
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of a PCcarbeneP cobalt(I) complex: the missing link in the cobalt PXP pincer series (X = B, C, N)†
Simon
Sung
 a,
Qingyang
Wang
a,
Tobias
Krämer
b and
Rowan D.
Young
*a
a,
Qingyang
Wang
a,
Tobias
Krämer
b and
Rowan D.
Young
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: rowan.young@nus.edu.sg
bDepartment of Chemistry, Maynooth University, Maynooth, Ireland
First published on 6th September 2018
Abstract
We report the first example of a cobalt PCcarbeneP pincer complex (1) featuring a central alkylidene carbon donor accessed through the dehydration of an alcoholic POP proligand. Complex 1 shares bonding similarities with cobalt PBP and PNP pincer complexes where the donor atom engages in π-bonding with the cobalt centre, and thus completes the PXP (X = B, C, N) pincer ligand series for cobalt (for X donors that partake in M–L π-bonding). As compared to PBP and PNP pincer complexes, which are known to be good hydride and proton acceptors (respectively), complex 1 is found to be an effective hydrogen atom acceptor. Complex 1 partakes in cooperative ligand reactivity, engaging in several small molecule activations with styrene, bromine, carbon disulphide, phenyl acetylene, acetonitrile, hydrogen, benzaldehyde and water (through microreversibility). The mechanism for the formation of complex 1 is studied through the isolation and computational analysis of key intermediates. The formation of 1 is found to avoid C–H activation of the proligand, and instead proceeds through a combination of O–H activation, hydrogen atom transfer, β-hydride elimination and hydrogen activation processes.
Introduction
Pincer ligands (i.e. tridentate meridional ligands) form a fundamental scaffold upon which many transition metal complexes are based. Pincer ligands have been pivotal in the development of C–C, C–H, C–O, N–H and O–H activation chemistries (inter alia), and are increasingly being used as supports in transition metal catalysis.1 As chemists place increasing importance on base metal catalysis in developing sustainable, cheap and non-toxic catalysts, PXP (X = B or N) pincer systems that are complicit in metal–ligand cooperativity have emerged as versatile supporting ligands for a wide array of chemical transformations.In particular, recent developments of cobalt PBP and PNP pincer systems have allowed advances in olefin hydrogenation,2 ester hydrogenation,3 ketone alkylation,4 carbon dioxide hydrogenation,5 transfer hydrogenation of alkynes, olefins and nitriles,6 amine alkylation,7 dinitrogen reduction,8 and C–H borylation9 (inter alia). The unique activity of these systems is in large part due to π-interaction between the cobalt metal centre and the N/B donor. This interaction is possible through the sp2 hybridisation of the N/B donor atoms observed in molecular structures of cobalt PNP and PBP complexes.
Although there are known examples of sp2 hybridised carbon PCP systems for cobalt, up to now they are restricted to carbon donors housed in aryl systems.10 Such complexes are poor analogues of PBP and PNP systems, in that they interact predominantly with the cobalt centre through a C–Co σ-coordination. In contrast, PBP and PNP systems are able to act as π-acids and π-bases respectively, somewhat akin to Fischer and Schrock carbene analogues (Fig. 1). As such, PCP alkylidene (PCcarbeneP) systems, with a central carbene donor, are perhaps the most suitable description for an intermediary between PBP and PNP pincer systems, and they would prove valuable ligands in exploring the reactivity spectrum of PXP ligands on cobalt.11 Yet examples of PCcarbeneP ligands for first row transition metals are largely unknown, except for a recent report concerning PCcarbeneP nickel complexes by Piers.12
 | ||
| Fig. 1 Cobalt PXP pincer ligand architectures involving Co–X π-interaction based on boron, carbon and nitrogen central X donors. PBP and PNP systems are widely reported (see ref. 2–9 for examples). | ||
The near absence of first row PCcarbeneP complexes can primarily be attributed to the restricted synthetic methodology available to access such systems. Early PCcarbeneP systems, based on aliphatic backbones, were reported by Shaw and then Gusev.13 Ozerov, and later Piers, introduced β-hydride elimination resistant systems.14 All of these systems are reliant upon double C–H activation, and are thus restricted to metals capable of facile C–H activation (i.e. noble metals). Indeed, prior reports of the installation of methylene bridged bisphosphino ligands on electron rich cobalt centres failed to provide evidence of double C–H activation.15 Piers, and later Iluc, developed double dehydrohalogenation methodologies for group 10 metal dihalides, which proceeded via electrophilic C–H activation then hydrohalide abstraction with a suitably strong base.12a,16 Thus, over the previous 40 years, only Os, Ru, Ir, Rh, Pd and Ni PCcarbeneP systems have been reported.
Metals supported by PCcarbeneP ligands have shown the ability to partake in a variety of ligand cooperativity reactions,17 including 1,2-bond addition,12a,13a,14b [2 + 2] cycloaddition,18 Frustrated Lewis pair type reactivity,16b radical reactivity,16c,d redox reactivity,14c,16e chalcogen abstraction14f,16f and ligand directed bond-activation.19 Thus, a variety of bond activations, including H–H, C–H, N–H, O–H and N–O bonds (inter alia), have demonstrated the potential of these systems for future catalytic purposes. However, routine access to these ligands continues to limit progress in the field, especially in the area of PCcarbeneP base metal complex reactivity.
