
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitation of ERK1/2 inhibitor cellular target occupancies with a reversible slow off-rate probe†
Honorine
Lebraud
*a,
Olga
Surova
ab,
Aurélie
Courtin
a,
Marc
O'Reilly
a,
Chiara R.
Valenzano
a,
Pär
Nordlund
b and
Tom D.
Heightman
 *a
*a
aAstex Pharmaceuticals, 436 Cambridge Science Park, Cambridge, CB4 0QA, UK. E-mail: honorine.lebraud@astx.com; tom.heightman@astx.com
bDepartment of Oncology-Pathology, Karolinska Institute, CCK R8:01, Karolinska Hospital, 171 76 Stockholm, Sweden
First published on 17th September 2018
Abstract
Target engagement is a key concept in drug discovery and its direct measurement can provide a quantitative understanding of drug efficacy and/or toxicity. Failure to demonstrate target occupancy in relevant cells and tissues has been recognised as a contributing factor to the low success rate of clinical drug development. Several techniques are emerging to quantify target engagement in cells; however, in situ measurements remain challenging, mainly due to technical limitations. Here, we report the development of a non-covalent clickable probe, based on SCH772984, a slow off-rate ERK1/2 inhibitor, which enabled efficient pull down of ERK1/2 protein via click reaction with tetrazine tagged agarose beads. This was used in a competition setting to measure relative target occupancy by selected ERK1/2 inhibitors. As a reference we used the cellular thermal shift assay, a label-free biophysical assay relying solely on ligand-induced thermodynamic stabilization of proteins. To validate the EC50 values measured by both methods, the results were compared with IC50 data for the phosphorylation of RSK, a downstream substrate of ERK1/2 used as a functional biomarker of ERK1/2 inhibition. We showed that a slow off-rate reversible probe can be used to efficiently pull down cellular proteins, significantly extending the potential of the approach beyond the need for covalent or photoaffinity warheads.
Introduction
Attrition in drug discovery projects has been attributed to a lack of efficacy, insufficient mechanistic understanding, and safety issues.1,2 Poor target engagement may underlie both efficacy failure and toxicity in clinical trials, due to the need for high doses to meet the desired pharmacological profile.2 In target validation studies using chemical probes, demonstration of target engagement is critical in building confidence that the observed phenotype is a consequence of modulating the intended target. Hence, target occupancy has been described as one of the four pillars3 representing a bridge between target and pharmacology, and quantitative cellular target engagement is a key parameter for effective optimization in a drug discovery project. Several techniques have been developed to measure target occupancy in cells, which may involve structural modifications or isotopic labelling of the protein, or the drug, or both.4,5 Click chemistry has demonstrated potential for protein enrichment (pull down) experiments with endogenous proteins. By introducing a clickable group on a covalent ligand, bound proteins can be pulled out and protein levels quantified.6,7 This technique has been extended to non-covalent ligands by introducing a photo-reactive group in addition to the clickable handle, allowing covalent protein–ligand cross linking upon UV irradiation.8 The Inverse Electron Demand Diels Alder (IEDDA) has been used only rarely for protein enrichment experiments compared with other bioorthogonal reactions.9,10 Nevertheless, this reaction benefits from significant advantages,11 among which its fast kinetics and quantitative nature make it attractive for chemical biology applications, such as target identification, cellular localisation and target occupancy, using biotinylated and fluorescent reporters, respectively.7 Recently, a new concept was introduced which allows measurement of target engagement in cells using endogenous protein and untagged inhibitors. The Cellular Thermal Shift Assay (CETSA) relies on the increased protein stability induced by drug binding which, under elevated temperature, translates into a shift in the protein's thermal denaturation.12,13 When performed at fixed temperature, CETSA can be used to explore the effects of compound concentration on target occupancy in an experiment referred to as isothermal dose–response (ITDR) CETSA. Both CETSA and ITDR-CETSA have been previously employed to determine cellular uptake, cellular availability, and intracellular binding of endogenous target proteins in their native environment14 but also to elucidate the differences in efficacy observed among inhibitors of the same protein in clinical trials.13 A wide range of targets have been assessed via CETSA illustrating the broad applicability and robustness of the method.12The extracellular signal regulated kinases (ERK) 1/2 form a key signalling node in the MAPK pathway cascade, which is frequently dysregulated in cancer, for example by activating Ras mutations which are present in ∼30% of all human cancers.15 ERK1/2 are activated by MEK1/2 via phosphorylation, resulting in the activation of a number of cytosolic and nuclear substrates leading to cell proliferation and differentiation.16 Several ERK1/2 inhibitors have been developed and are currently under clinical investigation. Two classes have emerged with distinct pharmacology (Fig. 1A). ATP-competitive (Type I) ligands such as GDC-0994![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and LY321499618 inhibit ERK1/2 enzymatic activity, suppressing phosphorylation of downstream substrates, but concomitantly triggering feedback loops which result in partial re-activation of the RAF-MEK-ERK cascade.19 On the other hand, the tool inhibitor SCH772984 (Fig. 1B) not only inhibits the enzymatic activity of ERK1/2 but also prevents its phosphorylation by MEK,20 possibly resulting from conformational changes to the ERK P-loop as observed in X-ray crystallographic studies.21
17 and LY321499618 inhibit ERK1/2 enzymatic activity, suppressing phosphorylation of downstream substrates, but concomitantly triggering feedback loops which result in partial re-activation of the RAF-MEK-ERK cascade.19 On the other hand, the tool inhibitor SCH772984 (Fig. 1B) not only inhibits the enzymatic activity of ERK1/2 but also prevents its phosphorylation by MEK,20 possibly resulting from conformational changes to the ERK P-loop as observed in X-ray crystallographic studies.21
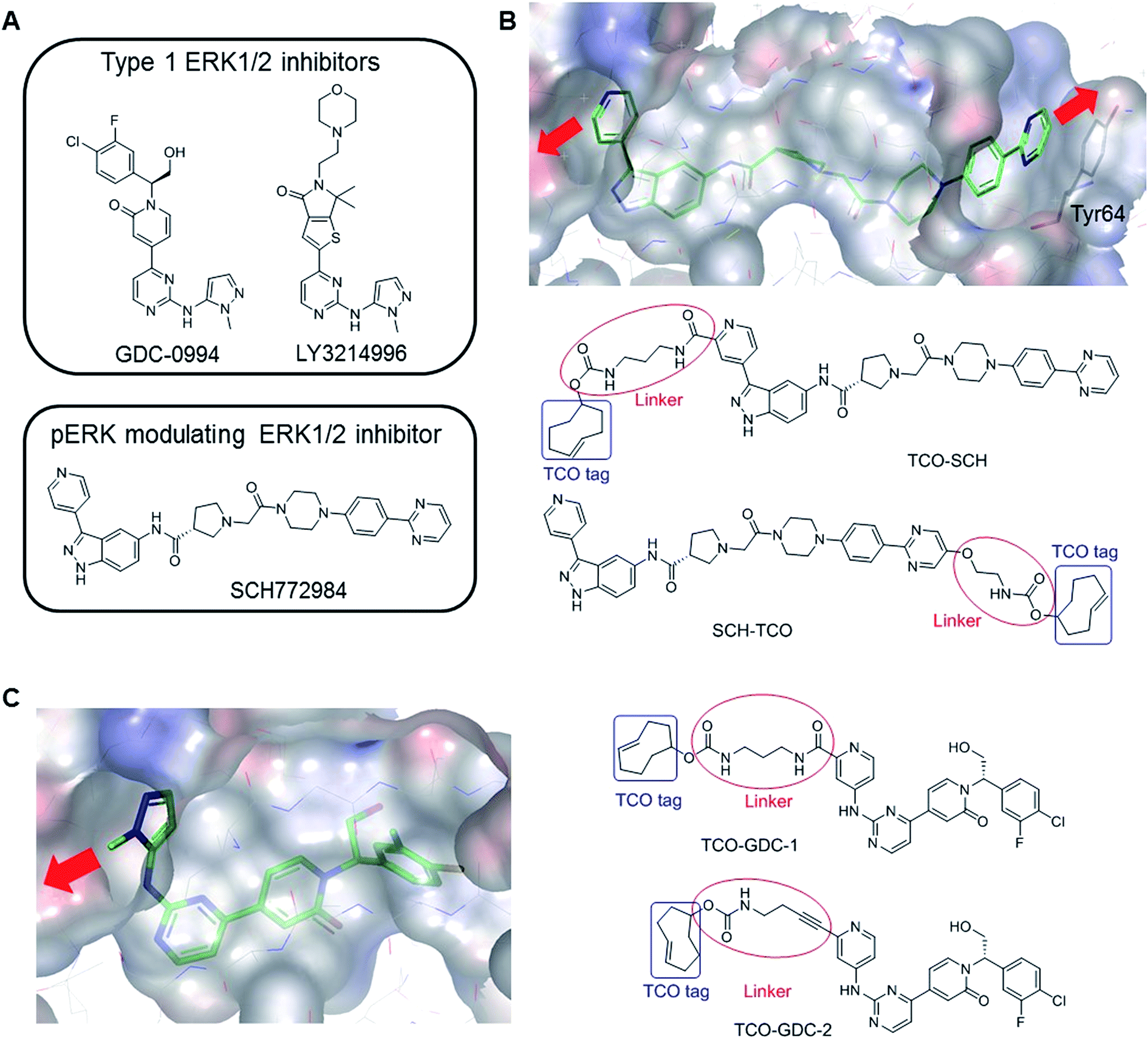 | ||
| Fig. 1 (A) Examples of ERK1/2 inhibitors classified in two categories: Type 1 and pERK modulating inhibitors. (B) Co-crystal structure of SCH772984 with ERK2 (pdb:4QTA).21 The inhibitor binds to an altered conformation of ERK2 allowing interaction with the Tyr64 side chain. The two sites identified to extend the molecule and insert the TCO tag are highlighted (red arrows). The structural design of the two probes, TCO-SCH and SCH-TCO are shown. (C) Co-crystal structure of GDC-0994 with ERK2 (pdb:5K4I)17 showing the vector for the extension of the molecule (red arrow) and structural design of the two probes, TCO-GDC-1 and TCO-GDC-2. | ||
Here, we report the design and synthesis of clickable trans-cyclooctene (TCO) tagged ERK1/2 probes, which were used to evaluate the target occupancies of published ERK1/2 inhibitors in competition mode. ERK1/2 bound to the TCO probe were combined with tetrazine-modified agarose beads, leading to attachment via the IEDDA reaction, and allowing analysis by immunoblotting in which protein levels were quantified by densitometry. We also measured the target occupancies of the same ERK1/2 inhibitors using the cellular thermal shift assay (CETSA) method. Following determination of the melting temperatures of ERK1 and 2, ITDR-CETSA was performed in cells and the levels of ERK1/2 in the soluble fractions were quantified by densitometry from the Western Blot analysis. This parallel evaluation enhanced our understanding of the relative pros and cons of the two techniques, and comparison of the results gave increased confidence in the values generated.
Results and discussion
Design and synthesis of TCO-tagged ERK1/2 probes
Covalent attachment of affinity probes to their target protein via a reactive electrophilic warhead or a photo-reactive group22–24 is usually required in pull down experiments in order to prevent dissociation of the probe–protein complex. Both techniques suffer from drawbacks. Probes containing an electrophilic warhead can only be used to target proteins with a suitably located reactive nucleophilic amino acid side chain such as cysteine, and can also react with other cellular proteins that contain this residue. The use of photo-reactive groups such as benzophenone or diazirine is generally associated with low labelling yields due to poor light penetration into cells, and the photo-activated carbenes can be formed and sequestered when not bound to their intended target, leading to high non-specific binding. Other factors such as conformational flexibility can also limit the cross-linking efficiency.25We previously reported the design and synthesis of a TCO tagged covalent probe which exploits the presence of a reactive cysteine residue in the active site.26 However, to circumvent the limitations described above and have a fair representation of the target occupancy of inhibitors studied in competition mode, we decided to design new non-covalent clickable probes and evaluate their potential to pull down ERK1/2. We designed TCO-tagged analogues of GDC-0994, an ATP-competitive type I ERK1/2 inhibitor, and SCH772984, a phospho-ERK modulating ERK1/2 inhibitor described as a slow off-rate ligand.21 Based on the co-crystal structure of SCH772984 with ERK2, we identified two sites on the inhibitor which appeared amenable to modifications, extending either from the pyridyl ring which binds close to the hinge region of ERK1/2 (TCO-SCH) or from the pyrimidine ring which interacts with Tyr64 on the C-α helix of ERK1/2 (SCH-TCO) (Fig. 1B). In each case the length of the linker was designed to position the TCO tag into the solvent, to minimise interference with the binding mode of the inhibitor (Fig. 1B). Both TCO-SCH and SCH-TCO were synthesised following a convergent route as described in ESI Schemes 1 and 2.