
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine-mediated bioorthogonal prodrug–prodrug activation†
Kevin
Neumann‡§
 *,
Alessia
Gambardella§
,
Annamaria
Lilienkampf
and
Mark
Bradley
*
*,
Alessia
Gambardella§
,
Annamaria
Lilienkampf
and
Mark
Bradley
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, King's Buildings, David Brewster Road, EH9 3FJ Edinburgh, UK. E-mail: mark.bradley@ed.ac.uk
First published on 12th July 2018
Abstract
The selective and biocompatible activation of prodrugs within complex biological systems remains a key challenge in medical chemistry and chemical biology. Herein we report, for the first time, a dual prodrug activation strategy that fully satisfies the principle of bioorthogonality by the symbiotic formation of two active drugs. This dual and traceless prodrug activation strategy takes advantage of the INVDA chemistry of tetrazines (here a prodrug), generating a pyridazine-based miR21 inhibitor and the anti-cancer drug camptothecin and offers a new concept in prodrug activation.
Introduction
Conventional prodrug activation strategies typically rely on physiological changes e.g. pH around a tumour or a specific biological stimulus, for example the expression of an enzyme, to “switch-on” or activate a prodrug.1 An alternative approach2,3 is the application of chemical reactions that can take place within a biological environment with high selectivity and biocompatibility,4 with such reactions typically being “unnatural” in origin. Bioorthogonal reactions have found applications in drug delivery and include examples of prodrug activation and even in situ drug synthesis.5 Examples of bioorthogonal prodrug activation include application of the Staudinger reaction and strain-promoted alkene–azide cycloaddition that have been used to activate prodrugs of doxorubicin.6–8 More broadly, bioorthogonal reactions have enabled the rapid and selective labelling of proteins,9,10 glycans,11 lipids12 and DNA13 under physiological conditions often in a pre-targeted imaging scenario.14–16Since the inverse electron demand Diels–Alder (INVDA) reaction between tetrazines and electron-rich dienophiles was first described as a bioorthogonal reaction,17 the tetrazine-promoted INVDA reaction has been the subject of intense interest. This includes a series of studies where tetrazine- quenched profluorophores undergo “switch-on” of fluorescence upon treatment with a dienophile,19–21 while tetrazine chemistry has been used to label pre-targeted antibodies with PET isotopes.22–24 Thus, tetrazine-mediated INVDA chemistry has been shown to offer high chemical selectivity and to be rapid, efficient and biologically compatible, undoubtedly enhanced by the acceleration shown in water for all Diels–Alder chemistries.18,25 Yet, despite their extensive use in imaging, examples of tetrazine-mediated prodrug activation are limited, but include a trans-cyclooctene–doxorubicin conjugate that liberates the decaged drug upon reaction with a tetrazine and subsequent oxidation of the resulting 1,4-dihydropyridazine to the pyridazine.26,27 This approach was recently adapted to allow the release of carbonyl sulphide (OCS) that was converted, via carbonic anhydrase, to the gasotransmitter H2S.28 In addition, tetrazine has been used for the targeted degradation of proteins.29 Recently, we and others, have shown that vinyl ethers undergo facile reaction with tetrazines resulting in elimination of the corresponding alkoxide or phenoxide.25,30–32 Thus, polymeric nanoparticles, bearing a vinyl ether caged linker were shown to liberate doxorubicin upon treatment with a tetrazine, resulting in “switch-on” of cytotoxicity.30
Here, we report a new concept in prodrug activation with the simultaneous, dual, and fully traceless (except the loss of N2) activation/generation of two different drugs. This chemistry utilises tetrazine as a masked prodrug, which removes the vinyl ether from a second prodrug and incorporated the structural elements of the vinyl group into its own structure, giving rise to two active drugs (Fig. 1A). The chemistry explored used a tetrazine as a prodrug of a pyridazine (a common scaffold found in many drugs such as apresoline®, sulfamethoxypyridazine® and cadralazine®) and, in our case, generated the known microRNA 21 (miR21) inhibitor 2,33,34 leading to downregulation of oncogenic miR21 and consequently “switch-on” of apoptosis. The other prodrug (the dienophile) was the vinyl ether masked-camptothecin 3 that liberated the anticancer drug 4, upon reaction with the tetrazine 1 (Fig. 1B). Notably, for the first time, the tetrazine scaffold can be considered as a protecting group for bioactive pyridazines.