Recently while exploring the mechanism of transfer hydrogenation mediated by acidic rhodium hydrides,20 we realized that facile access to PCcarbeneP systems could be achieved directly via dehydration of PCP α-hydroxyalkyl ligands.18,19 As such ligands can be generated by carbonyl insertion into metal hydrides, C–H activation of the proligand can be avoided completely.
Herein, we report the extension of this method to cobalt PCcarbeneP systems. The mechanism of formation is interrogated to distinguish between possible C–H and O–H activation pathways, and the PCcarbeneP system is compared to reported PBP and PNP cobalt systems to evaluate the electronic properties of the PCcarbeneP ligand on cobalt. The central alkylidene donor of the PCcarbeneP ligand enables a wide array of reactivity with cobalt including C–H bond activation, C–C bond activation, C–C bond formation, C–N bond activation, H–H bond activation and hydrogen atom transfer.
Results and discussion
Cobalt(I) PCcarbeneP complex 1 was generated directly from the reaction of alcoholic proligand A with [Co(PMe3)4][BArF4] {ArF = 3,5-(CF3)2C6H3} in a one-step process (Scheme 1). Previously, we accessed a related cationic rhodium PCcarbeneP system via a two-step synthesis involving C–H activation of A by [RhCl(PPh3)(COD)], then salt metathesis with Na[BArF4] that induced ligand dehydration.18 | ||
| Scheme 1 Synthesis of the cationic PCcarbeneP cobalt complex 1. Compound 1 was obtained in 47% isolated yield. | ||
In contrast to the reaction of A with rhodium as described above, formation of 1 required overnight heating. Upon cooling of the reaction solution, compound 1 crystallizes in 47% yield. A molecular structure of 1 obtained from an X-ray diffraction study of a crystal sourced directly from the reaction solution revealed the alkylidene linkage within the PCcarbeneP ligand (Fig. 2). The Co1–C1 distance of 1.892(3) Å in 1 falls in the range of previously reported cobalt Fischer carbenes, supporting a Co(I) assignment,21 and comparison of M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distances between 1 and Piers' Ni(II) PCcarbeneP examples are within experimental error (6σ).12a The angle sum of atoms subtending the carbene donor, C1, is 359.7(3)°, indicative of an sp2 carbon hybridisation. A 13C NMR signal was observed at 216.6 ppm (ddt, 2JPC = 33.5 (d), 33.5 (d), 18.2 (t) Hz), much further downfield than Piers' Ni(II) PCcarbeneP complex (δC 181.8). The majority of non-heteroatomic stabilised cobalt alkylidene complexes reported to date are paramagnetic,22 precluding suitable comparison of the carbenic 13C NMR resonance within this class of compounds, however, unsurprisingly the 13C NMR resonance of 1 is upfield of reported oxygen and nitrogen stabilised Fischer carbene examples.21a,c
C distances between 1 and Piers' Ni(II) PCcarbeneP examples are within experimental error (6σ).12a The angle sum of atoms subtending the carbene donor, C1, is 359.7(3)°, indicative of an sp2 carbon hybridisation. A 13C NMR signal was observed at 216.6 ppm (ddt, 2JPC = 33.5 (d), 33.5 (d), 18.2 (t) Hz), much further downfield than Piers' Ni(II) PCcarbeneP complex (δC 181.8). The majority of non-heteroatomic stabilised cobalt alkylidene complexes reported to date are paramagnetic,22 precluding suitable comparison of the carbenic 13C NMR resonance within this class of compounds, however, unsurprisingly the 13C NMR resonance of 1 is upfield of reported oxygen and nitrogen stabilised Fischer carbene examples.21a,c
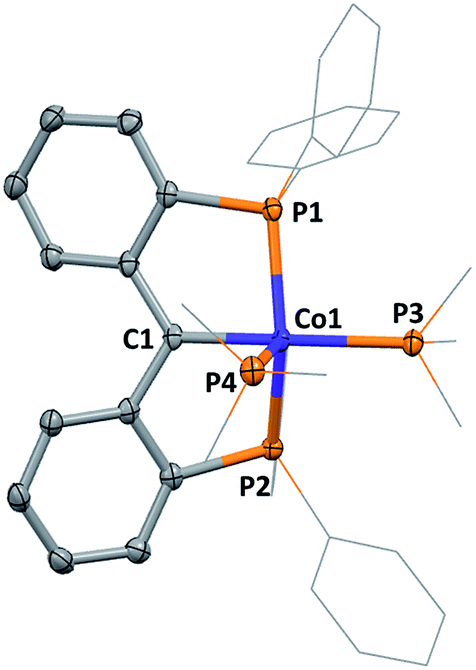 | ||
| Fig. 2 Molecular structure of 1. Hydrogen atoms and anion omitted, thermal ellipsoids shown at 50%. | ||
The coordination sphere of cobalt is completed by two PMe3 ligands, arranging themselves in axial and equatorial positions within the distorted square pyramidal geometry of the cobalt centre. The PMe3 ligand in the pseudo-trans position to C1 is elongated relative to the PMe3 in the axial position, with respective bond lengths of 2.254(1) Å (Co1–P3) and 2.224(1) Å (Co1–P4).