† The co-crystal structure of GDC-0994 with ERK2 suggested only a single vector from which to extend the molecule towards the solvent (Fig. 1C). As the 2-position of the pyrimidine ring of GDC-0994 showed tolerance towards other groups than pyrazole,17 we introduced a pyridyl heterocycle in our design, facilitating the introduction of the linker and TCO tag (Fig. 1C). Linkers containing amide (TCO-GDC-1) or alkyne (TCO-GDC-2) functionality were evaluated due to the limited space available to extend from the ATP pocket without perturbing the interaction of the inhibitor with Lys114. TCO-GDC-1 and TCO-GDC-2 were synthesised following similar routes, as illustrated in ESI Scheme 3.†
Pharmacological evaluation of the TCO-tagged probes
All TCO-tagged probes were tested in an ERK2 biochemical assay to assess the effects of linker attachment on inhibitory potency. Both inhibitors bearing an amide group on the pyridyl ring close to the hinge region of ERK1/2 (TCO-SCH and TCO-GDC-1) suffered a significant loss in potency compared with their parent inhibitors (ca. 20-fold and 8-fold respectively, Table 1). This was attributed to a steric clash between the protein and the carbonyl group of the amide linker, confirmed in the co-crystal structure of TCO-SCH with ERK2 (Fig. 2A and B). The alkyne containing analogue TCO-GDC-2 exhibited good activity against ERK2 with only a 2-3-fold decrease in potency (IC50 = 0.008 μM, Table 1) compared with GDC-0994 (IC50 = 0.0037 μM, Table 1). The co-crystal structure of TCO-GDC-2 and ERK2 confirmed the suitability of the more slender alkyne moiety as a linker to pass through the narrow channel from the ATP binding site to solvent (Fig. 3B). Attachment of the linker to the pyrimidine ring of SCH772984 (SCH-TCO) was well tolerated, showing only a modest drop off compared with the untagged SCH772984 (IC50 = 0.0053 and 0.0013 μM respectively, Table 1).| Compounds | Bioassay IC50 (μM) | Anti-proliferative IC50 (μM) | |
|---|---|---|---|
| A375 | HCT116 | ||
| a Geometric means. | |||
| SCH772984 | 0.0013 (2) | 0.037 (>10) | 0.05 (>10) |
| TCO-SCH | 0.022 (2) | — | — |
| SCH-TCO | 0.0053 (2) | 0.060 (2) | 0.10 (2) |
| Biotin clicked SCH-TCO | 45% @ 0.003 μM (2) | — | — |
| GDC-0994 | 0.0037 (2) | 0.13 (9) | 1.00 (8) |
| TCO-GDC-1 | 0.028 (2) | — | — |
| TCO-GDC-2 | 0.008 (3) | 0.93 (2) | 1.00 (2) |
| Biotin-clicked TCO-GDC-2 | 0.009 (2) | — | — |
| Biotin-PEG4-Tz | 8.1 (2) | — | — |
| LY3214996 | 62% @ 0.003 μM (2) | 0.35 (9) | 0.48 (3) |
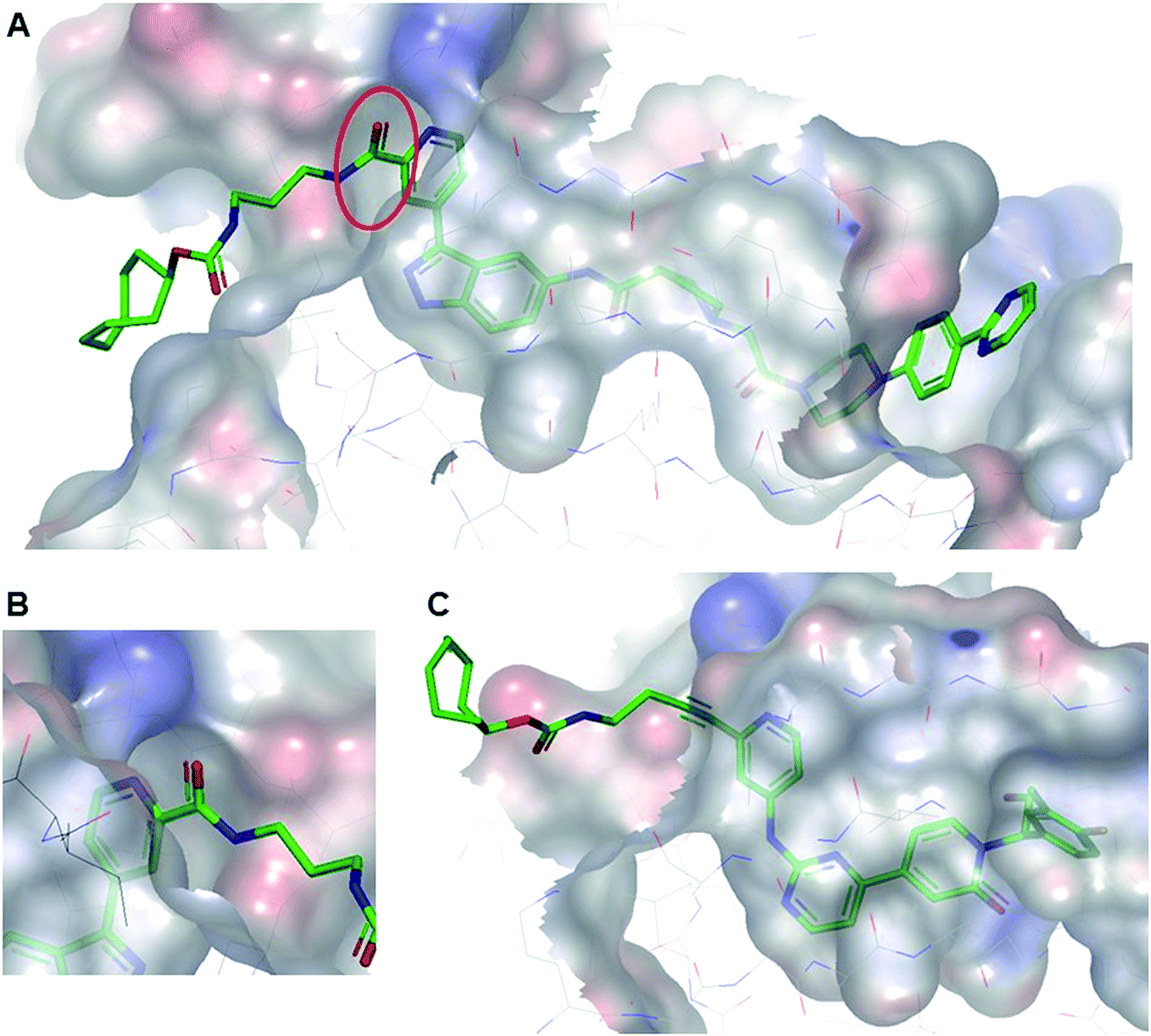 | ||
| Fig. 2 (A) Co-crystal structure of TCO-SCH with ERK2 (pdb:6GJD). The binding mode of the inhibitor core was conserved compared with the unmodified inhibitor SCH772984. (B) The amide linker of TCO-SCH likely encountering a steric clash with the protein is highlighted. (C) Co-crystal structure of TCO-GDC-2 with ERK2 (pdb:6GJB). The more slender alkyne linker provides a suitable vector to position the TCO tag in the solvent with a minimal steric clash with the protein. | ||
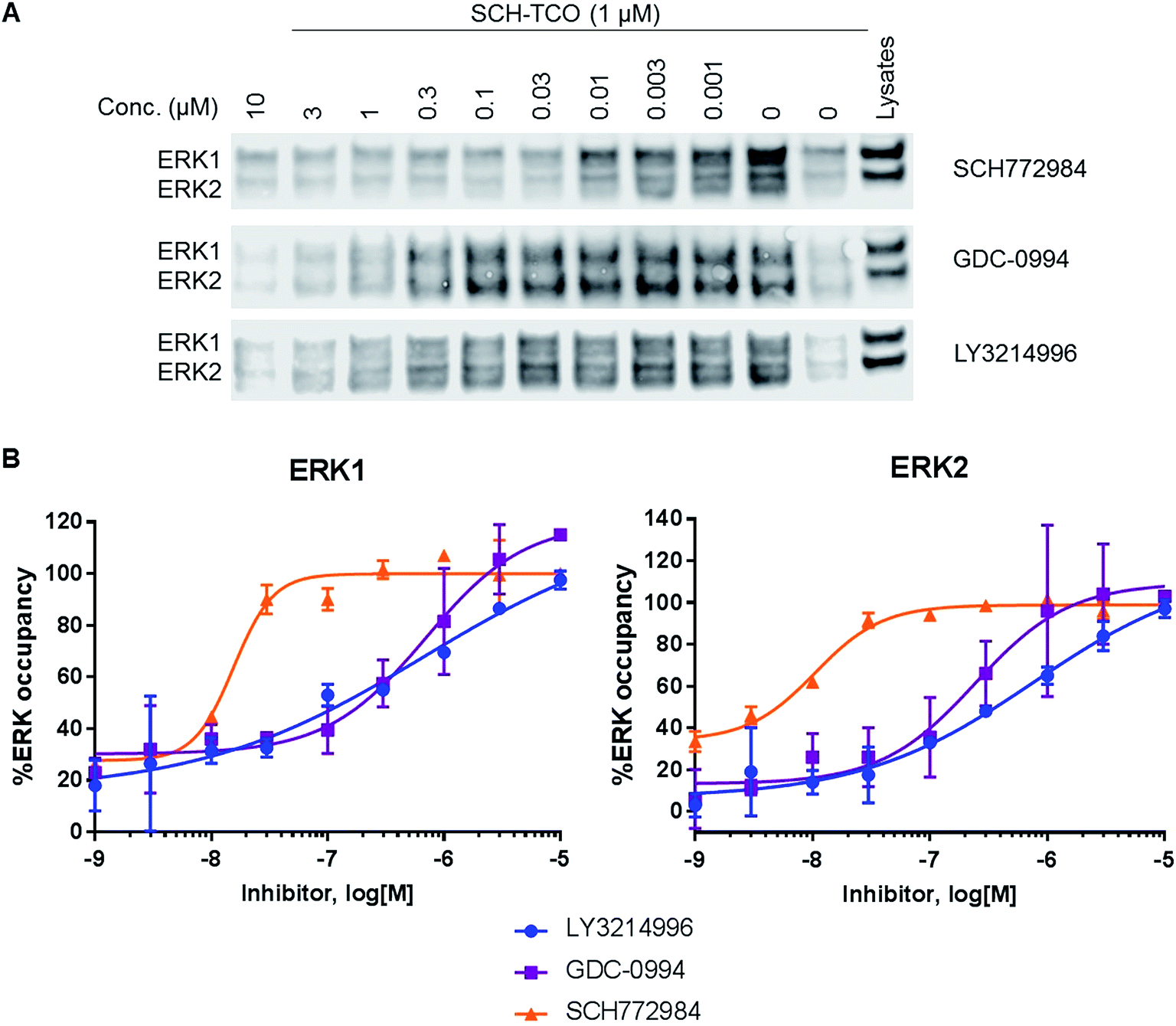 | ||
| Fig. 3 (A) Immunoblot analysis of ERK1/2 from the pulled down fractions obtained after competition experiments between SCH-TCO and a range of concentrations of selected ERK1/2 inhibitors in HCT116 cells. (B) Graphs showing the target occupancies of LY3214996, GDC-0994 and SCH772984 determined in competition mode with SCH-TCO in HCT116 cells. | ||
Based on these initial results, SCH-TCO and TCO-GDC-2 were selected for further investigation. The TCO probes were reacted with a tetrazine containing biotin conjugate (biotin-PEG4-Tz, ESI Fig. 1 and 2†) and the resulting clicked products were evaluated in the ERK2 bioassay. As expected, the clicked products exhibited similar potencies to their corresponding TCO precursors, demonstrating that the click derivatisation of the probes did not significantly interfere with their binding to ERK2. We also confirmed that biotin-PEG4-tetrazine does not appreciably inhibit ERK2 (Table 1). Next, the cellular activities of SCH-TCO and TCO-GDC-2 were evaluated in antiproliferative assays using A375 BRAFV600E mutant melanoma and HCT116 KRAS mutant colorectal cells. SCH-TCO showed good retention of cellular potency in both cell lines compared with its parent compound SCH772984 (Table 1). TCO-GDC-2 suffered a more significant drop-off in cellular IC50 values compared with its parent GDC-0994, consistent with the biochemical assay results. These data highlighted the importance of designing more than one probe in order to identify the position on the molecule which best tolerates modifications and select the probe which demonstrates the closest biological profile to its parent inhibitor, particularly for inhibitors which are somewhat enclosed when binding to their target protein.