 | ||
| Fig. 1 (A) INVDA reaction between a vinyl ether masked drug (inactive) and the tetrazine masked drug (inactive) leads to an active drug pair (a pyridazine and an alcohol). (B) Reaction between the tetrazine prodrug 1 (masked pyridazine-based miR21 inhibitor 2) and the vinyl-O-camptothecin 3 (caged camptothecin 4) showing the dual and traceless prodrug–prodrug generation of 2 and 4. The inhibition of microRNA 21 and topoisomerase would lead to cell death. | ||
Results and discussion
Synthesis of tetrazine-prodrug
Short non-coding microRNAs (miRNA) play a critical role in several biological processes with dysregulation of levels miRNA being associated with numerous diseases, in particular cancer.35,36 Oncogenic miR21 downregulates apoptosis with miRNA inhibition resulting in notable increase in apoptosis. Pyridazine 2, an miR21 inhibitor,33,34 was readily synthesized in two steps, starting from 2,5-dichloropyridazine 5, via 2-chloro-5-thiomethoxidepyridazine 6 (generated by reaction with sodium thiomethoxide) followed by a Suzuki coupling with 3-nitrophenylboronic acid (Scheme 1B). Pyridazines37 can also be formed viaINVDA reaction from the corresponding tetrazines and activated alkenes (Fig. 1). The synthesis of tetrazine 1 was achieved using 3-nitrophenyl imidoester 7 as a precursor, which was readily accessible from 3-nitrobenzonitrile 8. In a facile route to tetrazines, compound 7 was treated with methyl thiocarbohydrazidium S7 to give 2,4-dihydrotetrazine that was oxidised in situ with amyl nitrite to give the tetrazine prodrug 1 (Scheme 1A). Importantly, in the case of 2, the corresponding tetrazine prodrug 1 bears electron withdrawing and donating moieties which are known to increase reactivity and elimination of the alkoxide in INVDA chemistries.38 | ||
Scheme 1 (A) (i) HCl, EtOH/dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). (ii) Methyl thiocarbohydrazidium S7, pyridine, DMF. (iii) Amyl nitrite, CH2Cl2. (B) (i) NaSCH3, NEt3 (ii) 3-nitrophenylboronic acid, Na2CO3, Pd(dppf), dioxane/H2O (4:1). (C) (i) 1,2-dibromoethane, NaH, DMF. (ii) PhSeH, CsOH·H2O. (iii) (1) NaIO4, NaHCO3, CH3OH/H2O (5:1); (2) DIPEA, CH3CN. :1). (ii) Methyl thiocarbohydrazidium S7, pyridine, DMF. (iii) Amyl nitrite, CH2Cl2. (B) (i) NaSCH3, NEt3 (ii) 3-nitrophenylboronic acid, Na2CO3, Pd(dppf), dioxane/H2O (4:1). (C) (i) 1,2-dibromoethane, NaH, DMF. (ii) PhSeH, CsOH·H2O. (iii) (1) NaIO4, NaHCO3, CH3OH/H2O (5:1); (2) DIPEA, CH3CN. | ||
The miR21 inhibitor 2 and the tetrazine prodrug 1, were evaluated for their activity on breast, prostate and brain cancer cells (SK-BR3, PC3 and U87-MG, respectively), which all express miR21 providing a broad platform of cell lines for cytotoxicity assays.39–41 Whereas miR21 is seen as an oncogenic factor in glioblastoma pathogenesis and breast cancer progression, its role in the progression of prostate cancer is not fully understood.42 No influence on cell viability was observed when the cells were treated with up to 10 μM of the tetrazine prodrug 1; however, the same concentration of miR21 inhibitor 2 resulted in reduced cell viability in all three cell lines (MTT assay, Fig. 2B). Additionally, an Annexin V assay confirmed the results by indicating early apoptotic or dead SK-BR3 cells after treatment with the miR21 inhibitor 2 (Fig. 2C).