In CD2Cl2 solution, two broad signals of equal integration are observed in the 31P NMR spectrum of 1 at 52.8 and 2.2 ppm, corresponding to the PCcarbeneP and PMe3 phosphorus ligands respectively. Cooling a sample of 1 in CD2Cl2 to 198 K resolves the PMe3 phosphorus signals into a triplet of doublets at −9.2 ppm (2Jpp = 58.5 (t), 22.3 (d) Hz) and a triplet of doublets at 15.0 ppm (2JPP = 31.0 (t), 25.6 (d) Hz). The PCcarbeneP ligand's phosphorus signal at 198 K is observed as a doublet of doublets at 54.7 ppm (2JPP = 58.5, 31.0 Hz).
Direct comparison of 1 to the isoelectronic PNP cobalt(I) complex, [PNPCo(PMe3)2] (PNP = κ3-P′,N,P′′-{(Ph2PC6H4)2N}) reported by Sun (Fig. 3), demonstrates the structural similarities between PNP and PCcarbeneP ligands.23 For example, Sun's PNP complex displays an angle sum of 359.7(5)° around the nitrogen donor atom and a Co–N bond length of 2.026(3) Å. The carbene ligand in 1 also displays an enhanced trans influence compared to its PNP congener with a longer Co–PMe3 bond length trans to the pincer central donor of 2.254(1) Å {cf. 2.163(1) Å}.
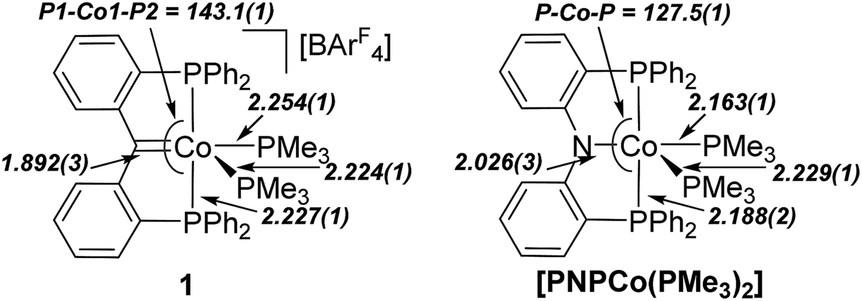 | ||
| Fig. 3 Comparison of the molecular metrics of the congeners 1 (left) and [(κ3-P′,N,P′′-{(Ph2PC6H4)2N})Co(PMe3)2]22 (right). Selected bond distances (Å) and angles (°) displayed. | ||
The PBP congener of 1 is not reported, thus cannot be directly compared, however, it is noted that isoelectronic iridium PBP and PCcarbeneP complexes {viz. [(κ3-P′,B,P′′-{(iPr2PC6H4)2B})Ir(CO)2] and [(κ3-P′,C,P′′-{(iPr2PC6H4)2C})Ir(CO)2]+}14e,24 have been reported by Ozerov (PBP) and Piers (PCcarbeneP), and that the trans influence of the PCcarbeneP ligand is greater than that of the PBP ligand in these systems. Comparison of the Co1–C1 bond length in 1 to a Co–B distance of 1.946(1) Å in Peters' PBP cobalt(I) complex [PBPCo(N2)]2a {PBP = κ3-P,B,P′-B(NCH2PtBu2)2C6H4}, indicates enhanced higher bonding in the carbon–cobalt bond of the PCcarbeneP ligand on cobalt(I) as compared to boron and nitrogen interactions in similar PBP and PNP ligand scaffolds.
Calculated iso-surfaces for the HOMO-1 and LUMO-1 orbitals of 1 show the LUMO to be predominantly located on the carbene ligand (Fig. 4). This observation is in agreement with the Fischer type alkylidene reactivity observed for compound 1 with a range of nucleophiles (vide infra).
 | ||
| Fig. 4 Calculated structure of 1 showing HOMO (left) and LUMO (right) iso-surface (0.05 a.u. cutoff). | ||
Addition of one equivalent of B(C6F5)3 to a solution of 1 results in sequestration of one of the PMe3 ligands as [Me3P–B(C6F3)3]25 and formation of complex 2 (Fig. 5). The use of less acidic BPh3 was also found to generate 2, although with a much lower conversion than B(C6F5)3. The stability of complex 2 was found to be solvent dependent, with the coordinating solvent, MeCN, stabilizing 2 in solution as [2·MeCN]. Indeed, the dissolution of 1 in MeCN generates an equilibrium between 1 and [2·MeCN]. In non-coordinating solvents, 2 was found to decompose over time, excluding the possibility of obtaining a molecular structure of the four-coordinate PCcarbeneP cobalt complex, however, the identity of 2 was confirmed by the addition of an equivalent of PMe3, which reformed 1 quantitatively.