Target engagement of ERK1/2 inhibitors using chemical probes
We first investigated the potential of the two selected chemical probes, SCH-TCO and TCO-GDC-2, to pull down ERK1/2 from cell lysates. The assays being conducted in competition mode, we systematically varied the probe concentration as well as its incubation time, as both parameters need to be tuned to allow the probe to engage all (or most) of the ERK without competing off the test inhibitor. HCT116 cell lysates were incubated with SCH-TCO at different concentrations followed by incubation with pre-coupled Tz-beads (prepared from streptavidin agarose beads and biotin-PEG4-Tz). The quantities of ERK1/2 pulled down were analysed by immunoblotting and were found to increase with probe concentrations (ESI Fig. 3A†). Similar levels of proteins were observed at 10, 3 and 1 μM for SCH-TCO suggesting that the majority of ERK1/2 protein was being pulled down across this concentration range. We then varied the incubation time of SCH-TCO at fixed concentrations (3 and 1 μM) with cell lysates. After 30 min incubation, the detection signal was found to be optimal (ESI Fig. 3B†). As such, 1 μM for 30 min was found to give an optimal compromise for SCH-TCO.Following the same procedures, the ability of TCO-GDC-2 to pull down ERK1/2 was studied in HCT116 lysates. TCO-GDC-2 showed a weaker ability to retrieve ERK1/2 than SCH-TCO, with incomplete pull-down observed at 1 μM after 30 min incubation (ESI Fig. 3C†). However, we considered that using a higher concentration of TCO probe during the competition experiment would overly compete with the inhibitor for ERK1/2 binding, and the results obtained would not be representative of the real target engagement of the inhibitor studied. We therefore used the same concentration and incubation time for TCO-GDC-2 as for SCH-TCO in the following target engagement experiments.
These preliminary pull-down studies showed that SCH-TCO offered more robust binding to ERK1/2 compared with TCO-GDC-2. Using SPR experiments with ERK2, we observed slow off-rate kinetics and high binding affinity for SCH-TCO (t1/2 = 233 min, KD = 0.17 nM), while TCO-GDC-2 showed relatively fast dissociation (t1/2 = 4 min, KD = 46 nM) (ESI Fig. 4†). The slow off-rate kinetics observed for SCH-TCO are consistent with published data for the untagged compound SCH772984 (t1/2 = 25–80 min),21 and are likely to favourably retard the dissociation of the probe/protein complex during the pull-down step. To further explore the potential of these non-covalent probes in pull-down experiments, the target occupancies of LY3214996, GDC-0994 and SCH772984 were assessed in A375 and HCT116 cell lysates, through competition with SCH-TCO or GTCO-GDC-2. Typically, cell lysates were incubated with the ERK1/2 inhibitor at different concentrations (1 h) followed by incubation with the probe (30 min), then Tz-beads (30 min), followed by filtration to separate the proteins retained by the beads from the rest of the lysates. Bound fractions were visualised by immunoblotting, alongside unbound fractions for control. The levels of ERK1/2 in the pulled down fractions (bound fractions) were inversely proportional to the test inhibitor concentration: the more inhibitor present in the incubation, the less ERK1/2 was available for binding to SCH-TCO. The levels of ERK1/2 in the unbound fractions, representing ERK1/2 bound to the test inhibitor, increased with the inhibitor concentration (ESI Fig. 5A†). The intensities of the bands from the immunoblots of the bound fractions were quantified by densitometry. The data were then plotted against the inhibitor concentration and EC50 values (concentration required to achieve 50% target engagement) were determined. At high compound concentration, residual signal was observed, attributed to unspecific binding of ERK1/2 protein to agarose beads. From these first experiments, similar EC50 values were obtained in both A375 and HCT116 cell lysates for SCH772984 and LY3214996 (ESI Fig. 5B and C†). A 2-3-fold difference was observed between the results obtained in A375 and HCT116 lysates for GDC-0994 (ESI Fig. 5B and C†). Overall, all compounds appeared to engage either ERK1 or 2 to a similar extent, consistent with the high homology for these two ERK isoforms.
We then investigated the target engagement of GDC-0994 and LY3214996 in HCT116 lysates by using TCO-GDC-2 which should offer an easier competition setting than SCH-TCO. The experiments revealed a ∼2-fold difference between the EC50 values observed for GDC-0994 and LY3214996 in competition with TCO-GDC-2 as compared with SCH-TCO (ESI Fig. 6†).
To study target engagement in competitive mode in intact cells, we prioritized SCH-TCO as the probe most likely to show sufficiently tight binding to ERK1/2 to survive the cell lysis step. We started by evaluating the ability of SCH-TCO to bind to ERK1/2 in HCT116 cells and pull down the proteins after lysis. The experiment showed that 15 min incubation at 1 μM followed by cell lysis was enough for a good signal by immunoblotting (ESI Fig. 7†). We then proceeded with the competition experiments, where typically the cells were treated with the test ERK1/2 inhibitor at a range of concentrations for 1 h, followed by the probe (1 μM) for 15 min. Following cell lysis, the lysates were treated as previously described to generate concentration–occupancy curves and allow calculation of target engagement EC50 values (Fig. 3, Table 2). In this setting, SCH772984 showed EC50 values of 16 and 11 nM against ERK1 and 2 respectively, in keeping with its potent growth inhibitory IC50 of 50 nM. GDC-0994 and LY3214996 showed weaker target engagement EC50s, again in keeping with their weaker growth inhibitory values of 1.0 and 0.48 μM respectively. Repeating the competition experiment with a longer incubation time of 90 min did not significantly change the EC50 values (ESI Fig. 8†).
| Inhibitors | Anti-proliferative IC50 (μM) | Pull downb – EC50 (μM) | CETSAc – EC50 (μM) | Pharmacodynamic IC50d (pRSK, μM) | ||
|---|---|---|---|---|---|---|
| ERK1 | ERK2 | ERK1 | ERK2 | |||
| a Geometric means. b In HCT116 cells with SCH-TCO. c In HCT116 cells. d In HCT116 cells with 2 h drug treatment. | ||||||
| SCH772984 | 0.05 (>10) | 0.016 (2) | 0.011 (2) | 0.063(3) | 0.015 (3) | 0.009 (3) |
| GDC-0994 | 1.00 (8) | 0.7 (2) | 0.24 (2) | 0.58 (3) | 0.61 (3) | 0.14 (2) |
| LY3214996 | 0.48 (3) | 0.67 (2) | 0.77 (2) | 0.30 (3) | 0.33 (3) | — |
In this work, we used X-ray crystallography to design a non-covalent clickable probe which efficiently pulled down drug-free ERK1/2 proteins, allowing the measurement of competitive target engagement of selected ERK1/2 inhibitors. Under competition conditions, inhibitor target occupancies are relative measurements which depend on the avidity and other properties of the probe used, making the selection and characterization of the probe of key importance. Here, the preliminary work conducted in cell lysates with both TCO-GDC-2 and SCH-TCO confirmed the importance of the kinetic profile of the probe. The slow off-rate kinetics of SCH-TCO engendered a robust probe-protein interaction, capable of mimicking a covalent probe in the pull-down process. This translated well to whole cell experiments, in which SCH-TCO was successfully used to quantify target engagement for the three selected ERK1/2 inhibitors, giving EC50 values which agree well with the other intact cell methods described herein (Table 3). Differences between the EC50 values obtained in lysates and in cells for LY3214996 and GDC-0994 might be ascribed to partial dissociation of the ERKi/ERK complex during the cell lysis step (ESI Fig. 9†) but also to incomplete cell permeation of the two drugs.