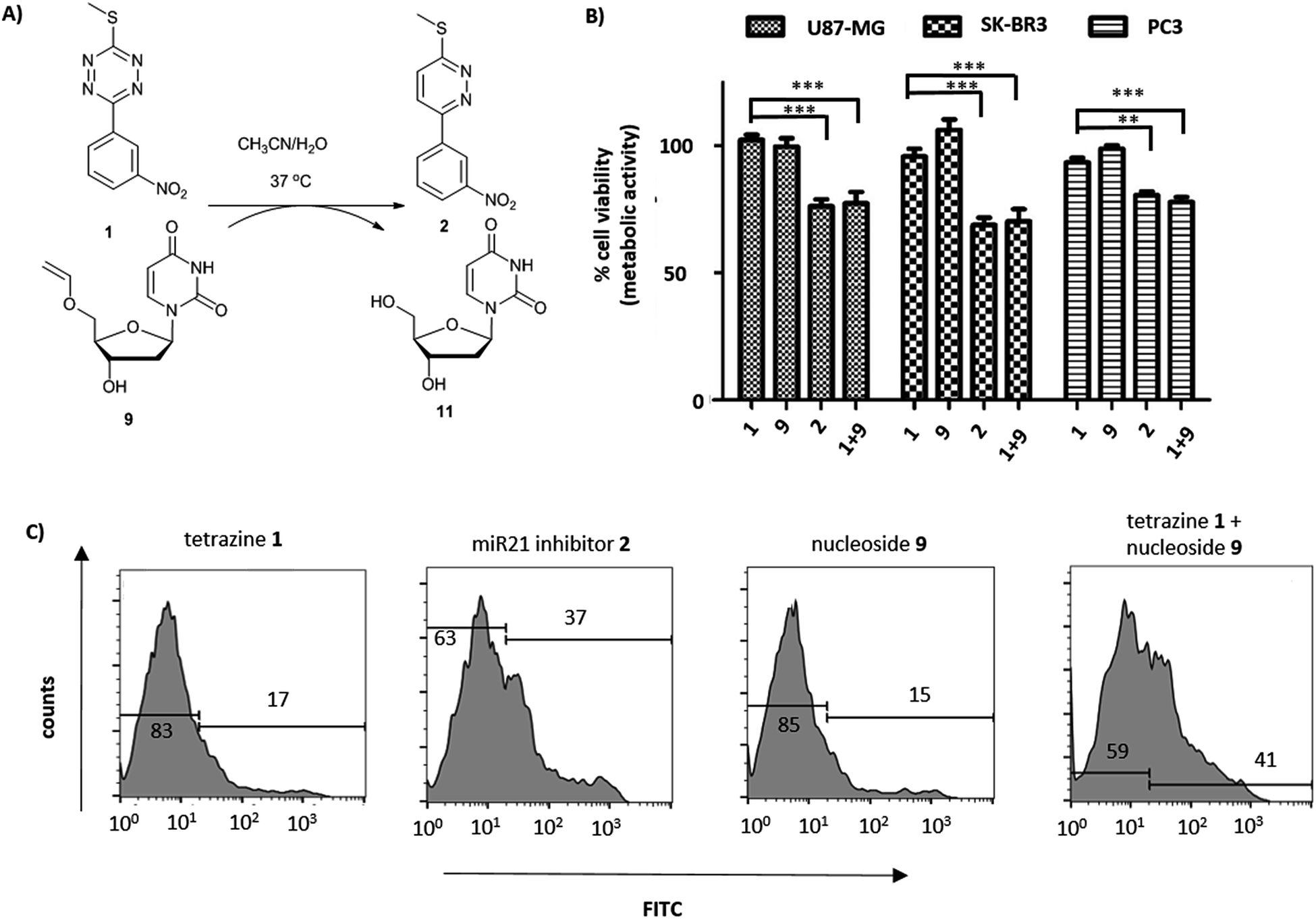 | ||
| Fig. 2 (A) Reaction between tetrazine 1 and 5′-O-vinyl deoxyuridine 9 (see ESI† for HPLC analysis and reaction kinetics). (B) U87-MG, SK-BK3 and PC3 cells incubated with tetrazine 1 (10 μM), 5′-O-vinyl deoxyuridine 9 (20 μM), miR21 inhibitor 2 (10 μM) and tetrazine 1 (10 μM) with 5′-O-vinyl deoxyuridine 9 (20 μM). Cell viability measured after 72 h (MTT assay, n = 3). ***P < 0.001 and **P < 0.01 by one-way ANOVA with Tukey post-test. No cytotoxicity was observed for 9 up to 20 μM; (C) flow cytometry histograms of Annexin V assay (FITC labelled) with tetrazine 1 (10 μM), miR21 inhibitor 2 (10 μM), 5′-O-vinyl deoxyuridine 9 (20 μM) and tetrazine 1 (10 μM) with 5′-O-vinyl deoxyuridine 9 (20 μM) after 14 h of incubation with SK-BR3. | ||
The activation of the tetrazine prodrug 1 with a vinyl ether containing small molecule was initially investigated. We postulated that 5′-O-vinyl deoxyuridine 9 would be a biocompatible, non-toxic dienophile, since the resulting alcohol is a naturally occurring nucleoside. Thus, deoxyuridine 11 was selectively alkylated with 1,2-dibromoethane to give 5′-O-bromoethyldeoxyuridine 10. Substitution of the bromine with caesium phenylselenolate gave the phenylselenyl ether 12,43 with oxidation with periodate giving 5′-O-vinyl deoxyuridine 9 (Scheme 1C).
Cellular incubation of the 5′-O-vinyl nucleoside 9 (20 μM) confirmed the biocompatibility of the vinyl ether with no apoptosis of SK-BR3 cells observed. The addition of tetrazine prodrug 1 (10 μM) with 9 (20 μM), however, gave equivalent levels of cell death as induced by the addition of 10 μM of pure inhibitor 2 with 30% of cells being positive in an Annexin assay (Fig. 2 and S1†), thus demonstrating in situ prodrug activation. The biological results are in accordance with the literature,41 showing a slightly enhanced effect of cancer progression by miR21 inhibition in glioblastoma and breast cancer cell lines compared with prostate cancer cell lines.