 | ||
| Fig. 5 Products from reactivity of 1 and 2 with various small molecules. Phenyl and methyl hydrogen atoms, and [BArF4] anions omitted for simplification. Thermal ellipsoids shown at 50%, selected bond distances (Å) and angles (°) displayed. | ||
Small molecule activation
The reactivity of 1 and 2 was explored with a number of reagents and is summarized in Fig. 5. As stated above, cobalt(I) PCcarbeneP complexes may be described within the cobalt(I) PBP-PNP spectrum (Fig. 1). It is noted that cobalt(I) PBP complexes have been shown to be good hydride acceptors, and cobalt(I) PNP complexes have been utilized as proton acceptors.2a,26 Considering this, we tested the reactivity of 1 and 2 with various hydrogen sources.When 1 is treated with 1 atm of H2 in PhF, reduction of the carbene anchor point occurs, presumably to generate [Co{κ2-P,P′-(Ph2PC6H4)2CH2}(PMe3)2], which is unstable under these conditions and results in the observation of liberated bisphosphine, (Ph2PC6H4)2CH2, (as confirmed by 1H, 31P, 13C NMR spectroscopy and ESI-MS).27 When identical reaction conditions were applied using MeCN solvent {i.e. with in situ generated [2·MeCN]}, apart from the reduced pincer ligand, a mass fragment corresponding to MeCN being incorporated into the reduced ligand was observed in the ESI-MS mass spectrum of the crude reaction mixture. Addition of hydrogen to 1 or [2·MeCN] in MeCN in a more controlled manner, via the use of NaH, resulted in the generation of 3 in 52% yield, featuring a coordinated enamine motif (Fig. 5). In non-coordinating solvent (e.g. PhF), 1 failed to react with NaH, even after prolonged heating.
Cobalt NNP and CCC pincer complexes have recently been shown to be efficient catalysts towards nitrile reduction. The isolation of 3, may represent the interception of an intermediate in this process arising from 1,2-insertion of MeCN into a Co–H bond, and may shed light onto the mechanism of reported cobalt pincer complex catalysed nitrile reductions.28
When 1 was treated with an equivalent of [H(OEt2)2][BArF4] in PhF, [HPMe3][BArF4] and 2 were generated. In contrast to reaction with B(C6F5)3 (vide supra), this reaction only proceeded to 22% conversion. Although 1 failed to incorporate proton or hydride sources directly, it was found to act as a hydrogen atom acceptor in hydrogen atom transfer (HAT) reactions. For example, reaction of [CoH(N2)(PPh3)3] or HSnBu3 with 1 in PhF resulted in complete consumption of 1 and generation of 4, as determined by ESI-MS and EPR spectroscopy. Compound 4 features a PCsp3P pincer ligand where the carbene linkage present in 1 has been transformed into an alkyl donor. Although the incorporated hydrogen was spectrometrically and crystallographically observed (i.e. located in a Fourier difference map), the sp3 nature of the carbon linkage is evident from the angle sum of the non-hydrogen atoms sub-tending the carbon donor (C1) {angle Σ = 333.4(4)°}, deviating greatly from sp2 geometry. The ability of 1 to act as a potent hydrogen atom acceptor is in sharp contrast to reactivity of rhodium and iridium PCcarbeneP complexes, and is likely a result of the stability of cobalt(II).
In a related reaction, 1 was able to homolytically cleave elemental bromine to generate 5, featuring a cobalt(II) centre. In this instance, the bromine atom is transferred to the cobalt centre (cf. the alkylidene carbon site) and a PMe3 ligand migrates to the alkylidene carbon position to again generate a PCsp3P type ligand. A mixture of compounds 4 and 5 could be generated through the reaction of hydrogen bromide with 1 (as indicated by ESI-MS), however, no reaction between 1 and hydrogen chloride was observed. Hydrogen bromide is known to generate hydrogen and bromine radicals (through homolytic bond cleavage), and the contrast between hydrogen chloride and hydrogen bromide reactivities supports the formation of 4 and 5via one-electron oxidative processes.
Although complex 2 was found to be stable in coordinating solvents, its enhanced reactivity (cf. compound 1) in non-coordinating solvents was revealed in the presence of small donor molecules. For example, generation of 2 (by addition of B(C6F5)3 to 1) in the presence of styrene led to the formation of complex 6 in PhF. No reaction between 1 and styrene was observed in the absence of added Lewis acid. Periodically obtained ESI-MS spectrometric data from the reaction mixture suggest that β-hydride elimination of an initial intermediate metallabutacycle generates a Co(III) allyl hydride, which then slowly loses hydrogen to generate 6. Olefin metathesis with cobalt alkylidenes is rare, but reported for fluorinated carbene groups.29 Indeed, isolated perfluorometallabutacycles (arising from [2 + 2] cycloaddition) have been postulated to form intermediate cobalt allyl complexes upon β-fluoride activation.29a
Similarly, although 1 underwent no reaction with benzaldehyde, reaction of 2 {generated in situ from 1 and B(C6F5)3} with benzaldehyde led to the formation of 7. Compound 7 is another example of the ability of the PCcarbeneP ligand to enhance the C–H activating capability of cobalt. In the formation of 7, C–H activation likely proceeds via β-hydride elimination after the formation of an oxabutacycle (in a similar manner to the formation of 6), rather than direct concerted C–H cleavage of benzaldehyde, as is common for noble metals. The formation of metallaoxacyclobutanes is an important step in epoxide carbonylations and carboxylations, epoxide isomerisations, epoxide reductive couplings, and olefin epoxidation (inter alia).30 Compound 7 features a coordinated η3-C,C′,O-enolato motif, arising from incorporation of benzoyl into the pincer ligand. The C2–O1 bond distance of 1.325(9) Å, and C1–C2 bond distance of 1.455(10) Å suggest that there is some multiple bond character in both the C–O and C–C bonds of the enolate ligand.