The use of the TCO-GDC-2 probe resulted in apparently exaggerated EC50s values in the low nanomolar range for the two weaker inhibitors LY3214996 and GDC-0994, which could be explained by their increased ability to displace the less avid probe, but also the more facile dissociation of the ERK/TCO-GDC-2 complex during the pull-down process: with less ERK being pulled down, higher apparent target occupancy was achieved by the test inhibitors (ESI Fig. 10†).
Target engagement of ERK1/2 inhibitors using CETSA
Following our studies using the pull-down method, we investigated the potential of CETSA to quantify the target occupancies of GDC-0994, LY3214996 and SCH772984, to allow comparison of the data generated by the two methods. CETSA is a broadly applicable biophysical technique allowing detection of changes in ligand/drug binding interactions directly in intact cells, tissues or lysates. The technique is based on the thermal shift assay (TSA) concept where ligand binding affects protein stability. When a protein binds to a ligand, the melting temperature (Tm) is typically shifted to a higher temperature, producing a thermal shift (ΔTm). CETSA represents a novel, label-free strategy for the direct determination of drug binding and offers an easy and fast approach for the assessment of target engagement. With CETSA relying on thermal stabilization of the protein upon ligand binding, it is crucial to identify the melting temperature (Tm) at which the stabilization will be best observed. We first looked at the denaturation pattern of ERK1/2 by measuring the protein stability in lysates and in intact cells. Following a heating step, which caused protein unfolding and therefore precipitation, the remaining soluble fractions (i.e. folded proteins) were isolated and analyzed by immunoblotting. The levels of ERK1/2 were quantified by densitometry and subsequently plotted against the temperature, providing the CETSA melting curves. In CETSA experiments in lysates, ERK1 and 2 exhibited different thermal stability profiles with ERK2 being more stable (Tm = 58–60 °C) than ERK1 (Tm = 51–53 °C) in both lysates and cells (ESI Fig. 11† (DMSO)). While the melting profiles of ERK2 were similar in both lysates and cells, ERK1 appeared to be less thermally stable in cells (ESI Fig. 11†). Distinct thermal stability profiles were observed for ERK1 and ERK2, and consequently each isoform was studied separately.The same CETSA experiments were conducted with HCT116 cells pre-treated with ERK1/2 inhibitors (10 μM, 1 h) in order to confirm ligand binding. Treatment with SCH772984, GDC-0994 and LY3214996 led to a temperature shift in the melting curves relative to DMSO control, validating the binding interaction between the inhibitors and ERK1/2 both in cells and lysates (ESI Fig. 11†). Similar affinity trend for ERK1 and 2 was observed for all inhibitors, with SCH772984 being the most potent ligand, followed by LY3214996 and GDC-0994; results in accordance with the enzymatic data (IC50 data, Table 1).
After demonstrating the potential of CETSA to monitor the direct target binding of ERK inhibitors in both lysates and cells, we used a dose–response measurement assay (ITDR-CETSA) to assess the target occupancy of each ligand. ITDR-CETSA is performed at a fixed temperature, usually the melting temperature at which protein stabilization upon ligand binding will be best observed. The EC50 determination from ITDR-CETSA measurements has a softness in the response depending on the temperature used for the isotherm and is typically used as a relative measurement and upper estimate for EC50.12 The lowest temperature allowing significant protein level measurement is expected to give the best EC50 estimates.
We first studied the target engagement of the three ERK1/2 inhibitors in HCT116 lysates for ERK1 and ERK2 separately since the two proteins unfold at different temperature. HCT116 lysates were incubated at their respective Tm with the selected inhibitor (0–100 μM) for 15 min followed by heat challenge for 3 min (Tm = 53 °C (ERK1) and 57 °C (ERK2)), centrifugation to isolate the soluble protein fractions, and immunoblot analysis. The protein levels were quantified by densitometry and the data were plotted against the inhibitor concentration (ESI Fig. 12†). As expected from the CETSA melting curves, SCH772984 showed the strongest target occupancy.
The same ITDR-CETSA experiments were performed in HCT116 cells which were treated with the inhibitors at concentrations ranging from 0 to 50 μM. The melting temperatures were adjusted based on the previous CETSA analysis (Tm = 52 °C (ERK1) and 58 °C (ERK2)). The results showed similar engagement by GDC-0994 and LY3214996 with both ERK1 and ERK2; while a 4-fold difference was observed between the engagement of ERK1 and ERK2 by SCH772984, with apparently higher affinity for ERK2 (Fig. 4, Table 2). A similar scenario, in which SCH772984 shows preferential binding to ERK2, was also seen in the lysate CETSA experiment (ESI Fig. 12†).
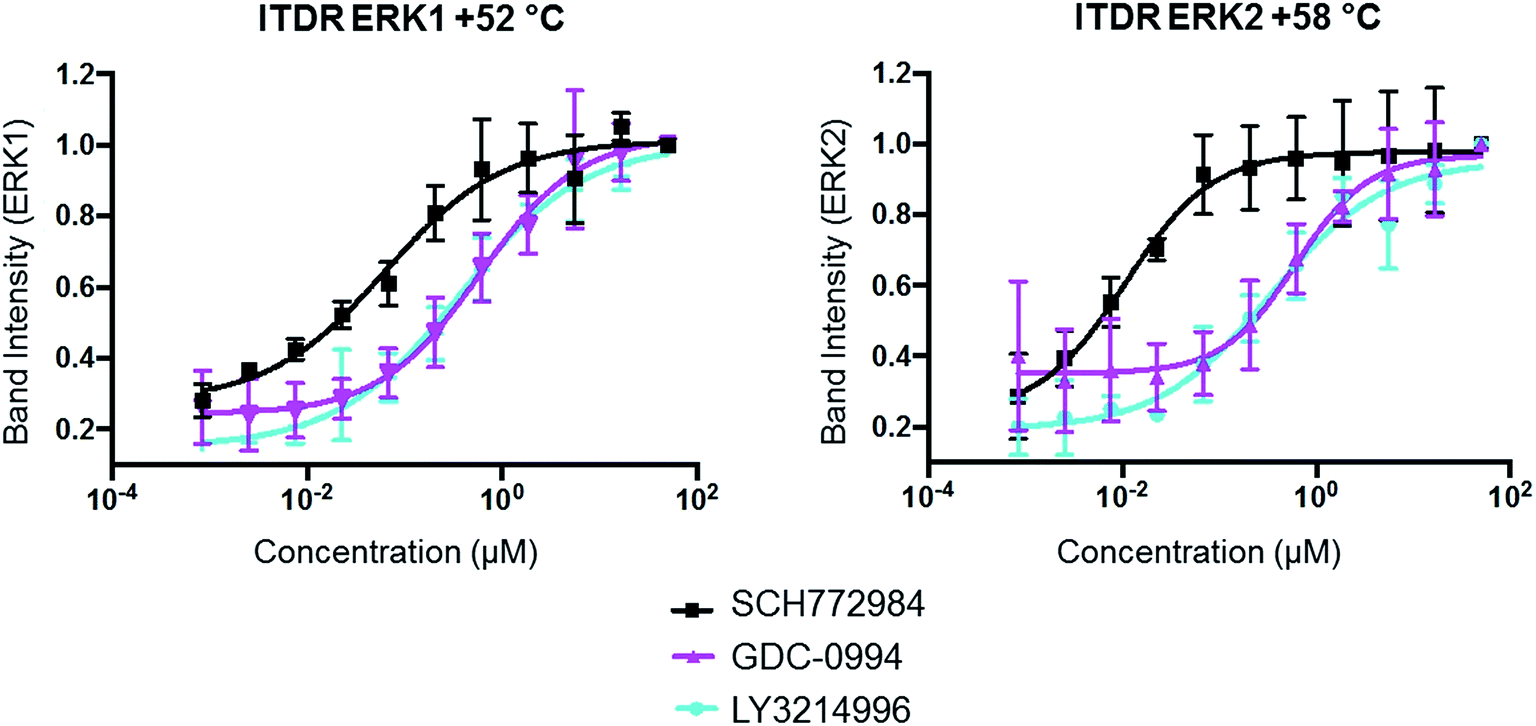 | ||
| Fig. 4 In-cell ERK1 and ERK2 occupancies of SCH772984, GDC-0994 and LY3214996 determined by ITDR-CETSA. HCT116 cells in suspension were treated with the appropriate ERK inhibitor for 1 h at 37 °C followed by heating at either +52° or +58 °C for 3 min. | ||
As a reference for target engagement using our new probes, CETSA was performed to evaluate the target occupancy of the selected ERKi both in lysates and in cells. The EC50 values measured in HCT116 cells for the three ERK inhibitors using the CETSA technique correlated well with those measured with the pull-down approach (Table 2). The ERK1/2 target engagement EC50 values obtained for SCH772984 and GDC-0994 are broadly similar to their respective pRSK IC50 values and lower than their anti-proliferative IC50 values, consistent with the notion that more than 50% of ERK needs to be occupied and inhibited to observe a 50% reduction in proliferation (Table 2). The target engagement EC50 values for LY3214996 are similar to those obtained for GDC-0994, as is its anti-proliferative IC50 value; however, in our hands this compound failed to show convincing suppression of pRSK in HCT116 cells, preventing a correlation of the target engagement and pharmacodynamic response.