Hydrolytic stability is a critical parameter for any tetrazine targeted for biological applications and the half-life of prodrug 1 was determined to be 2.2 ± 0.04 days in DMSO/PBS, some 10-fold higher than the widely used 3,6-di-2-pyridinyltetrazine S5 (t1/2 = 0.31 ± 0.03 days in DMSO/PBS) (Fig. S2–S5†). Tetrazine 1 also exhibited reasonable stability in the presence of glutathione (5 mM GSH in DMSO/H2O) with 77% of 1 remaining after 3 days vs. 88% remaining without GSH (Fig. S6†).
The reactivity of tetrazine 1 in INVDA chemistry was investigated by determining the second order rates constants using two different dienophiles (see ESI, Scheme S2†). The reaction between tetrazine 1 and water-soluble vinyl ether S1 displayed a significant water acceleration, which is in accordance with the literature and confirmed the biocompatibility of this reaction (Fig. S2–S5†).18,25
Prodrug–prodrug activation
Camptothecin 4 is a topoisomerase I inhibitor that induces S-phase specific cell death. Since its discovery in the 1960's, several camptothecin derivatives and prodrugs have been reported with the aim of overcoming the drawbacks associated with camptothecin such as solubility and the stability of the lactone, which has been shown to play a crucial role in inhibiting topoisomerase I.43,44 In particular, it has been shown that alkylation or acetylation of the hydroxy group at the C20 position enhances the stability of the lactone;45,46 however, masking the hydroxy group of camptothecin causes a loss of its therapeutic efficiency with only a few examples known where the protecting group can be cleaved (usually by enzymatic triggering) without loss of activity.47,48Vinyl-O-camptothecin 3 was synthesised in a single step procedure by slightly modifying a reported iridium-catalysed trans-vinylation reaction49 using 1,4-dioxane to overcome the poor solubility of camptothecin 4 and an excess of vinylacetate (Fig. 3A). As postulated, masking the hydroxy group of camptothecin with a vinyl ether, caused a significant reduction in cytotoxicity, increasing the IC50 from 0.15 μM to 4.6 μM for PC3 cells and from 0.18 μM to 4.9 μM for SK-BR3 cells (Fig. 3 and S11†).