Compound 1 was found to activate a CS bond in carbon disulfide in PhF to generate 8a, with an equivalent of concomitant SPMe3 formed during the reaction. Although no other products were identified, presumably the sacrificial PMe3 (forming SPMe3) originated from compound 1. Thus, this reaction was optimized through the addition of an extra equivalent of PMe3, resulting in an isolated yield of 91%. Interestingly, the addition of an equivalent of PPh3 as a sacrificial phosphine (in place of PMe3) led to compound 8b, where PPh3 ligates the cobalt centre, and SPMe3 is again generated as a reaction by-product. In contrast to similar reactivity reported for rhodium PCcarbeneP complexes, where (thio)carbonyl attack of the PCcarbeneP alkylidene forms a central η2-(thio)ketene,14e,18 compound 7 features a coordinated thioenolate that has presumably been generated by the attack of an η2-thioketene by an auxiliary PMe3 ligand. Generation of the thioenolate ligand demonstrates greater bond activation by cobalt as compared to our previously reported activation of CS2 with a PCcarbeneP rhodium complex, with respective C–S bond lengths of 1.743(6) Å and 1.582(4) Å in the cobalt thioenolate and rhodium thioketene complexes. Thiocarbonyl chemistry has been explored with cobalt, but is difficult to access directly from CS2 (c.f. Ir and Rh thiocarbonyl congeners).31 To the best of our knowledge η3-thioenolates of cobalt are unreported.
In a similar manner to that reported for rhodium PCcarbeneP complexes,181 was found to react with phenyl acetylene at room temperature to generate 9, where the acetylene motif has combined with the carbene carbon atom to generate an allylic ligand, and one of the pincer phosphino donors has migrated to the terminal allylic carbon position. Similar phosphine migration has been reported between phosphines and metal vinyliums,32 and reports exist of phosphine nucleophilic attack on metallacyclobutadienes (formed from alkylidyne/alkyne [2 + 2] addition),33 but the combination of such reactivity is unprecedented for alkylidene moieties, apart from a previous example reported earlier by us.18 The reaction pathway for the formation of the resulting phosphino-allyl ligand present in 9 is subject to a more in-depth, ongoing investigation by our group.
In an analogous manner, the nitrile moiety of acetonitrile was found to insert across the carbon and one of the phosphorus donors of the PCcarbeneP ligand in 1 to generate compound 10. As stated above, [2·MeCN] is stable in solution at room temperature, and thus the formation of 10 required heating of compound 1 at 80 °C for 24 hours in acetonitrile solvent. Complex 10 displays many structural similarities to 9, and if it forms via a metallabutacyle intermediate (i.e. [2 + 2] addition between a nitrile and alkylidene), it would mirror the [2 + 2] addition chemistry observed in recently reported alkyne/nitrile metathesis catalysts.34
The generation of compounds 3–10 likely proceeds via ligand cooperative homolysis, [2 + 2] cycloaddition or migratory insertion. However, examples of isolable 1,2-addition products could not be generated with 1. This stands in stark contrast to other reported PCcarbeneP systems capable of ligand cooperativity that tend to activate substrates via 1,2-addition reactions (Scheme 2, top).12a,b,14b,16a,d,h,i,17 Indeed, compound 1 was found to react with neither hydrochloric acid (vide infra) nor triethyl hydrosilane (both reagents are prone to oxidative addition), suggesting that 1,2-additions may be less favoured for this system. Despite the inability of 1 or 2 to partake in 1,2-addition reactions, challenging room temperature C–H activation chemistry of benzaldehyde and styrene was found to proceed, via metallacycle formation and subsequent β-hydrogen elimination (Scheme 2, bottom), representing a new ligand cooperative pathway for bond activation in PCcarbeneP complexes.
 | ||
| Scheme 2 (Top) Previously reported ligand cooperative C–H activation in PCcarbeneP complexes via 1,2-addition.11a,b,13b,15,16a,d,h,i (bottom) PCcarbeneP ligand cooperative C–H activation of styrene and benzaldehyde via β-hydrogen elimination (this report). | ||
Mechanistic studies
The reaction components and conditions chosen for the generation of 1 presumed that cobalt might undergo a single C–H activation with proligand A, followed by dehydration to generate 1. In reality we found the reaction pathway for the generation of 1 to be much more complex than anticipated and to completely avoid C–H activation (Scheme 3). As stated above, access to 1 required overnight heating. However, it was found that at room temperature A very slowly displaced two equivalents of PMe3 in [Co(PMe3)4][BArF4], and led to the formation of III over the period of two weeks. Compound III could also be generated in a timelier manner via one electron oxidation of the cobalt(0) complex [CoA(PMe3)2] using [Fc][BArF4] as an oxidant (see ESI†). | ||
| Scheme 3 Mechanistic evidence for the formation of 1. [BArF4] anions and hydrogen atoms (except H11 on III) omitted for simplification. Thermal ellipsoids drawn at 50%, selected bond distances (Å) and angles (°) displayed. | ||
Complex III was characterized via X-ray diffractrometry, which revealed a molecular geometry best described as distorted square pyramidal around the cobalt centre (Scheme 3). Compound III was found to be paramagnetic (S = 1/2), indicating a low-spin cobalt(II) centre. A Fourier difference map failed to reveal any hydrogen atoms in the coordination sphere of III, and ESI-MS spectrometric data supported the loss of a hydrogen atom and two trimethyl phosphine molecules from the constituting reaction starting components (that generate III). Cobalt phosphorus and cobalt oxygen bond distances supported the assignment of III as a cobalt(II) complex, with comparison to DFT optimized geometries of postulated Co(I) (I), Co(III) (II) and Co(II) (III) complexes concurring that III exists as a Co(II) alkoxide (see ESI†).