As observed for the competitive displacement method, some differences were found between the EC50 values obtained in lysates and in cells, largely attributed to the dilution of important cofactors or protein partners in lysates, known to influence target engagement.
Indirect target engagement via PD response: phosphorylation of pRSK
Target engagement is commonly assessed indirectly as a pharmacodynamic response, in the case of a kinase by measuring the level of phosphorylation of a downstream substrate. RSK is a downstream substrate of ERK1/2 and the cellular phospho-RSK level has been commonly used as a biomarker to assess the target engagement of inhibitors. The concentration of inhibitor required to inhibit 50% of RSK phosphorylation (pRSK IC50) was determined for GDC-0994, LY3214996 and SCH772984 in HCT116 and A375 cells at different time points (Table 3). For GDC-0994 and SCH772984, similar pRSK IC50 values were obtained at either time point (2 or 7 h), demonstrating rapid target engagement in both cell lines. Surprisingly, LY3214996 showed a rather weak pharmacodynamic effect on pRSK, despite showing similar enzymatic and anti-proliferative IC50s to GDC-0994 (Table 1).Conclusion
Lack of efficacy, insufficient mechanistic understanding and safety issues are the three major reasons for attrition of drug development projects. Ensuring that a drug engages with its target is critical for a project's survival. Here we developed a new probe for quantitation of ERK1/2 inhibitor target engagement, and compared it to CETSA.While in the CETSA technique the protein stability is measured in the lysate or cell, the protein enrichment (pull-down) approach is a direct quantification of the level of unbound protein. A particular benefit of CETSA is that it allows direct measurement of the interaction between native protein and test inhibitors, regardless of their binding kinetics, and no target specific probes need to be developed. The observed target occupancy tends to be an upper estimate, but relative affects can be assessed for each target.
The protein enrichment approach is a straightforward and cost-efficient method. The use of the IEDDA in protein enrichment experiments has previously been demonstrated7 and was further validated in this work. Its fast reaction kinetics and quantitative yields as well as its practicality (no additional reagents) make this cycloaddition a bio-orthogonal reaction of choice for pull-down experiments. However, this approach requires the design and synthesis of chemical tools to be used in competition setting, which may perturb the real target engagement, depending on the kinetics of binding of the studied inhibitors.
Given the different advantages and drawbacks of our protein enrichment approach and CETSA, we evaluated them in parallel to increase confidence in the results. The target engagement of GDC-0994, LY3214996 and SCH772984 were quantified using both methods. Although, as expected, there were some quantitative differences, the EC50 values determined from the experiments conducted with SCH-TCO in cells and those obtained with CETSA both showed a clear difference in the target engagement of SCH772984 compared with GDC-0994 and LY3214996, consistent with its higher potency in biochemical and functional cellular assays. With this study, SCH-TCO was validated as a valuable tool compound for the study of target occupancy of ERK1/2 inhibitors.
Previously, the use of chemical probes in protein enrichment assays has required inclusion of a covalent warhead to ensure efficient pull down of the chosen protein. However, developing a chemical probe with a covalent warhead can be challenging; while the addition of a photoreactive group may perturb the physico-chemical properties of the probe, impairing cell permeability and solubility, and the low yield of photo-crosslinking can render the analysis challenging due to low concentration of protein pulled down. Here, we show that a reversible probe with a relatively slow off-rate (t1/2 ∼4 h) allows efficient protein pull-down, mimicking the avidity of an electrophilic covalent probe but avoiding the requirement for a reactive amino acid residue (such as cysteine) in the target protein and issues of electrophile reactivity. Interestingly, probes with slow dissociation kinetics (up to 1 h) were recently described in competitive target engagement studies against MDM2 using fluorescence imaging.27
Since most drug discovery projects necessarily develop non-covalent ligands, we envision that clickable slow off-rate probes might provide a widely applicable complementary approach to covalent tagging for chemical biology studies, including competitive target engagement and protein pull-down.
Materials and methods
Materials
A375 and HCT116 cells were purchased from American Type Culture Collection, Teddington, UK. The high capacity streptavidin agarose beads (20359) and the centrifuge columns (89868) were purchased from ThermoScientific. Biotin-PEG4-tetrazine was bought from Conju-Probe. The ERK1/2 antibody was bought from Cell Signaling Technology (9102S).
ERK2 bioassay
Activity of ERK2 enzyme (Life Technologies) was determined using a time-resolved fluorescence format measuring the phosphorylation of a truncated version of activating transcription factor 2 labelled with green fluorescent protein (ATF2-GFP) (Life Technologies). Assay reactions containing 50 mM Tris pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% Triton X-100, 1 mM DTT, 2.5% DMSO, 0.4 μM ATF2-GFP, 20 μM ATP and 0.25 nM ERK2 were set up in the presence of Tz-thalidomide and allowed to proceed for 30 min at room temperature. Reactions were then stopped using TR-FRET dilution buffer (Life Technologies), 25 mM EDTA and 2 nM Tb-Anti-pATF2 (Thr71) (Life Technologies). After a further incubation period of at least 30 minutes, fluorescence was read on a Pherastar reader (Lanthascreen optic module; excitation 340 nm, emission 520 nm (channel A), 495 nm (channel B)). The ratio between A and B counts was used to calculate signal. IC50 values were calculated using a sigmoidal dose–response equation (Prism GraphPad software, La Jolla, CA, USA).Cell culture
A375 and HCT116 cells (purchased from ATCC) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS (Gibco, Life Technologies) and were grown at 37 °C with 5% CO2.Cell proliferation assay
Cell proliferation assays were carried out using Alamar Blue (ThermoFisher Scientific, Waltham, Massachusetts, USA) as described previously.28 Briefly, 5 × 103 cells were seeded in complete culture medium into flat-bottomed 96-well plates, one day before the drug treatment. Cells were incubated with compound in 0.1% (v/v) dimethyl sulfoxide (DMSO) for 96 hours before viability was assessed using Alamar blue. IC50 values were calculated using a sigmoidal dose–response equation (Prism GraphPad software, La Jolla, CA, USA).Preparation of cell lysates
A375 and HCT116 cells were lysed with lysis buffer (TG lysis buffer supplemented with cOmplete EDTA-free protease inhibitor cocktail (Roche)) and kept on ice for 20 min. The cell lysates were centrifuged at 14000 rpm for 10 min at 4 °C and the protein concentration was determined by a Pierce™ BCA Protein Assay Kit. The concentrations were adjusted to 1.5 mg mL−1.