 | ||
| Fig. 3 (A) (i) Camptothecin 4, vinyl acetate, Na2CO3, [Ir(cod)Cl]2, 1,4-dioxane, 100 °C, 4 h. The reaction between tetrazine 1 and vinyl-O-camptothecin 3 gave >85% conversion (CH3OH/CH3CN/H2O) within 5 days as determined by HPLC. (B) Cell viability of PC3 cells after incubation with vinyl-O-camptothecin 3 (IC50 = 4.64 ± 1.13 μM) and camptothecin 4 (IC50 = 0.15 ± 0.06 μM) for 72 h at 37 °C; insert is non-linear fit used to determine IC50 values (MTT assay, n = 3). | ||
Treatment of vinyl-O-camptothecin 3 with the tetrazine prodrug 1 showed (monitored by HPLC) the generation of the active parent drug camptothecin 4 alongside the miR21 inhibitor 2. HPLC analysis also indicated the formation of small quantities of the oxidised tetrazine and a peak assigned to the oxidised pyridazine (Scheme S3, Fig. S14†). Thus, this demasking generates two active drugs and resulted in controlled switch-on of cytotoxicity (Fig. 4 and S12†). Importantly, co-treatment of PC3 cells with 2 and 4 showed an additive effect beyond the decaging/activation of 1 alone with increased levels of dead cells compared to treatment with 2 or 4 (Fig. S15†). In addition, masking the hydroxyl moiety not only increased the IC50 value but also compound stability. We assume that the enhanced stability of prodrug 3 leads eventually to a higher concentration of the active drug 4 at the target site, or at least longer duration of the active drug. Although the activation was slow in solution, the biological experiments suggest that the slow but constant release overtime together with the enhanced stability of 3 results in an efficient drug activation system that even with incomplete conversion delivers cytotoxicity comparable to that of the free drug. Future research will focus on prodrug pairs with even higher reactivity.
 | ||
| Fig. 4 Cell viability after treatment with tetrazine 1 (10 μM) 95 ± 14%, vinyl-O-camptothecin 3 (0.5 μM) 101 ± 10%, co-treatment of tetrazine 1 (10 μM) and vinyl-O-camptothecin 3 (0.5 μM) 47 ± 8%, camptothecin 4 (0.5 μM) 38 ± 5%, (PC3, MTT-assay, n = 3) ***P < 0.001 by one-way ANOVA with Tukey post-test. | ||
Thus, the herein presented prodrug–prodrug activation leads to an increased therapeutic window providing a concept that we believe will find wide application in drug delivery.