Complex III was confirmed as an intermediate enroute to 1 by heating isolated samples of complex III in toluene at 100 °C overnight. Under these conditions, a reduced yield of 1 was obtained (33%).
Competing O–H and C–H activation pathways have been reported for similar iridium complexes resulting in formation of PCcarbeneP complexes. However, reactions of [Co(PMe3)4][BArF4] with oxygen protected variations of the alcohol proligand, AMe (methyl protected) and ATIPS (triisopropylsilyl protected), failed to react after heating at 100 °C for 14 hours or produce any trace of III or 1 (i.e. no C–H activation was observed). Thus, it is likely that III is generated via O–H activation (forming II) followed by loss of half an equivalent of hydrogen.
Interestingly, the (non-coordinated) keto bridged diphosphine A-H2 was observed in solution during reactions that generated 1 from A and [Co(PMe3)4][BArF4]. Thus, it was hypothesized that III re-enters a Co(I)/Co(III) pathway via β-hydride elimination (producing IV) and loss of another half equivalent of H2, generating an η2-keto POP cobalt(I) complex (V). Indeed, precedence for the formation of PCcarbeneP ligands from hydrogen and η2-keto ligands has been reported in iridium systems.14f,19
To test this hypothesis, complex V was independently generated by addition of keto proligand A-H2 to [Co(PMe3)4][BArF4] in toluene at room temperature (Scheme 3). Compound V exists in equilibrium with its parent reagents, thus repeated dissolution and evaporation of the mixture was required to remove liberated PMe3 and drive the reaction towards V. A molecular structure of V (Scheme 3) confirmed the κ3-P,(η2-C,O),P′ coordination mode of the keto ligand. The ketone C1–O1 distance of 1.348(5) Å also suggests significant retrodonation from the cobalt centre to the η2 carbonyl group {C–O = 1.213(3) Å in proligand}. Indeed, the C1–O1 bond distance is much greater than those reported by Moret for the related η2-carbonyl nickel(0) and nickel(I) complexes, [Ni(A-H2)(PPh3)] {C–O = 1.310(2) Å} and [NiCl(A-H2)] {C–O = 1.330(3) Å}.35 NBO analyses of V and Moret's nickel complexes suggest the longer C–O distance in V is a result of significantly more retrodonation to a CO π* orbital (see ESI†).
Although 1 was not directly detected by 31P NMR after addition of H2 to V at 100C°, its formation was inferred by further reactivity with H2 to generate the liberated reduced ligand (Ph2PC6H4)2CH2 (vide supra). Additionally, compound V was observed to be formed and consumed (by 31P NMR spectroscopy) when heating isolated samples of III in toluene at 100C°, which ultimately generated compound 1.
The observation of any intermediates between V and 1 could not be accomplished. However, based on previously reported iridium and rhodium systems,14f,18,19 the addition of hydrogen to Vvia oxidative addition (forming VI) and subsequent hydride migration to generate an α-hydroxyalkyl complex (VII) prior to elimination of water to form 1 would constitute a valid reaction pathway.
In support of a HAT pathway mediating a Co(I)/Co(II) pathway, complex 4 (Fig. 5) was subsequently isolated from the supernatant of a reaction forming 1 from A and [Co(PMe3)4][BArF4]. Given that 1 has been shown to act as a good hydrogen atom acceptor from cobalt hydrides {viz. [CoH(N2)(PPh3)3]}, it is likely that in situ generated 1 may also react with cobalt hydrides II or IV to form III, accounting for the relatively moderate yields of 1 isolated after the reaction is completed (as outlined in Scheme 1).
Conclusions
We have demonstrated facile access to a PCcarbeneP cobalt complex (1) from an alcoholic POP proligand (A). Access to PCcarbeneP base metal systems is rare and synthetically difficult using traditional synthetic methods.Complex 1 completes the triad of cobalt PXP pincer complexes featuring first-row main group X donors that form M–L π interactions (X = B, C, N). As such, complex 1 displays structural similarities to its PBP and PNP analogues. In line with this analogy, PCP acts as a good hydrogen atom acceptor, whereas PBP and PNP cobalt complexes are known to be good hydride and proton acceptors (respectively).
The enhanced reactivity of 1, induced by metal–alkylidene ligand cooperativity, enables the activation of a number of small molecules. In contrast to previously reported PCcarbeneP complexes, 1 does not show a tendency to partake in 1,2-addition chemistry, but is capable of homolysis, migratory insertion reactions (enabling the formation of C–C bonds), and [2 + 2] cycloaddition (allowing novel ligand cooperative C–H activation). The availability of a stable cobalt(II) oxidation state may further enhance the reactivity of 1, with compounds 4, 5, 6 and 7 all examples of compound 1 accessing a cobalt(II) oxidation state.