Coupling of streptavidin beads with biotin-PEG4-tetrazine
The beads (400 μL of 50% slurry) were first washed twice with 200 μL of lysis buffer. The beads were then re-suspended in 200 μL of lysis buffer and 10 μL of Biotin-PEG4-tetrazine (from a 30 mM stock solution in DMSO) were added. The suspension was incubated on ice for 30 min with shaking. The suspension was then centrifuged (using centrifuge columns) at 1200 rpm for 1 min and washed twice with lysis buffer (2 × 200 μL, 1200 rpm, 1 min). The Tz-beads were re-suspended in 800 μL of lysis buffer to give a 25% slurry.In-lysate ERK1/2 pull down
The lysates (120 μL) were incubated with the appropriate inhibitor (1.2 μL from a 100× DMSO stock solution for a final concentration ranging from 1 nM to 10 μM) for 1 h on ice with shaking. The TCO probe (1.2 μL from a 100× DMSO stock solution for a final concentration of 1 μM) was then added and the lysates were incubated for 30 min on ice with shaking. In parallel, 60 μL of Tz-beads (25% slurry) were added to centrifuge columns, the suspension was centrifuged (1200 rpm, 1 min) and the buffer was removed. The Tz-beads were then incubated with the lysates on ice for 30 min with shaking (60 μL of 25% slurry for 120 μL of lysates). The suspensions were then transferred into centrifuge columns and centrifuged (1200 rpm, 1 min). The unbound fractions were kept and the beads were washed twice with 100 μL of lysis buffer. 60 μL of elution buffer (2× LDS sample buffer) were added to each columns which were incubated for 30 min at room temperature. The columns were then centrifuged (5000 rpm, 1 min) and the elution fractions were collected. Samples were separated on 4–12% NuPAGE gels (Life Technologies) and transferred onto a nitrocellulose membrane (Novex). The membrane was blocked in blocking buffer (Odyssey) at r.t. for 1 hour and subjected to immunodetection using a total ERK1/2 primary antibody (p44/42 MAPK ERK1/2, Cell Signaling Technologies®, 1:1000) in blocking buffer, at 4 °C overnight (or alternatively, at room temperature for 1 hour). After washing 3× with a Tris-buffered saline (TBS) with 0.1% Tween-20 solution (TBST), the membrane was incubated with fluorescently labelled secondary antibody (IRDye800CW Donkey Anti-Rabbit, 1:10000 and IRDye680RD Donkey Anti-Mouse, 1:10000) for 1 hour at r.t. in the dark. After washing 2× with a TBS solution, the membrane was imaged on an Li-Cor Biosciences Odyssey system in the 800 nm and 700 nm channels. The intensities of the bands observed were determined by densitometry using the Odyssey software and were used to calculate target engagement. EC50 values were calculated using a sigmoidal dose–response equation (Prism GraphPad software, La Jolla, CA, USA).
In-cell ERK1/2 pull down
HCT116 cells were seeded in 6-well plates at 4.0 × 105 cells per mL with 2 mL per well and allowed to attach overnight. The appropriate inhibitor was added from a 1000× stock solution in DMSO (2 μL for a final concentration ranging from 1 nM to 10 μM) and the cells were incubated at 37 °C in an atmosphere of 5% CO2 and air for 1 h. Then the TCO probe was added from a 1000× stock solution in DMSO (2 μL for a 1 μM final concentration) and the cells were incubated at 37 °C in an atmosphere of 5% CO2 and air for 15 min. The media was removed, the cells were washed with PBS (2 mL per well) and lysed with TG lysis buffer supplemented with cOmplete EDTA-free protease inhibitor cocktail (Roche) (120 μL per well) for 20 min on ice. The lysates were centrifuged (14000 rpm, 5 min, 4 °C) and the protein concentration was determined by a Pierce™ BCA Protein Assay Kit. Samples were normalised to 1.5 mg mL−1 and treated with Tz-beads following the procedure described above.
CETSA melting curves
In order to identify ERK1/2 melt temperature (Tm) for dose–response studies, HCT116 cells were harvested, re-suspended in McCoy's 5A Modified Media (Sigma Aldrich) at a concentration of 3 × 106 cells per mL and aliquoted into PCR strip tubes (99 μL per tube). Cells suspensions were incubated with either DMSO or the selected inhibitor (10 μM) at 37 °C for 1 hour. Following drug treatment, the cells were heated at 12 different temperatures in Applied Biosystems Verti 96 well Thermal Cycler (37–70 °C gradient) for 3 minutes. Then a cOmplete EDTA-free protease inhibitor cocktail (Roche) was added and the cells were lysed by 3 freeze–thaw cycles in liquid nitrogen. The soluble protein fractions were obtained by centrifugation of lysates for 20 min at 20000g. The supernatants were analysed by immunoblotting using a total ERK1/2 primary antibody (p44/42 MAPK ERK1/2, Cell Signaling Technologies®, 1:1000) in 5% milk solution, at 4 °C overnight. The membrane was afterwards incubated with an anti-Rabbit IgG HPR Conjugated (Promega, W401B, 1:10000) for 1 hour at r.t. The amount of proteins was determined by densitometry using the Image Lab™ Software (Bio-Rad) and used for melting curves plotting. For the lysate experiments, the HCT116 cells were lysed as described above using TG lysis buffer. The lysates were then incubated with 10 μM of the selected inhibitor for 15 min at RT with mild shaking. Same protocol for heating and analysis was applied.
Isothermal dose–response (ITDR) CETSA in HCT116 cells and lysate
For in-cell experiments, the HCT116 cells were harvested, re-suspended in McCoy's 5A Modified Media (Sigma Aldrich) at a concentration of 3 × 106 cells per mL, aliquoted in PCR strip tubes (99 μL per tube). Cells in suspension were treated with 12 concentrations of ERK inhibitors from 0 to 50 μM, at 37 °C for 1 hour. The inhibitors were diluted in DMSO prior to addition to cells, and DMSO concentration was kept at 1% for all treatments. Treated cells were heated at +52 °C or +58 °C for 3 min. Cells were lysed by 3 freeze/thaw cycles using liquid nitrogen. Samples were centrifuged for 20 min at 20000g and were analysed by Western Blot as described above. The amount of proteins was determined by densitometry using the Image Lab™ Software (Bio-Rad) and was used to assess target engagement. EC50 values were calculated using a sigmoidal dose–response equation (Prism GraphPad software). For the ITDR-CETSA lysate experiments, the HCT116 cells were lysed as described above using TG lysis buffer. The lysates were incubated with 12 concentrations of ERK inhibitors from 0 to 100 μM for 15 min at r.t. with mild shaking. Same protocol for heating and analysis was applied.
Quantification of pRSK by MSD
A375 or HCT116 cells were seeded at 1.5 × 104 cells per well into 96-well plates and allowed to recover for 16 h, prior to the addition of compounds (in 0.1% DMSO v/v) and incubation for a further 2 or 7 h. Cells were lysed by adding cell lysis buffer (Cell Signaling Technology, Massachusetts, USA) and incubating at room temperature for 20 minutes. Custom MSD plates (Meso Scale Discovery, Maryland, USA) pre-coated with anti-pRSK antibody (Cell Signaling Technology, Massachusetts, USA) were blocked with kit blocking buffer for 1 hour at room temperature, prior to washing. Equivalent amounts of protein lysate were added to the blocked plates and incubated for 3 hours at room temperature. After washing, plates were incubated for 1 hour at room temperature with sulfo-tag conjugated anti-RSK detection antibodies (R&D Systems, Minneapolis, USA). Plates were washed, and read buffer added before reading the plate on a MESO QuickPlex SQ 120 (Meso Scale Discovery, Maryland, USA). IC50 values were calculated using a sigmoidal dose–response equation (Prism GraphPad software, La Jolla, CA, USA).SPR binding measurements
All SPR experiments were carried out on a BIAcore S200 (GE Healthcare) at 25 °C. ERK2 protein with an N-terminal 12His-tag was immobilised on an NTA chip (GE Healthcare) using the capture-coupling method as described by the manufacturer to achieve an immobilisation level of 3000–4000 resonance units (RU). The immobilisation buffer was 50 mM TRIS/HCl, 300 mM NaCl, 5 mM MgCl2, 2 mM TCEP, 0.05% (v/v) surfactant P20 at pH 8. Compound binding measurements were performed at a flow rate of 50 μL min−1 in the same running buffer used for the immobilisation supplemented with 2% DMSO. GDE-TCO-2 was tested in a in a multi-cycle format (concentration series from 5000 nM, as eight point three-fold serial dilutions). Each concentration was injected for 240 s and the dissociation was monitored for 900 s. The binding kinetics for SCH-TCO were measured in a single-cycle format and the dissociation was monitored for 3600 s. Five concentrations of SCH-TCO, prepared in three-fold serial dilution from 50 nM, were injected for 240 s. Solvent corrected, and background subtracted binding curves were analyzed using the Biacore insight evaluation software following standard procedures and data were fitted to a 1:1 binding model.