Conclusions
In summary, we report for the first time a symbiotic prodrug–prodrug activation strategy that fully complies with the principle of bioorthogonality. To illustrate the power of this new strategy, we showed that a tetrazine prodrug scaffold was converted into a pyridazine-based miR21 inhibitor upon reaction and decaging of a vinyl ether masked camptothecin. This demasking takes advantage of the water acceleration effect (for water dependency of kinetics see Fig. S1†), which has been widely exploited and acknowledged in tetrazine chemistry18,25 and results in the activation of two drugs without the generation of by-products, such as the phosphine oxide seen in the Staudinger ligation. Since drug resistance is a major concern in anti-cancer therapy, which has been linked to an overexpression of miRNA,50 activation of a conventional anti-cancer drug such as camptothecin in concert with a miR21 inhibitor, offers a new bioorthogonal prodrug–prodrug activation strategy and is an exceptionally atom efficient method of prodrug activation. The dual/traceless prodrug–prodrug activation strategy opens up new possibilities and directions in the field of drug delivery, in particular in the field of combination therapy (administration of two or more drugs) that is the most common clinical used strategy in cancer therapy.It should be noted that the herein presented prodrug–prodrug activation is not only suitable for hydroxyl and pyridazine containing drugs. One could imagine, for example that the traceless Staudinger ligation could be utilised in a similar manner leading to free drugs containing amines and organo phosphorous moieties, e.g. cyclophosphamides. In a broader context, the prodrug–prodrug approach presented here is not limited to the treatment of cancer and could be useful as a combination approach in other therapeutic areas.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council (Advanced Grant ADREEM ERC-2013-340469).Notes and references
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef PubMed.
- J. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef PubMed.
- K. Gorska, A. Manicardi, S. Barluenga, N. Winssinger, J. J. Hyldig-Nielsen, G. Zon, D. H. Ly, S. K. Kim, B. Norden and P. E. Nielsen, Chem. Commun., 2011, 47, 4364–4366 RSC.
- R. van Brakel, R. C. M. Vulders, R. J. Bokdam, H. Grüll and M. S. Robillard, Bioconjugate Chem., 2008, 19, 714–718 CrossRef PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- T. Peng and H. C. Hang, J. Am. Chem. Soc., 2016, 138, 14423–14433 CrossRef PubMed.
- M. C. Uzagare, I. Claußnitzer, M. Gerrits and W. Bannwarth, ChemBioChem, 2012, 13, 2204–2208 CrossRef PubMed.
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930–2944 CrossRef PubMed.
- A. B. Neef and C. Schultz, Angew. Chem., Int. Ed., 2009, 48, 1498–1500 CrossRef PubMed.
- C.-X. Song and C. He, Acc. Chem. Res., 2011, 44, 709–717 CrossRef PubMed.
- B. L. Oliveira, Z. Guo, O. Boutureira, A. Guerreiro, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2016, 55, 14683–14687 CrossRef PubMed.
- B. M. Zeglis, K. K. Sevak, T. Reiner, P. Mohindra, S. D. Carlin, P. Zanzonico, R. Weissleder and J. S. Lewis, J. Nucl. Med., 2013, 54, 1389–1396 CrossRef PubMed.
- L. Carroll, H. L. Evans, E. O. Aboagye and A. C. Spivey, Org. Biomol. Chem., 2013, 11, 5772–5781 RSC.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef PubMed.
- A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef PubMed.
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef PubMed.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- B. M. Zeglis, C. Brand, D. Abdel-Atti, K. E. Carnazza, B. E. Cook, S. Carlin, T. Reiner and J. S. Lewis, Mol. Pharmaceutics, 2015, 12, 3575–3587 CrossRef PubMed.
- J.-P. Meyer, J. L. Houghton, P. Kozlowski, D. Abdel-Atti, T. Reiner, N. V. K. Pillarsetty, W. W. Scholz, B. M. Zeglis and J. S. Lewis, Bioconjugate Chem., 2016, 27, 298–301 CrossRef PubMed.
- C. Denk, D. Svatunek, S. Mairinger, J. Stanek, T. Filip, D. Matscheko, C. Kuntner, T. Wanek and H. Mikula, Bioconjugate Chem., 2016, 27, 1707–1712 CrossRef PubMed.
- M. Staderini, A. Gambardella, A. Lilienkampf and M. Bradley, Org. Lett., 2018, 20, 3170–3173 CrossRef PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef PubMed.
- R. Rossin and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, 161–169 CrossRef PubMed.
- A. K. Steiger, Y. Yang, M. Royzen and M. D. Pluth, Chem. Commun., 2017, 53, 1378–1380 RSC.
- H. Lebraud, D. J. Wright, C. N. Johnson and T. D. Heightman, ACS Cent. Sci., 2016, 2, 927–934 CrossRef PubMed.