The formation of 1 is found to completely avoid C–H activation, and instead proceeds via O–H activation of alcoholic proligand A. Intermediate III suggests that a cobalt(I/II/III) reaction pathway exists meditated by molecular hydrogen. The isolation and subsequent reactivity of compound V implies insertion of keto ligand A-H2 into a Co–H bond to form an α-hydroxyalkyl intermediate may represent a valid reaction pathway in forming 1.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National University of Singapore and the Singapore Ministry of Education for financial support WBS R-143-000-666-114, R-143-000-697-114 and R-143-000-A05-112. TK wishes to acknowledge the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.Notes and references
- (a) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Topics in Organometallic Chemistry, Springer, Berlin, vol. 40, 2013 Search PubMed; (b) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959 RSC.
- (a) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310 CrossRef PubMed; (b) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672 CrossRef PubMed.
- J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 7, 3735 CrossRef.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080 CrossRef PubMed.
- H. Ge, Y. Jing and X. Yang, Inorg. Chem., 2016, 55, 12179 CrossRef PubMed.
- (a) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588 CrossRef PubMed; (b) G. Zhang, Z. Yina and J. Tana, RSC Adv., 2016, 6, 22419 RSC; (c) Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653 CrossRef PubMed.
- (a) Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546 CrossRef; (b) G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300 CrossRef PubMed.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291 CrossRef PubMed.
- B. A. Schaefer, G. W. Margulieux, B. L. Small and P. J. Chirik, Organometallics, 2015, 34, 1307 CrossRef.
- (a) Z. Lian, G. Xua and X. Lia, Acta Crystallogr., 2010, E66, m636 Search PubMed; (b) T. J. Hebden, A. J. S. John, D. G. Gusev, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, Angew. Chem., Int. Ed., 2011, 50, 1873 CrossRef PubMed; (c) M. A. Kent, C. H. Woodall, M. F. Haddow, C. L. McMullin, P. G. Pringle and D. F. Wass, Organometallics, 2014, 33, 5686 CrossRef.
- The discussion in this paper excludes cobalt pincer complexes that contain NHC-type carbene donors due to the lack of π-interaction between cobalt and the carbene in such mesomerically stabilised donors. For examples of such pincers, see: (a) B. Liu, X. Liu, C. Chen, C. Chen and W. Chen, Organometallics, 2012, 31, 282 CrossRef; (b) C. F. Harris, C. S. Kuehner, J. Bacsa and J. D. Soper, Angew. Chem., Int. Ed., 2018, 57, 1311 CrossRef PubMed; (c) C. F. Harris, M. B. Bayless, N. P. van Leest, Q. J. Bruch, B. N. Livesay, J. Bacsa, K. I. Hardcastle, M. P. Shores, B. de Bruin and J. D. Soper, Inorg. Chem., 2017, 56, 12421 CrossRef PubMed; (d) A. A. Danopoulos, J. A. Wright, W. B. Motherwell and S. Ellwood, Organometallics, 2004, 23, 4807 CrossRef; (e) A. D. Ibrahim, K. Tokmic, M. R. Brennan, D. Kim, E. M. Matson, M. J. Nilges, J. A. Bertkea and A. R. Fout, Dalton Trans., 2016, 45, 9805 RSC.
- (a) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776 CrossRef PubMed; (b) E. A. LaPierre, W. E. Piers, D. M. Spasyuk and W. Bi, Chem. Commun., 2016, 52, 1361 RSC; (c) A Ferraquinone complex was reported by Milstein, but was not structurally characterised, and spectroscopic evidence suggested it retained a degree of aromatic character, see: A. Dauth, U. Gellrich, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 2799 CrossRef PubMed.
- (a) H. D. Empsall, E. M. Hyde, R. Markham, W. McDonald, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Chem. Commun., 1977, 589 RSC; (b) D. G. Gusev and A. J. Lough, Organometallics, 2002, 21, 2601 CrossRef.
- (a) W. Weng, S. Parkin and O. V. Ozerov, Organometallics, 2006, 25, 5345 CrossRef; (b) R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949 CrossRef; (c) R. J. Burford, W. E. Piers and M. Parvez, Eur. J. Inorg. Chem., 2013, 3826 CrossRef; (d) J. R. Logan, W. E. Piers, J. Borau-Garcia and D. M. Spasyuk, Organometallics, 2016, 35, 1279 CrossRef; (e) J. D. Smith, J. R. Logan, L. E. Doyle, R. J. Burford, S. Sugawara, C. Ohnita, Y. Yamamoto, W. E. Piers, D. M. Spasyuk and J. Borau-Garcia, Dalton Trans., 2016, 45, 12669 RSC; (f) L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187 CrossRef PubMed.
- H. Zhao, H. Sun, L. Wang and X. Li, Acta Chim. Sin., 2015, 73, 1307 CrossRef.
- (a) C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059 CrossRef; (b) P. Cui, C. C. Comanescu and V. M. Iluc, Chem. Commun., 2015, 51, 6206 RSC; (c) C. C. Comanescu, M. Vyushkova and V. M. Iluc, Chem. Sci., 2015, 6, 4570 RSC; (d) C. C. Comanescu and V. M. Iluc, Organometallics, 2015, 34, 4684 CrossRef; (e) P. Cui, M. R. Hoffbauer, M. Vyushkova and V. M. Iluc, Chem. Sci., 2016, 7, 4444 RSC; (f) P. Cui and V. M. Iluc, Chem. Sci., 2015, 6, 7343 RSC; (g) P. E. Rothstein, C. C. Comanescu and V. M. Iluc, Chem.–Eur. J., 2017, 23, 16948 CrossRef PubMed; (h) C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048 RSC; (i) C. C. Comanescu and V. M. Iluc, Polyhedron, 2018, 143, 176 CrossRef.
- K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540 RSC.
- S. Sung, T. Joachim, T. Krämer and R. D. Young, Organometallics, 2017, 36, 3117 CrossRef.
- S. Sung and R. D. Young, Dalton Trans., 2017, 46, 15407 RSC.
- S. Sung, J. K. Boon, J. J. C. Lee, N. A. Rajabi, S. A. Macgregor, T. Krämer and R. D. Young, Organometallics, 2017, 36, 1609 CrossRef.
- (a) C. C. Tso and A. R. Cutler, Polyhedron, 1993, 12, 149 CrossRef; (b) W. D. Wulff, S. R. Gilbertson and J. P. Springer, J. Am. Chem. Soc., 1986, 108, 520 CrossRef PubMed; (c) A. C. Filippou and E. Herdtweck, J. Organomet. Chem., 1988, 355, 437 CrossRef; (d) F. Car, G. Cerveau, E. Colomer, R. J. P. Corriu, J. C. Young, L. Ricard and R. Weiss, J. Organomet. Chem., 1979, 179, 215 CrossRef.
- (a) H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518 CrossRef PubMed; (b) S. L. Marquard, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2013, 135, 6018 CrossRef PubMed; (c) J. A. Bellow, S. A. Stoian, J. van Tol, A. Ozarowski, R. L. Lord and S. Groysman, J. Am. Chem. Soc., 2016, 138, 5531 CrossRef PubMed.
- H. Zhao, X. Li, S. Zhang and H. Sun, Z. Anorg. Allg. Chem., 2015, 641, 2435 CrossRef.
- W.-C. Shih and O. V. Ozerov, Organometallics, 2017, 36, 228 CrossRef.
- G. C. Welch, T. Holtrichter-Roessmann and D. W. Stephan, Inorg. Chem., 2008, 47, 1904 CrossRef PubMed.
- (a) G. Zhang and S. K. Hanson, Org. Lett., 2013, 15, 650 CrossRef PubMed; (b) S. S. Rozenel, R. Padilla, C. Camp and J. Arnold, Chem. Commun., 2014, 50, 2612 RSC.
- W. Lesueur, W. Solari and C. Floriani, Inorg. Chem., 1997, 36, 3354 CrossRef PubMed.
- (a) A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2015, 137, 8888 CrossRef PubMed; (b) K. Tokmic, B. J. Jackson, A. Salazar, T. J. Woods and A. R. Fout, J. Am. Chem. Soc., 2017, 139, 13554 CrossRef PubMed.
- (a) D. J. Harrison, G. M. Lee, M. C. Leclerc, I. Korobkov and R. T. Baker, J. Am. Chem. Soc., 2013, 135, 18296 CrossRef PubMed; (b) G. M. Lee, D. J. Harrison, I. Korobkov and R. T. Baker, Chem. Commun., 2014, 50, 1128 RSC; (c) D. J. Harrison, A. L. Daniels, I. Korobkov and R. T. Baker, Organometallics, 2015, 34, 5683 CrossRef; (d) M. Goswami, B. de Bruin and W. I. Dzik, Chem. Commun., 2017, 53, 4382 RSC.
- (a) C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153 CrossRef PubMed; (b) A. Dauth and J. A. Love, Chem. Rev., 2011, 111, 2010 CrossRef PubMed.
- (a) H. Werner and O. Kolb, Angew. Chem., Int. Ed., 1979, 18, 865 CrossRef; (b) C. Bianchini, A. Meli and G. Scapacci, Organometallics, 1983, 2, 1834 CrossRef.
- S. A. Fairhurst, D. L. Hughes, K. Marjani and R. L. Richards, J. Chem. Soc., Dalton Trans., 1998, 1899 RSC.
- (a) L. L. Padolik, J. C. Gallucci and A. Wojcicki, J. Am. Chem. Soc., 1993, 115, 9986 CrossRef; (b) V. Plantevin and A. Wojcicki, J. Organomet. Chem., 2004, 689, 2000 CrossRef.
- A. M. Geyer, R. L. Gdula, E. S. Wiedner and M. J. Johnson, J. Am. Chem. Soc., 2007, 129, 3800 CrossRef PubMed.
- B. W. H. Saes, D. G. A. Verhoeven, M. Lutz, R. J. M. Klein Gebbink and M.-E. Moret, Organometallics, 2015, 34, 2710 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1848387–1848397. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02782j |
| This journal is © The Royal Society of Chemistry 2018 |