Conflicts of interest
HL, AC, MOR, CRV and TDH are employees of Astex Pharmaceuticals which has a commercial interest in the development of ERK1/2 inhibitors. PN is a director of Pelago Biosciences, which is a commercial company developing CETSA.Acknowledgements
We thank Charlotte E. East and Sharna J. Rich for the biological evaluation of the ERK1/2 inhibitors and TCO probes. The authors also thank Dr Torren M. Peakman for the NMR characterisation of the TCO probes and Stuart Whibley for his assistance with LC-MS studies. HL and OS were funded by the Astex Sustaining Innovation Postdoctoral program.References
- P. Morgan, P. H. Van Der Graaf, J. Arrowsmith, D. E. Feltner, K. S. Drummond, C. D. Wegner and S. D. A. Street, Drug Discovery Today, 2012, 17, 419–424 CrossRef PubMed.
- D. Cook, D. Brown, R. Alexander, R. March, P. Morgan, G. Satterthwaite and M. N. Pangalos, Nat. Rev. Drug Discovery, 2014, 13, 419–431 CrossRef PubMed.
- M. E. Bunnage, E. L. P. Chekler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef PubMed.
- G. M. Simon, M. J. Niphakis and B. F. Cravatt, Nat. Chem. Biol., 2013, 9, 200–205 CrossRef PubMed.
- M. Schürmann, P. Janning, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef PubMed.
- A. Rutkowska, D. W. Thomson, J. Vappiani, T. Werner, K. M. Mueller, L. Dittus, J. Krause, M. Muelbaier, G. Bergamini and M. Bantscheff, ACS Chem. Biol., 2016, 11, 2541–2550 CrossRef PubMed.
- J.-P. Schülke, L. A. McAllister, K. F. Geoghegan, V. Parikh, T. A. Chappie, P. R. Verhoest, C. J. Schmidt, D. S. Johnson and N. J. Brandon, ACS Chem. Biol., 2014, 9, 2823–2832 CrossRef PubMed.
- H. Xu, A. Gopalsamy, E. C. Hett, S. Salter, A. Aulabaugh, R. E. Kyne, B. Pierce and L. H. Jones, Org. Biomol. Chem., 2016, 14, 6179–6183 RSC.
- H. Xu, L. R. Roberts, S. Chou, B. Pierce, A. Narayanan and L. H. Jones, MedChemComm, 2017, 8, 767–770 RSC.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef PubMed.
- D. M. Molina and P. Nordlund, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 141–161 CrossRef PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef PubMed.
- B. X. Tan, C. J. Brown, F. J. Ferrer, T. Y. Yuen, S. T. Quah, B. H. Chan, A. E. Jansson, H. L. Teo, P. Nordlund and D. P. Lane, Sci. Rep., 2015, 5, 12116 CrossRef PubMed.
- G. Hatzivassiliou, B. Liu, C. O'Brien, J. M. Spoerke, K. P. Hoeflich, P. M. Haverty, R. Soriano, W. F. Forrest, S. Heldens, H. Chen, K. Toy, C. Ha, W. Zhou, K. Song, L. S. Friedman, L. C. Amler, G. M. Hampton, J. Moffat, M. Belvin and M. R. Lackner, Mol. Cancer Ther., 2012, 11, 1143–1154 CrossRef PubMed.
- J. Rodríguez and P. Crespo, Sci. Signaling, 2011, 4, re3 CrossRef PubMed.
- J. F. Blake, M. Burkard, J. Chan, H. Chen, K.-J. Chou, D. Diaz, D. A. Dudley, J. J. Gaudino, S. E. Gould, J. Grina, T. Hunsaker, L. Liu, M. Martinson, D. Moreno, L. Mueller, C. Orr, P. Pacheco, A. Qin, K. Rasor, L. Ren, K. Robarge, S. Shahidi-Latham, J. Stults, F. Sullivan, W. Wang, J. Yin, A. Zhou, M. Belvin, M. Merchant, J. Moffat and J. B. Schwarz, J. Med. Chem., 2016, 59, 5650–5660 CrossRef PubMed.
- G. S. Cortez, S. Joseph, J. A. McLean, W. T. McMillen, M. J. Rodriguez and G. Zhao, US Pat., 20160176896, 23 Jun 2016.
- S.-Y. Shin, O. Rath, S.-M. Choo, F. Fee, B. McFerran, W. Kolch and K.-H. Cho, J. Cell Sci., 2009, 122, 425–435 CrossRef PubMed.
- E. J. Morris, S. Jha, C. R. Restaino, P. Dayananth, H. Zhu, A. Cooper, D. Carr, Y. Deng, W. Jin, S. Black, B. Long, J. Liu, E. DiNunzio, W. Windsor, R. Zhang, S. Zhao, M. H. Angagaw, E. M. Pinheiro, J. Desai, L. Xiao, G. Shipps, A. Hruza, J. Wang, J. Kelly, S. Paliwal, X. Gao, B. S. Babu, L. Zhu, P. Daublain, L. Zhang, B. A. Lutterbach, M. R. Pelletier, U. Philippar, P. Siliphaivanh, D. Witter, P. Kirschmeier, W. R. Bishop, D. Hicklin, D. G. Gilliland, L. Jayaraman, L. Zawel, S. Fawell and A. A. Samatar, Cancer Discovery, 2013, 3, 742–750 CrossRef PubMed.
- A. Chaikuad, E. M. C. Tacconi, J. Zimmer, Y. Liang, N. S. Gray, M. Tarsounas and S. Knapp, Nat. Chem. Biol., 2014, 10, 853–860 CrossRef PubMed.
- A. L. MacKinnon and J. Taunton, in Current Protocols in Chemical Biology, John Wiley & Sons, Inc., 2009, DOI:10.1002/9780470559277.ch090167.
- A. Herner, J. Marjanovic, T. M. Lewandowski, V. Marin, M. Patterson, L. Miesbauer, D. Ready, J. Williams, A. Vasudevan and Q. Lin, J. Am. Chem. Soc., 2016, 138, 14609–14615 CrossRef PubMed.
- G. W. Preston and A. J. Wilson, Chem. Soc. Rev., 2013, 42, 3289–3301 RSC.
- A. Kawamura, S. Hindi, D. M. Mihai, L. James and O. Aminova, Bioorg. Med. Chem., 2008, 16, 8824–8829 CrossRef PubMed.
- H. Lebraud, D. J. Wright, C. E. East, F. P. Holding, M. O'Reilly and T. D. Heightman, Mol. BioSyst., 2016, 12, 2867–2874 RSC.
- P. L. D'Alessandro, N. Buschmann, M. Kaufmann, P. Furet, F. Baysang, R. Brunner, A. Marzinzik, T. Vorherr, T.-M. Stachyra, J. Ottl, D. E. Lizos and A. Cobos-Correa, Angew. Chem., Int. Ed., 2016, 55, 16026–16030 CrossRef PubMed.
- M. M. Nociari, A. Shalev, P. Benias and C. Russo, J. Immunol. Methods, 1998, 213, 157–167 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02754d |
| This journal is © The Royal Society of Chemistry 2018 |