemSusChem, 2015, 8, 990–993 CrossRef PubMed.
- D. Pankratov, G. Pankratov, T. P. Dyachkova, P. Falkman, H.-E. Akerlund, M. D. Toscano, Q. Chi and L. Gorton, ACS Energy Lett., 2017, 2, 2635–2639 CrossRef CAS.
- R. I. Pinhassi, D. Kallman, G. Saper, H. Dotan, A. Linkov, A. Kay, V. Liveanu, G. Schuster, N. Adir and A. Rothschild, Nature Commun., 2016, 7, 12552 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- C. E. Lubner, R. A. Grimme, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 404–414 CrossRef CAS PubMed.
- M. Ihara, H. Nishihara, K. L. Yoon, O. Lenz, B. Friedrich, H. Nakamoto, K. Kojima, D. Honma, T. Kamachi and I. Okura, Photochem. Photobiol., 2006, 82, 676–682 CrossRef CAS PubMed.
- C. E. Lubner, A. M. Applegate, P. Knorzer, A. Ganago, D. A. Bryant, T. Happe and J. H. Golbeck, Proc. Natl. Acad. Sci. USA, 2011, 108, 20988–20991 CrossRef CAS PubMed.
- I. Yacoby, S. Pochekailov, H. Toporik, M. L. Ghirardi, P. W. King and S. G. Zhang, Proc. Natl. Acad. Sci. US., 2011, 108, 20988–20991 CrossRef PubMed.
- R. A. Grimme, C. E. Lubner, D. A. Bryant and J. H. Golbeck, J. Am. Chem. Soc., 2008, 130, 6308–6309 CrossRef CAS PubMed.
- I. Iwuchukwu, M. D. Vaughn, N. Myers, H. O'Neill, P. Frymier and B. D. Bruce, Nat. Nanotechnol., 2010, 5, 73–79 CrossRef CAS PubMed.
- L. M. Utschig, N. M. Dimitrijevic, O. G. Poluektov, S. D. Chemerisov, K. L. Mulfort and D. M. Tiede, J. Phys. Chem. Lett., 2011, 2, 236–241 CrossRef CAS.
- L. M. Utschig, S. C. Silver, K. L. Mulfort and D. M. Tiede, J. Am. Chem. Soc., 2011, 133, 16334–16337 CrossRef CAS PubMed.
- S. C. Silver, J. Niklas, P. W. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, J. Am. Chem. Soc., 2013, 135, 13246–13249 CrossRef CAS PubMed.
- L. M. Utschig, S. R. Soltau and D. M. Tiede, Curr. Opin. Chem. Bio., 2015, 25, 1–8 CrossRef CAS PubMed.
- P. Setif, Biochim. Biophys. Acta, 2001, 1507, 161–179 CrossRef CAS.
- I. Grotjohann and P. Fromme, Photosynth. Res., 2005, 85, 51–72 CrossRef CAS PubMed.
- E. Greenbaum, Science, 1985, 230, 1373–1375 CrossRef CAS PubMed.
- E. Greenbaum, J. Phys. Chem., 1988, 92, 4571–4574 CrossRef CAS.
- B. R. Evans, H. M. O'Neill, S. A. Hutchens, B. D. Bruce and E. Greenbaum, Nano. Lett., 2004, 4, 1815–1819 CrossRef CAS.
- D. A. Berthold, G. T. Babcock and C. F. Yocum, FEBS Lett., 1981, 134, 231–234 CrossRef CAS.
- S. Chen and K. Kimura, J. Phys. Chem. B, 2001, 105, 5397 CrossRef CAS.
- Govindjee and R. Govindjee, Bioenergetics of Photosynthesis, Academic, New York, 1975 Search PubMed.
- H. Kirchhoff, U. Mukherjee and H.-J. Galla, Biochemistry, 2002, 41, 4872–4882 CrossRef CAS PubMed.
- J. W. Lee, C. V. Tevault, S. L. Balankinship, R. T. Collins and E. Greenbaum, Energy & Fuels, 1994, 8, 770–773 CrossRef CAS.
- J. W. Lee, R. T. Collins and E. Greenbaum, J. Phys. Chem. B, 1998, 102, 2095–2100 CrossRef CAS.
- J. W. Lee and E. Greenbaum, J. Phys.Chem. B, 2004, 108, 3935–3939 CrossRef CAS.
- L. M. Utschig, L. X. Chen and O. G. Poluektov, Biochemistry, 2008, 47, 3671–3676 CrossRef CAS PubMed.
- R. MacColl, J. Struct. Biol., 1998, 124, 311–334 CrossRef CAS PubMed.
- M. Medina, Febs J., 2009, 276, 3942–3958 CrossRef CAS PubMed.
- L. M. Utschig, D. M. Tiede and O. G. Poluektov, Biochemistry, 2010, 49, 9682–9684 CrossRef CAS PubMed.
- I. R. Vassiliev, M. L. Antonkine and J. H. Golbeck, Biochim. Biophys. Acta, 2001, 1507, 139–160 CrossRef CAS.
- W. Lubitz, H. Orgata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- J. A. Birrell, O. Rudiger, E. J. Reijerse and W. Lubitz, Joule, 2017, 1, 61–76 CrossRef CAS.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. white, O. A. Zadvornyy, J. B. Broderick and B. Bothner, et al. , J. Am. Chem. Soc, 2015, 137, 1809–1816 CrossRef CAS PubMed.
- A. Volgusheva, S. Styring and F. Mamedov, Proc. Natl. Acad. Sci. USA, 2013, 110, 7223–7228 CrossRef CAS PubMed.
- A. Dubini and M. L. Ghirardi, Photosyn. Res., 2015, 123, 241–253 CrossRef CAS PubMed.
- S. Kosourov, M. Jokel, E.-M. Aro and Y. Allahverdiyeva, Energy Environ. Sci., 2018, 11, 1431–1436 RSC.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. USA, 2009, 106, 20681–20686 CrossRef PubMed.
- J. Kalms, A. Schmidt, S. Frielingsdorf, T. Utesch, G. Gotthard, D. von Stetten, P. van der Linden, A. Royant, M. A. Mroginski, P. Carpentier, O. Lenz and P. Scheere, Proc. Natl. Acad. Sci. USA, 2018, 115, 2229–2237 CrossRef PubMed.
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauss, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- D. W. Wakerley, M. A. Gross and E. Reisner, Chem. Comm., 2014, 50, 15995–15998 RSC.
- S. R. Soltau, P. D. Dahlberg, J. Niklas, O. Poluektov, K. L. Mulfort and L. M. Utschig, Chem. Sci., 2016, 7, 7068–7078 RSC.
- H. K. Lichtenthaler, Meth. Enzymol., 1987, 148, 350–382 CrossRef CAS.
- W. C. Trogler, R. C. Stewart, L. A. Epps and L. G. Marzilli, Inorg. Chem., 1974, 13, 1564–1570 CrossRef CAS.
- G. Markl, G. Y. Jin and C. Schoerner, Tetrahedron Lett., 1980, 21, 1409–1412 CrossRef.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- H. L. Crespi, J. R. Norris, J. P. Bays and J. J. Katz, Ann. N. Y. Acad. Sci., 1973, 800–815 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary experimental procedures, additional EPR spectra, and time traces of photocatalysis. See DOI: 10.1039/c8sc02841a |
| ‡ Present address: S. R. S.: Department of Chemical Sciences, Bridgewater State University, Bridgewater, MA 02325, USA. |
| This journal is © The Royal Society of Chemistry 2018 |