- K. Neumann, S. Jain, A. Gambardella, S. E. Walker, E. Valero, A. Lilienkampf and M. Bradley, ChemBioChem, 2017, 18, 91–95 CrossRef PubMed.
- E. Jiménez-Moreno, Z. Guo, B. L. Oliveira, I. S. Albuquerque, A. Kitowski, A. Guerreiro, O. Boutureira, T. Rodrigues, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 243–247 CrossRef PubMed.
- H. Wu, S. C. Alexander, S. Jin and N. K. Devaraj, J. Am. Chem. Soc., 2016, 138, 11429–11432 CrossRef PubMed.
- Q. Huang, A. Deiters and K. Gumireddy, Microrna modulators and method for identifying and using the same, WO 2013019469 A1, 2012 Search PubMed.
- K. Gumireddy, D. D. Young, X. Xiong, J. B. Hogenesch, Q. Huang and A. Deiters, Angew. Chemie Int. Ed, 2008, 47, 7482–7484 CrossRef PubMed.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef PubMed.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef PubMed.
- C. G. Wermuth, C. Vergelli, C. Biancalani, N. Cesari, A. Graziano, P. Biagini, J. Gracia, A. Gavalda, V. D. Paz and C. Norton, MedChemComm, 2011, 2, 935 RSC.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang and J. Zhao, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef PubMed.
- G. Shi, D. Ye, X. Yao, S. Zhang, B. Dai, H. Zhang, Y. Shen, Y. Zhu, Y. Zhu and W. Xiao, Acta Pharmacol. Sin., 2010, 31, 867–873 CrossRef PubMed.
- L. Shi, J. Chen, J. Yanga, T. Pan, S. Zhang and Z. Wang, Brain Res., 2010, 1352, 255–264 CrossRef PubMed.
- L. X. Yan, Q. N. Wu, Y. Zhang, Y. Y. Li, D. Z. Liao, J. H. Hou, J. Fu, M. S. Zeng, J. P. Yun, Q. L. Wu, Y. X. Zeng and J. Y. Shao, Breast Cancer Res., 2011, 13, R2 CrossRef PubMed.
- M. Folini, P. Gandellini, N. Longoni, V. Profumo, M. Callari, M. Pennati, M. Colecchia, R. Supino, S. Veneroni, R. Salvioni, R. Valdagni, M. Grazia Daidone and N. Zaffaroni, Mol. Cancer, 2010, 9, 12 CrossRef PubMed.
- R. J. Cohen, D. L. Fox and R. N. Salvatore, J. Org. Chem., 2004, 69, 4265–4268 CrossRef PubMed.
- C. Jin, S. Wen, Q. Zhang, Q. Zhu, J. Yu and W. Lu, ACS Med. Chem. Lett., 2017, 8, 762–765 CrossRef PubMed.
- M. Deshmukh, P. Chao, H. L. Kutscher, D. Gao and P. J. Sinko, J. Med. Chem., 2010, 53, 1038–1047 CrossRef PubMed.
- R. P. Hertzberg, M. J. Caranfa, K. G. Holden, D. R. Jakas, G. Gallagher, M. R. Mattern, S. M. Mong, J. O. Bartus, R. K. Johnson and W. D. Kingsbury, J. Med. Chem., 1989, 32, 715–720 CrossRef PubMed.
- A. Gopin, S. Ebner, B. Attali and D. Shabat, Bioconjugate Chem., 2006, 17, 1432–1440 CrossRef PubMed.
- B. Schmid, D. Chung, A. Warnecke, I. Fichtner and F. Kratz, Bioconjugate Chem., 2007, 18, 702–716 CrossRef PubMed.
- Y. Okimoto, S. Sakaguchi and Y. Ishii, J. Am. Chem. Soc., 2002, 124, 1590–1591 CrossRef PubMed.
- J. Ma, C. Dong and C. Ji, Cancer Gene Ther., 2010, 17, 523–531 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02610f |
| ‡ Current address: Laboratorium fur Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland, kevin.neumann@org.chem.ethz.ch |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |