
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of a nickel(II) thioperoxide complex: demonstration of sulfide-mediated N2O reduction†
Nathaniel J.
Hartmann
,
Guang
Wu
and
Trevor W.
Hayton
 *
*
Department of Chemistry and Biochemistry, University of California, Santa Barbara, California, 93106 USA. E-mail: hayton@chem.ucsb.edu
First published on 27th June 2018
Abstract
The thiohyponitrite ([SNNO]2−) complex, [K(18-crown-6)][LtBuNiII(κ2-SNNO)] (LtBu = {(2,6-iPr2C6H3)NC(tBu)}2CH), extrudes N2 under mild heating to yield [K(18-crown-6)][LtBuNiII(η2-SO)] (1), along with minor products [K(18-crown-6)][LtBuNiII(η2-OSSO)] (2) and [K(18-crown-6)][LtBuNiII(η2-S2)] (3). Subsequent reaction of 1 with carbon monoxide (CO) results in the formation of [K(18-crown-6)][LtBuNiII(η2-SCO)] (4), [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)] (5), [K(18-crown-6)][LtBuNiII(κ2-CO3)] (6), carbonyl sulfide (COS) (7), and [K(18-crown-6)][LtBuNiII(S2CO)] (8). To rationalize the formation of these products we propose that 1 first reacts with CO to form [K(18-crown-6)][LtBuNiII(S)] (I) and CO2, via O-atom abstraction. Subsequently, complex I reacts with CO or CO2 to form 4 and 5, respectively. Similarly, the formation of complex 6 and COS can be rationalized by the reaction of 1 with CO2 to form a putative Ni(II) monothiopercarbonate, [K(18-crown-6)][LtBuNiII(κ2-SOCO2)] (11). The Ni(II) monothiopercarbonate subsequently transfers a S-atom to CO to form COS and [K(18-crown-6)][LtBuNiII(κ2-CO3)] (6). Finally, the formation of 8 can be rationalized by the reaction of COS with I. Critically, the observation of complexes 4 and 5 in the reaction mixture reveals the stepwise conversion of [K(18-crown-6)][LtBuNiII(κ2-SNNO)] to 1 and then I, which represents the formal reduction of N2O by CO.
Introduction
Nitrous oxide (N2O) features a long atmospheric lifetime and large global warming potential (ca. 300 times larger than CO2), making it an important greenhouse gas.1–4 Anthropogenic sources of N2O include agriculture, fossil fuel combustion, adipic acid synthesis, and nitric acid production.1,5 The latter two sources use on-site N2O mitigation to remove N2O from the effluent stream, either by decomposition to the elements6 or reduction to N2 and H2O, but neither of these methods is completely effective and some N2O is still released into the atmosphere.7Given the above considerations, the development of new catalysts for N2O reduction could help reduce its impact on global temperatures.1,8 Not surprisingly, a large number of heterogeneous systems have been developed to catalyze this reaction.9 Of most relevance to the current study are the catalyst systems used for automotive applications, which consist of nanoparticulate Pt and Rh on a ceramic support. This process uses partially oxidized fuel (i.e., CO) to reduce N2O, forming N2 and CO2.9 Sita and co-workers developed a homogeneous version of this transformation, mediated by the Mo(II) complex, Cp*Mo(NCN)(CO)2 (NCN = iPrNC(Me)NiPr).10 In this process, N2O oxidizes Cp*Mo(NCN)(CO)2 to form a Mo(IV) oxo, Cp*Mo(NCN)(O), which then reacts with CO to form CO2 and regenerate Cp*Mo(NCN)(CO)2. However, an N–N bond cleavage reaction, which results in irreversible formation of Cp*Mo(NCN)(NCO)(NO), was found to be competitive with oxo formation. Similarly, Limberg and co-workers reported the stoichiometric oxidation of a Ni(0) CO complex, [K]2[LtBuNi0(CO)]2, with N2O to form a carbonate complex, [K]6[LtBuNiII(CO3)]6, and N2.11 Subsequent release of carbonate from the metal center was not discussed. The homogeneous hydrogenation of N2O has also been explored.12,13 For example, in 2015 Piers and co-workers reported an Ir(III) pincer carbene complex that could hydrogenate N2O;14 however, this system was not reported to be catalytic. More recently, Milstein and co-workers reported that the Ru pincer complex, [(PNP)RuH(CO)(OH)] (PNP = 2,6-[CH2PiPr2]2(C5H3N)), was an effective catalyst for the hydrogenation of N2O, achieving a turnover number of ca. 400.15
Recently, we reported the activation of N2O by the “masked” terminal nickel sulfide complex, [K(18-crown-6)][LtBuNiII(S)] (I) (LtBu = {(2,6-iPr2C6H3)NC(tBu)}2CH), which yielded an unprecedented thiohyponitrite complex, [K(18-crown-6)][LtBuNiII(κ2-SNNO)] (II) (eqn (1)).16 Given the challenge of activating N2O,17 and the novelty of the [SNNO]2− ligand in II, we endeavored to explore its reactivity in greater detail. Herein, we describe the first reactivity study of the [SNNO]2− ligand in an effort to uncover new routes to N2O reduction.
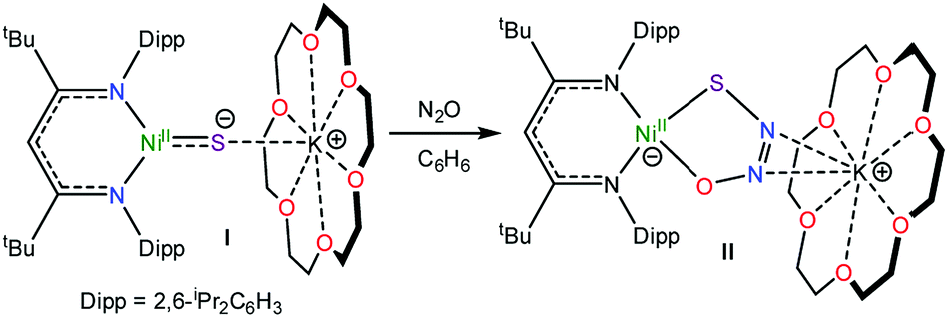 | (1) |
Results and discussion
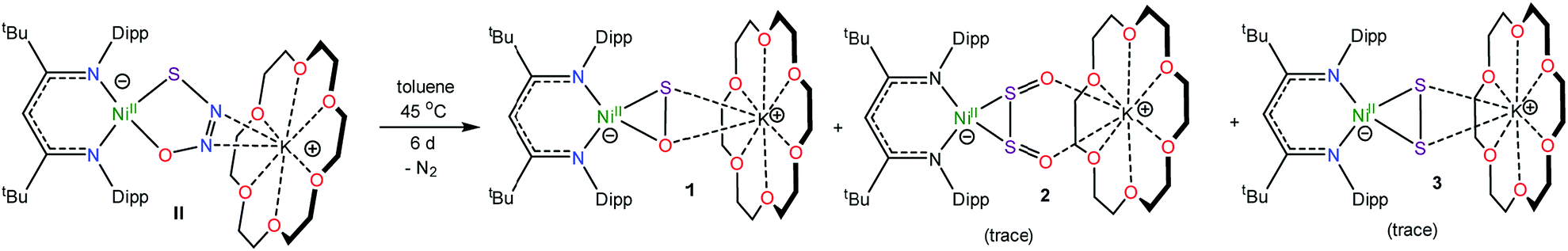
Synthesis of an [η2-SO]2− complex
Gentle heating of a toluene-d8 solution of [K(18-crown-6)][LtBuNiII(κ2-SNNO)] (II) at 45 °C results in the complete disappearance of II over the course of 6 d. A 1H NMR spectrum of this reaction mixture reveals the presence of a new γ-CH resonance at 5.43 ppm (Fig. S2 and 3†), which we have assigned to the thioperoxide complex, [K(18-crown-6)][LtBuNiII(η2-SO)] (1). A preliminary kinetic analysis suggests that the formation of 1 is first-order with respect to complex II, indicating that this transformation is unimolecular (Fig. S25†). Also present in these spectra are two minor γ-CH resonances. The first, observed at 5.53 ppm, has been tentatively assigned to the disulfur dioxide complex, [K(18-crown-6)][LtBuNiII(η2-OSSO)] (2), and the second resonance at 5.47 ppm, has been assigned to the disulfide complex, [K(18-crown-6)][LtBuNiII(η2-S2)] (3). Work-up of the reaction mixture affords [K(18-crown-6)][LtBuNiII(η2-SO)] (1) as an orange crystalline solid in 82% yield (eqn (2)). The solid state molecular structure of 1 is shown in Fig. 1. Complex 1 features a rare example of an η2-thioperoxide ([η2-SO]2−) ligand, which is formed via N2 extrusion from the thiohyponitrite fragment. The [η2-SO]2− ligand in 1 is disordered over two positions in a 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, which are related by a C2 rotation about the Ni–K axis. It possesses an S–O bond length of 1.656(3) Å, consistent with an S–O single bond.18 For comparison, the S–O distance in free S
:3 ratio, which are related by a C2 rotation about the Ni–K axis. It possesses an S–O bond length of 1.656(3) Å, consistent with an S–O single bond.18 For comparison, the S–O distance in free S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is substantially shorter (1.48108(8) Å), due to its higher bond order.19 The Ni–S (2.127(1) Å) and Ni–O (1.954(3) Å) distances in 1 are both consistent with single bonds and are comparable with those found in the starting material (II), while the Ni–N bond lengths (1.881(4) and 1.900(4) Å) are similar to those observed in other square planar LRNiII complexes.16,20
O is substantially shorter (1.48108(8) Å), due to its higher bond order.19 The Ni–S (2.127(1) Å) and Ni–O (1.954(3) Å) distances in 1 are both consistent with single bonds and are comparable with those found in the starting material (II), while the Ni–N bond lengths (1.881(4) and 1.900(4) Å) are similar to those observed in other square planar LRNiII complexes.16,20
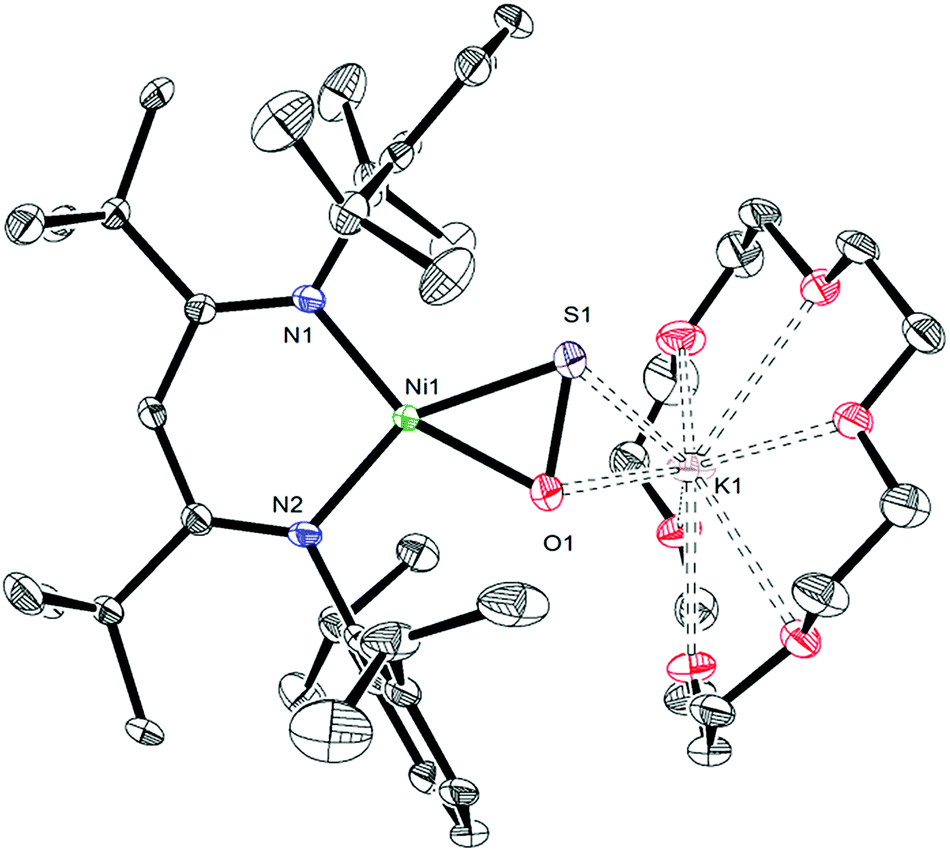 | ||
| Fig. 1 ORTEP drawing of [K(18-crown-6)][LtBuNiII(η2-SO)]·C7H8 (1·C7H8) shown with 50% thermal ellipsoids. Hydrogen atoms, a C7H8 solvate molecule, and one orientation of the disordered [η2-SO]2− ligand have been omitted for clarity. Selected metrical parameters: S1–O1 1.656(3) Å, Ni1–S1 2.127(1) Å, Ni1–O1 1.954(3) Å, Ni1–N1 1.881(4) Å, Ni1–N2 1.900(4) Å, S1–K1 3.162(2) Å, O1–K1 2.881(3) Å, N1–Ni1–N2 99.2(2)°, N1–Ni1–S1 110.0(1)°, N2–Ni1–O1 103.2(1)°, S1–Ni1–O1 47.65(9)°. | ||
The 1H and 13C{1H} NMR spectra of 1 are consistent with its formulation as a Cs symmetric, diamagnetic, square planar NiII complex. The 1H NMR spectrum of 1 in C6D6 features two tert-butyl resonances at 1.32 and 1.37 ppm and a single γ-CH resonance at 5.54 ppm. The IR spectrum (KBr pellet) of 1 reveals a strong νSO mode at 902 cm−1, which is consistent with values reported for other [η2-SO]2− ligands (883, 873 cm−1).21,22 Only a handful of structurally-characterized thioperoxide complexes are known,23–26 including [(triphos)Rh(μ-η2,η1-SO)2Rh(triphos)][BPh4]2 (triphos = CH3C(CH2PPh2)3), [{RhCl(μ-η2,η1-SO)(PPh3)2}2], and Fe3(μ3-SO)(S)(CO)9.21,27,28 The iron example is notable because it can be prepared by O-atom transfer to Fe3(S)2(CO)9,29 a manner of preparation that is similar to that of 1. Interestingly, Mankad and co-workers suggest that a transient SO complex is formed upon reaction of [(IPr*)Cu]2(μ-S) with N2O,30 a transformation that parallels our conversion of I to II and then 1.
As mentioned above, we also observe formation of [K(18-crown-6)][LtBuNiII(η2-OSSO)] (2), as a minor side product, during the conversion of [K(18-crown-6)][LtBuNiII(κ2-SNNO)] to 1. Despite its presence in trace amounts, we have been able to obtain a few single crystals of 2 as orange plates from the reaction mixture. The solid state molecular structure of 2 is shown in Fig. 2. It features the first example of a co-planar [OSSO]2− ligand (OSSO dihedral angle = 2°). The [η2-OSSO]2− ligand in 2 is bound to the Ni center in an η2 fashion, via both sulfur atoms, while the O atoms are bound to the [K(18-crown-6)]+ cation in a κ2 fashion. Its S–S distance is 2.093(3) Å, while the S–O distances are 1.485(5) and 1.496(7) Å. For comparison, the S–S (2.0245(6) Å) and S–O (1.458(2) Å) distances in free S2O2 are shorter than those observed for 2,31–33 consistent with the reduced S–S and S–O bond orders anticipated for the [OSSO]2− fragment in the former.31,34,35 Notably, complex 2 is only the third OSSO complex to be reported and only second to be structurally characterized.29,36–38
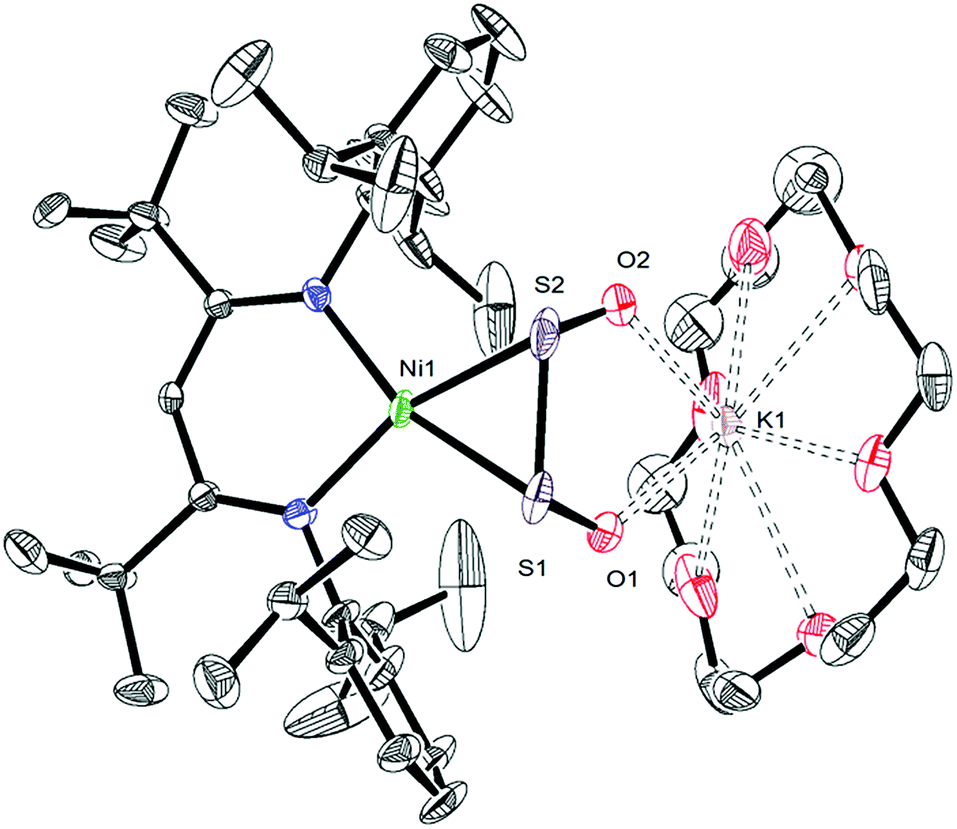 | ||
| Fig. 2 ORTEP drawing of [K(18-crown-6)][LtBuNiII(η2-OSSO)]·2C6H14 (2·2C6H14) shown with 50% thermal ellipsoids. Hydrogen atoms and C6H14 solvate molecules have been omitted for clarity. Selected metrical parameters: S1–S2 2.093(3) Å, S1–O1 1.485(5) Å, S2–O2 1.496(7) Å, Ni1–S1 2.181(2) Å, Ni1–S2 2.173(2) Å, Ni1–N1 1.920(4) Å, Ni1–N2 1.925(4) Å, O1–K1 2.747(4) Å, O2–K1 2.777(6) Å, N1–Ni1–N2 97.3(2)°, N1–Ni1–S1 102.1(1)°, N2–Ni1–S2 102.9(1)°, O1–S1–S2 107.4(2)°, O2–S2–S1 107.4(2)°. | ||
To account for the presence of 2 in the reaction mixture, we hypothesize that complex 1 undergoes a formal disproportionation, forming 2 and an equivalent of unobserved “[K(18-crown-6)][LtBuNi0]”. However, because of the low yield (typically less than 3% relative to complex 1, as assessed by 1H NMR spectroscopy), this transformation must be very inefficient. The low yield has also impeded our ability to fully characterize this complex.
 | (2) |
Synthesis of an [η2-S2]2− complex
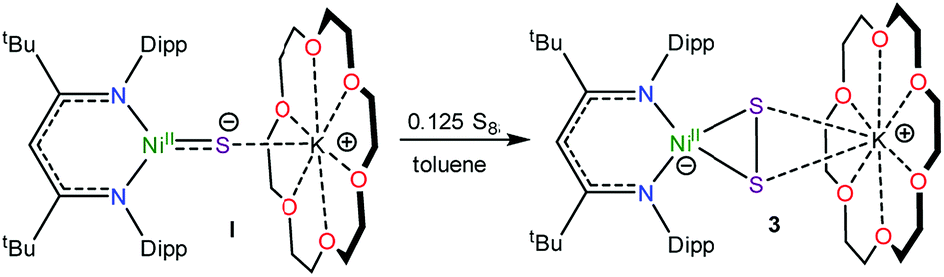
To further support the formation of the disulfide ([η2-S2]2−) complex, [K(18-crown-6)][LtBuNiII(η2-S2)] (3), during the synthesis of 1, we endeavored to independently synthesize 3. We, and others, have previously shown that terminal metal sulfides can react with S8 to form metal disulfides.39–41 Thus, we explored the reaction of [K(18-crown-6)][LtBuNiII(S)] (I) with elemental sulfur. Addition of 0.125 equiv. of S8 to a toluene solution of [K(18-crown-6)][LtBuNiII(S)] results in a rapid color change from brown to orange. Work-up of the reaction mixture affords [K(18-crown-6)][LtBuNiII(η2-S2)] (3), as an orange crystalline solid in 81% yield (eqn (3)). The solid state molecular structure of 3 is shown in Fig. 3. The disulfide (S22−) ligand in 3 has a S–S distance of 2.050(2) Å, consistent with a single bond.18 This distance is comparable to those reported for other NiII(η2-S2) complexes.42–49 The Ni–S distances (2.202(2) and 2.199(2) Å) in 3 are consistent with single bonds, and are much longer than the Ni–S bond length in the starting material (I, 2.064(2) Å). Finally, the Ni–N bonds in 3 are similar to those found in other square planar NiII β-diketiminate complexes.16,49,50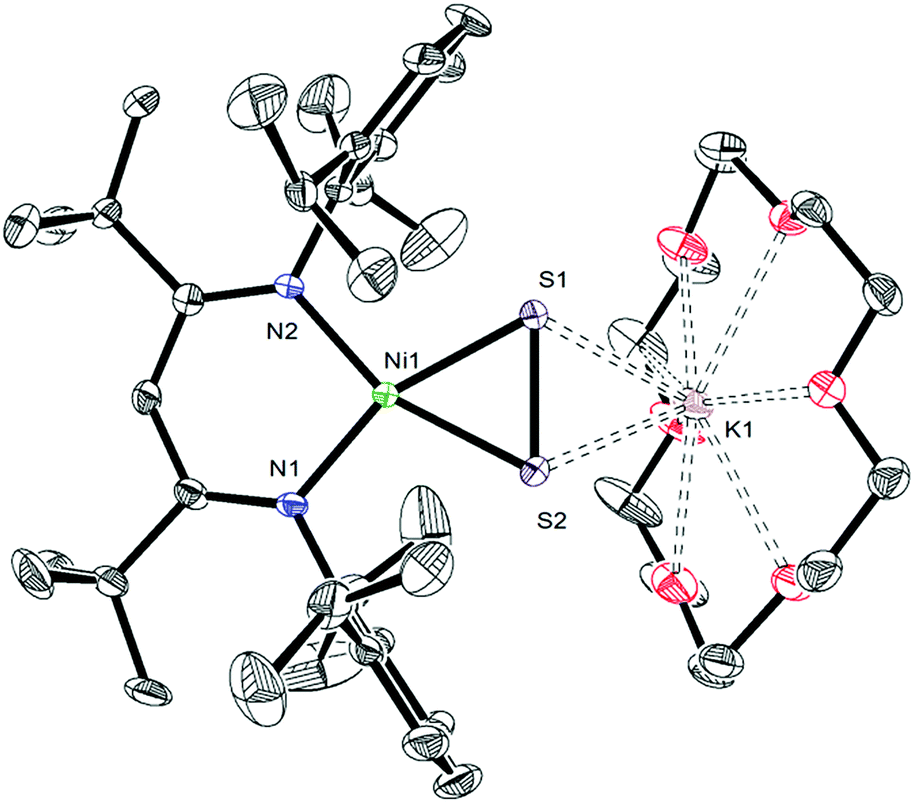 | ||
| Fig. 3 ORTEP drawing of [K(18-crown-6)][LtBuNiII(η2-S2)]·2C7H8 (3·2C7H8) shown with 50% thermal ellipsoids. Hydrogen atoms and C7H8 solvate molecules have been omitted for clarity. Selected metrical parameters: S1–S2 2.050(2) Å, Ni1–S1 2.202(2) Å, Ni1–S2 2.199(2) Å, Ni1–N1 1.900(4) Å, Ni1–N2 1.906(4) Å, S1–K1 3.248(2) Å, S2–K1 3.249(2) Å, N1–Ni1–N2 98.0(2)°, N1–Ni1–S2 103.1(1)°, N2–Ni1–S1 103.4(1)°. | ||
The 1H NMR spectrum of 3 in toluene-d8 (Fig. S7†) is consistent with a C2v symmetric, diamagnetic, square planar NiII complex and features one tert-butyl resonance at 1.30 ppm and a single γ-CH resonance at 5.46 ppm. Importantly, this latter resonance is also present in the in situ1H NMR spectrum of the thermolysis of II (Fig. S3†), confirming the formation of 3 during that reaction, via an as-yet-unknown mechanism.
 | (3) |
Reactivity of the [η2-SO]2− ligand
While the reactivity of the SO ligand has not been well established, it is known to react with phosphines. For example, Schmid and co-workers reported that [(diphos)2Ir(η2-OSSO)][Cl] reacted with PPh3 to form Ph3PO, Ph3PS, and [(diphos)2IrCl].36 Similarly, Rauchfuss and co-workers demonstrated that Cp2Nb(S2O)Cl reacted with Ph3P to form Cp2Nb(O)Cl and two equiv. of Ph3PS.29 Both transformations were presumed to proceed through an unobserved SO intermediate. More recently, Mizobe et al. reported that PPh3 could abstract an O-atom from the thioperoxide ligand in [(Cp′RuCl)2(SbCl2)(μ-Cl)(μ3:κ2-SO)] (Cp′ = C5Me4Et).25 In contrast, the reactivity of the SO ligand with CO has not been studied. Accordingly, we explored the reactivity of [K(18-crown-6)][LtBuNiII(η2-SO)] (1) with this substrate. Thus, exposure of a C6D6 solution of complex 1 to an atmosphere of 13CO results in complete consumption of 1 after 6 h. A 13C{1H} NMR spectrum (Fig. S11†) of the reaction mixture reveals the formation of several 13C-enriched products, indicating the incorporation of 13CO. Specifically, this spectrum features resonances at 214.7, 177.3, 165.3, and 152.9 ppm, which are assignable to [K(18-crown-6)][LtBuNiII(η2-SCO)] (4),51 [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)] (5), [K(18-crown-6)][LtBuNi(κ2-CO3)] (6), and SCO (7),52 respectively (Scheme 1). This spectrum also features a minor 13C-enriched resonance at 206.9 ppm, which we have tentatively assigned to [K(18-crown-6)][LtBuNiII(S2CO)] (8), on the basis of the similarity of its dithiocarbonate ([S2CO]2−) chemical shift with those reported for other dithiocarbonate complexes.52–54 | ||
| Scheme 1 | ||
A 1H NMR spectrum of the reaction mixture further supports these assignments. Specifically, an examination of the γ-CH region of this spectrum reveals overlapping resonances at 5.48 ppm (Fig. S10†), which are assignable to [K(18-crown-6)][LtBuNiII(η2-SCO)] (4)51 and [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)] (5), and a resonance at 5.42 ppm, assignable to [K(18-crown-6)][LtBuNi(κ2-CO3)] (6). This spectrum also contains a minor resonance at 5.57 ppm that has been tentatively assigned to [K(18-crown-6)][LtBuNiII(S2CO)] (8). Interestingly, at short reaction times, we observe the presence of a paramagnetic intermediate in the reaction mixture (Fig. S9†). We have identified this intermediate as the NiII sulfide, [K(18-crown-6)][LtBuNiII(S)] (I), on the basis of the similarity of its 1H NMR resonances with those of the previously characterized material.16 For example, this intermediate features diagnostic resonances at −130.25, −0.63, and 5.87 ppm, which are assignable to the γ-proton of the LtBu ligand, its tBu substituents, and one environment of its diastereotopic iPr methyl groups, respectively. For comparison, these resonances appear at −115.21, −0.88, and 6.56 ppm, respectively, for authentic I.16 This intermediate is quickly formed upon addition of 13CO, but its signals immediately begin to decay, and they are completely absent after 6 h (Fig. S9†).
We also characterized the products of the reaction of 1 and CO by IR spectroscopy. An IR spectrum of the reaction residue, dissolved in hexanes, reveals the presence of νCO modes at 2021, 1666, and 1620 cm−1 (Fig. S24†), which are assignable to the νCO modes of [LtBuNiI(CO)] (9),55 [K(18-crown-6)][LtBuNiII(η2-SCO)] (4),51 and [K(18-crown-6)][LtBuNiII(κ2-CO3)] (6), respectively. Curiously, though, we do not observe any signals in the 1H NMR spectrum of the reaction mixture that could be assigned to paramagnetic 9, suggesting that it is only a minor product of the reaction.
The 13C NMR spectrum of the in situ reaction mixture also features a minor 13C-enriched resonance at 178.5, as well as a major resonance at 191.4 ppm (Fig. S11†). While these two resonances remain unassigned, we know that neither of the peaks is assignable to [K(18-crown-6)][LtBuNiII(η2-CO2)] (10), as we have performed the independent synthesis of this complex for spectroscopic comparison (see below). We also do not observe a resonance that could be assignable to free CO2.
Finally, we observe no reaction between [K(18-crown-6)][LtBuNiII(η2-SO)] (1) and PPh3 in C6D6, according to 1H and 31P NMR spectroscopies. The lack of reactivity of the [SO]2− ligand in 1 with PPh3 is somewhat surprising on the basis of thermodynamic considerations,56 and could reflect steric shielding of the [SO]2− ligand by the bulky Dipp substituents.
Synthesis of an [S,O:κ2-SCO2]2− complex
To further support the formation of [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)] (5) upon reaction of 1 with CO, we pursued its synthesis via an independent route. Thus, exposure of a C6D6 solution of [K(18-crown-6)][LtBuNiII(S)] (I) to excess carbon dioxide (CO2) results in a rapid color change from deep brown to gold. The 1H NMR spectrum of the reaction mixture taken 15 min after addition of CO2 reveals full consumption of the starting material and formation of a new diamagnetic product whose spectroscopic signatures are consistent with a square planar NiII complex.51 Work-up of the reaction mixture provides 5 as a pale brown crystalline solid in 57% yield (eqn (4)). The solid state molecular structure of 5 is shown in Fig. 4. The thiocarbonate ([S,O:κ2-SCO2]2−) ligand in 5 features a μ:κ2,κ2 binding mode and is disordered over two positions, which are related by a C2 rotation about the Ni–K vector, in a 87:13 ratio. The S–C (1.756(4) Å) and O–C (1.279(5) and 1.238(4) Å) bond lengths in 5 are consistent with those observed for previously reported [SCO2]2− complexes,57,58 while the Ni–S and Ni–O distances are 2.234(1) Å and 1.922(3) Å, respectively. Moreover, the K–S and K–O distances are 3.531(1) Å and 2.715(3) Å, respectively, which are comparable to other K–S and K–O dative interactions.59,60 Finally, the Ni–N distances in 5 are comparable to those found in the starting material.16 To the best of our knowledge, complex 5 is the first structurally characterized transition metal complex containing the [SCO2]2− ligand. Other structurally characterized thiocarbonate complexes include [{((AdArO)3N)U}2(μ-η1,(O):κ2(O′,S)SCO2)], prepared by reaction of [{((AdArO)3N)U}2(μ-S)] with CO2, and [Cp*2Sm(μ-η1:κ2-SCO2)SmCp*2], prepared via reaction of [(Cp*2Sm)2(μ-O)] with COS.57,58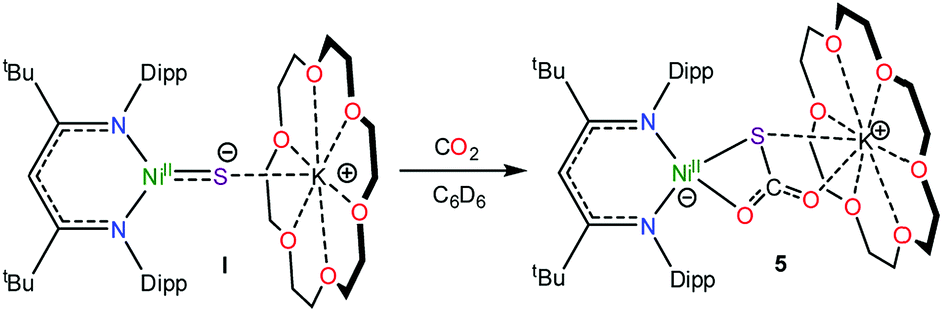 | (4) |
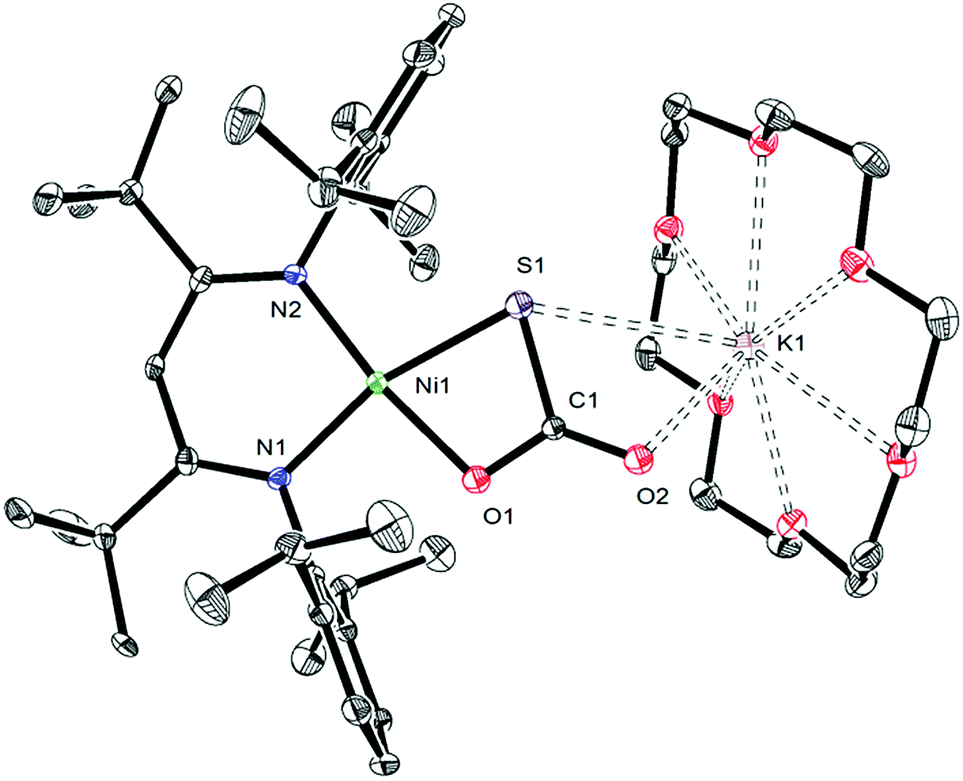 | ||
| Fig. 4 ORTEP drawing of [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)]·1.5C7H8 (5·1.5C7H8) shown with 50% thermal ellipsoids. Hydrogen atoms, C7H8 solvate molecules, and one orientation of the disordered [S,O:κ2-SCO2]2− ligand have been omitted for clarity. Selected metrical parameters: S1–C1 1.756(4) Å, O1–C1 1.279(5) Å, O2–C1 1.238(4) Å, Ni1–S1 2.234(1) Å, Ni1–O1 1.922(3) Å, Ni1–N1 1.904(3) Å, Ni1–N2 1.899(3) Å, S1–K1 3.531(1) Å, O2–K1 2.715(3) Å, S1–C1–O1 108.0(3)°, S1–C1–O2 126.2(3)°, O1–C1–O2 125.9(4)°, N1–Ni1–N2 96.7(1)°, N1–Ni1–O1 91.5(1)°, N2–Ni1–S1 99.22(9)°. | ||
The 13C{1H} NMR spectrum of 5 in benzene-d6 features a resonance at 177.3 ppm, which we have assigned to the [S,O:κ2-SCO2]2− moiety (Fig. S13†). This chemical shift is identical to the resonance assigned to this complex in the in situ13C NMR spectrum of the reaction of 1 with CO (Fig. S11†). Moreover, the 1H NMR spectrum of 5 in C6D6 features a γ-CH resonance at 5.48 ppm. This resonance is also present in the in situ1H NMR spectrum of the reaction mixture of 1 and 13CO (Fig. S10†), further confirming its formation in that transformation. Overall, these data conclusively demonstrate that complex 5 is formed during reduction of [K(18-crown-6)][LtBuNiII(η2-SO)] (1) with CO.
Synthesis of an [κ2-CO3]2− complex
To further support the formation of [K(18-crown-6)][LtBuNi(κ2-CO3)] (6) upon reaction of 1 with CO, we pursued its synthesis via an independent route. The hexameric nickel carbonate complex, [K]6[LtBuNiII(κ2-CO3)]6,11 first reported by Limberg and coworkers in 2012, was found to serve as a convenient starting material for the synthesis of [K(18-crown-6)][LtBuNiII(κ2-CO3)] (6). Addition of 6 equiv. of 18-crown-6 to a suspension of [K]6[LtBuNiII(κ2-CO3)]6 results in the formation of complex 6 in 52% yield (eqn (5)). Its solid state molecular structure is shown in Fig. 5. The carbonate (CO32−) ligand in 6 features a μ:κ2,η1 binding mode, identical to that observed for the trithiocarbonate (CS32−) ligand in [K(18-crown-6)][LtBuNi(κ2-CS3)].50 The O1–C1 (1.306(7) Å), O2–C1 (1.309(7) Å), and O3–C1 (1.242(7) Å) bond lengths in 6 are consistent with those reported for [K]6[LtBuNiII(κ2-CO3)]6,11 while the Ni–O1 and Ni–O2 distances are 1.882(4) and 1.901(4) Å, respectively, which are similar to those reported for the starting material.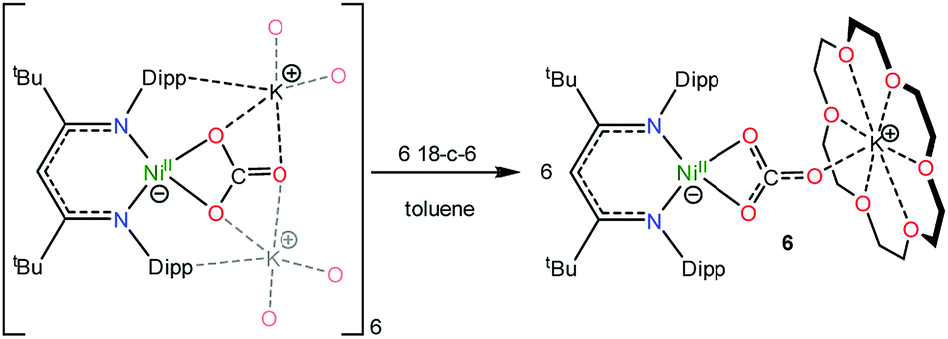 | (5) |
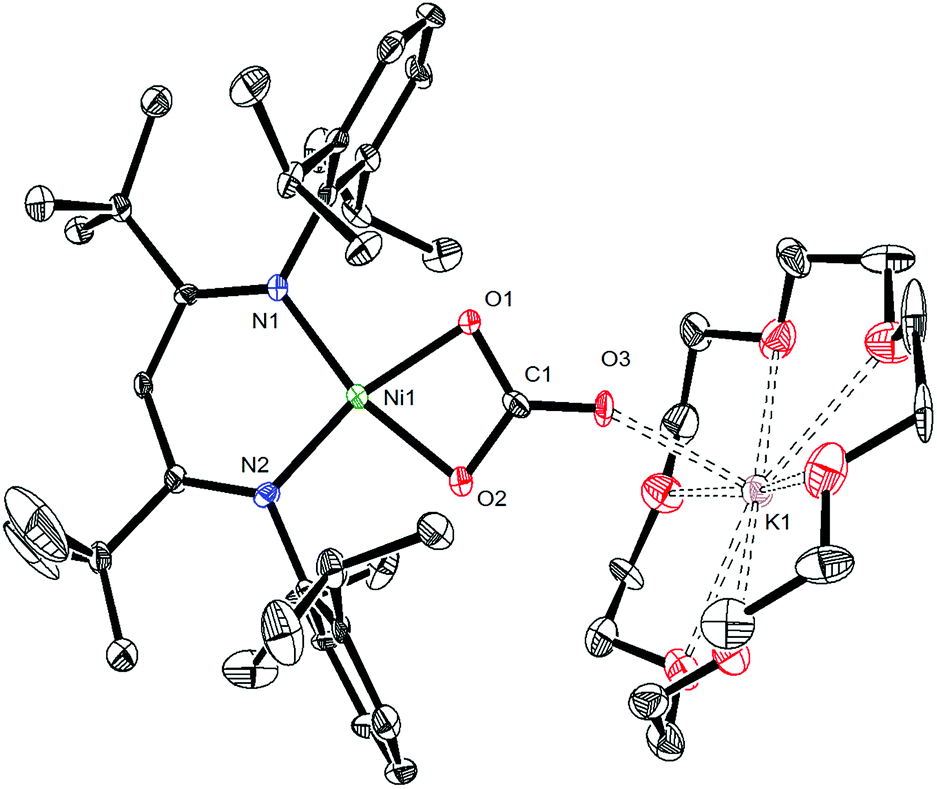 | ||
| Fig. 5 ORTEP drawing of [K(18-crown-6)][LtBuNiII(κ2-CO3)]·0.5C5H12 (6·0.5C5H12) shown with 50% thermal ellipsoids. Hydrogen atoms, a C5H12 solvate molecule, and a second independent molecule of [K(18-crown-6)][LtBuNiII(κ2-CO3)] have been omitted for clarity. Selected metrical parameters: C1–O1 1.306(7) Å, C1–O2 1.309(7) Å, C1–O3 1.242(7) Å, Ni1–O1 1.882(4) Å, Ni1–O2 1.901(4) Å, Ni1–N1 1.883(5) Å, Ni1–N2 1.879(5) Å, O3–K1 2.510(4) Å, O1–C1–O2 110.8(5)°, O1–C1–O3 125.0(6)°, N1–Ni1–N2 97.9(2)°, N1–Ni1–O1 96.6(2)°, N2–Ni1–O2 96.5(2)°. | ||
The 13C{1H} NMR spectrum of 6 in C6D6 features a resonance at 165.3 ppm, which is assignable to the [CO3]2− moiety (Fig. S15†). This chemical shift matches the resonance assigned to this complex in the in situ13C NMR spectrum of the reaction mixture of 1 and 13CO (Fig. S11†). In addition, the 1H NMR spectrum of 6 in C6D6 features a γ-CH resonance at 5.42 ppm, which is present in the in situ1H NMR spectrum of the reaction mixture of 1 and 13CO (Fig. S10†). The IR spectrum (hexanes solution) of 6 features a strong νCO mode at 1620 cm−1, which is also present in a solution IR spectrum of the reaction mixture formed upon addition of CO to 1 (Fig. S24†). Overall, these data conclusively demonstrate that complex 6 is formed during reduction of [K(18-crown-6)][LtBuNiII(η2-SO)] (1) with CO.
Synthesis of an [η2-CO2]2− complex
In an effort to assign the resonance at 191.4 ppm in the in situ13C{1H} NMR spectrum of the reaction of 1 and 13CO, we endeavored to independently synthesize the carbon dioxide complex, [K(18-crown-6)][LtBuNiII(η2-CO2)] (10). We rationalized that 10 was a plausible reaction product, given the formation of CO2 during the reaction (see below). Several previously reported Ni(CO2) complexes have been synthesized by reaction of CO2 with a Ni0 precursor.61–64 In a similar vein, the Ni(0)–N2 complex, [K]2[LtBuNi0(μ-η1:η1-N2)Ni0LtBu], previously reported by Limberg and co-workers in 2009,65 was found to serve as an effective Ni0 source for the synthesis of 10. Thus, exposure of [K]2[LtBuNi0(μ-η1:η1-N2)Ni0LtBu] to two equiv. of CO2, followed by addition of 18-crown-6, resulted in the formation of 10 (eqn (6)), which was isolated as pale orange plates in 41% yield after work-up. Its formulation was confirmed by X-ray crystallography and its solid state molecular structure is shown in Fig. 6.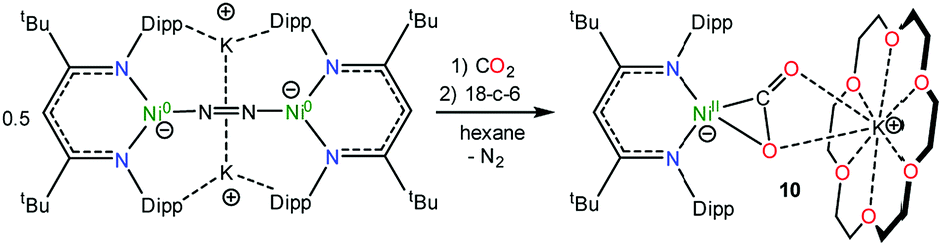 | (6) |
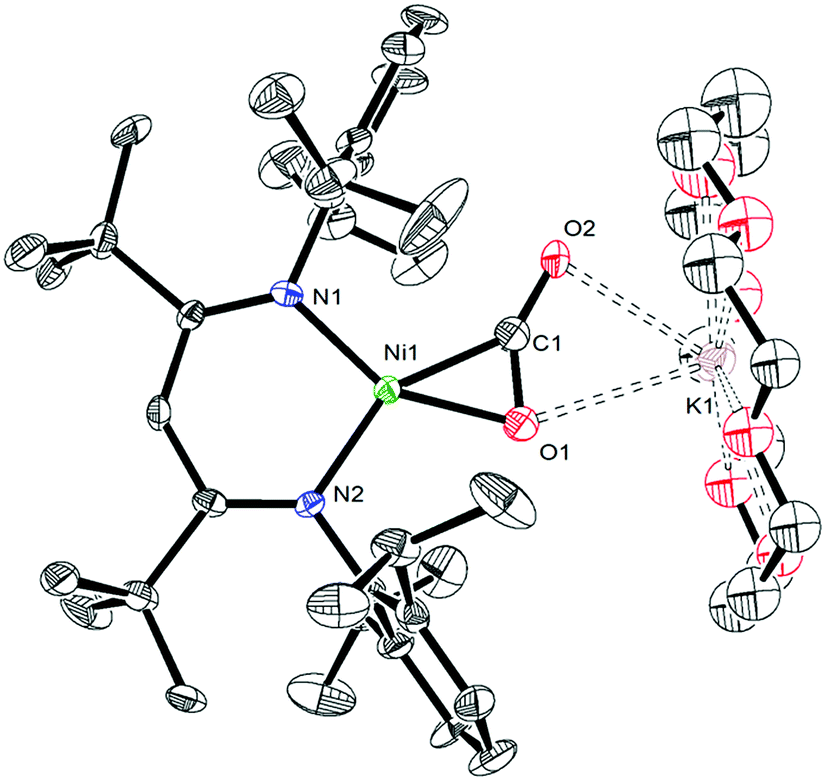 | ||
| Fig. 6 ORTEP drawing of [K(18-crown-6)][LtBuNiII(η2-CO2)]·2C6H6 (10·2C6H6) shown with 50% thermal ellipsoids. Hydrogen atoms, C6H6 solvate molecules, and second orientations of the CO2 and 18-crown-6 fragments have been omitted for clarity. Selected metrical parameters: C1–O1 1.231(9) Å, C1–O2 1.22(1) Å, Ni1–C1 1.890(6) Å, Ni1–O1 1.897(6) Å, Ni1–N1 1.901(6) Å, Ni1–N2 1.896(5) Å, O1–K1 2.980(6) Å, O2–K1 2.71(1) Å, O1–C1–O2 144.0(8)°, N1–Ni1–N2 99.2(2)°, N1–Ni1–C1 112.2(3)°, N2–Ni1–O1 110.7(3)°. | ||
Complex 10 features a square planar NiII center ligated by the β-diketiminate ligand and a [CO2]2− ligand. The [CO2]2− ligand in 10 features a μ:η2,κ2 binding mode, similar to that observed for the [COS]2− ligand in complex 4. The [CO2]2− ligand in 10 is disordered over two positions, in a 76:24 ratio, which are related by a C2 rotation axis about the Ni–K vector. The Ni1–O1 (1.897(6) Å) and Ni1–C1 (1.890(6) Å) distances are consistent with those previously reported for the Ni(η2-CO2) fragment.61,62,64,66,67 Additionally, the Ni–N bonds in 10 are consistent with those found in other square planar NiII β-diketiminate complexes.16,49,50
The 1H NMR spectrum of 10 in C6D6 is consistent with that expected for a Cs symmetric, square planar NiII complex. It features two tert-butyl resonances at 1.42 and 1.34 ppm, and a single γ-CH resonance at 5.42 ppm. Its 13C{1H} NMR spectrum in C6D6 features a resonance at 167.2 ppm, which we have assigned to the [η2-CO2]2− ligand. This chemical shift is consistent with those reported for previously isolated Ni(η2-CO2) complexes.62–64 Most importantly, however, these resonances are not observed in the in situ13C{1H} and 1H NMR spectra of the reaction between 1 and 13CO (Fig. S10 and 11†). Thus, we can definitively conclude that complex 10 is not being formed in that reaction. Finally, complex 10 features a νCO mode at 1664 cm−1 in its IR spectrum (KBr pellet), which is similar to those reported for other nickel CO2 complexes.62–64 This vibration is also not present in the in situ IR spectrum (recorded in hexanes) of the reaction residue formed upon reaction of 1 with CO (Fig. S24†).
Mechanistic considerations
To rationalize the formation of complexes 4 and 5, we propose that CO initially reacts with 1 to form CO2 and [K(18-crown-6)][LtBuNiII(S)] (I) (Scheme 2). Complex I then reacts with either CO or CO2 to yield [K(18-crown-6)][LtBuNiII(η2-SCO)] (4) or [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)] (5), respectively. Significantly, their presence, along with the observation of [K(18-crown-6)][LtBuNiII(S)] (I) in the reaction mixture, demonstrates the formal reduction of N2O by CO, as originally envisioned. That said, the reaction rates of I with CO and CO2 are qualitatively similar to the reaction rate of I with N2O. As a consequence, I is unlikely to be an effective catalyst for N2O reduction because off-cycle reactions with CO and CO2 would be competitive with the desired N2O capture reaction (Scheme 2).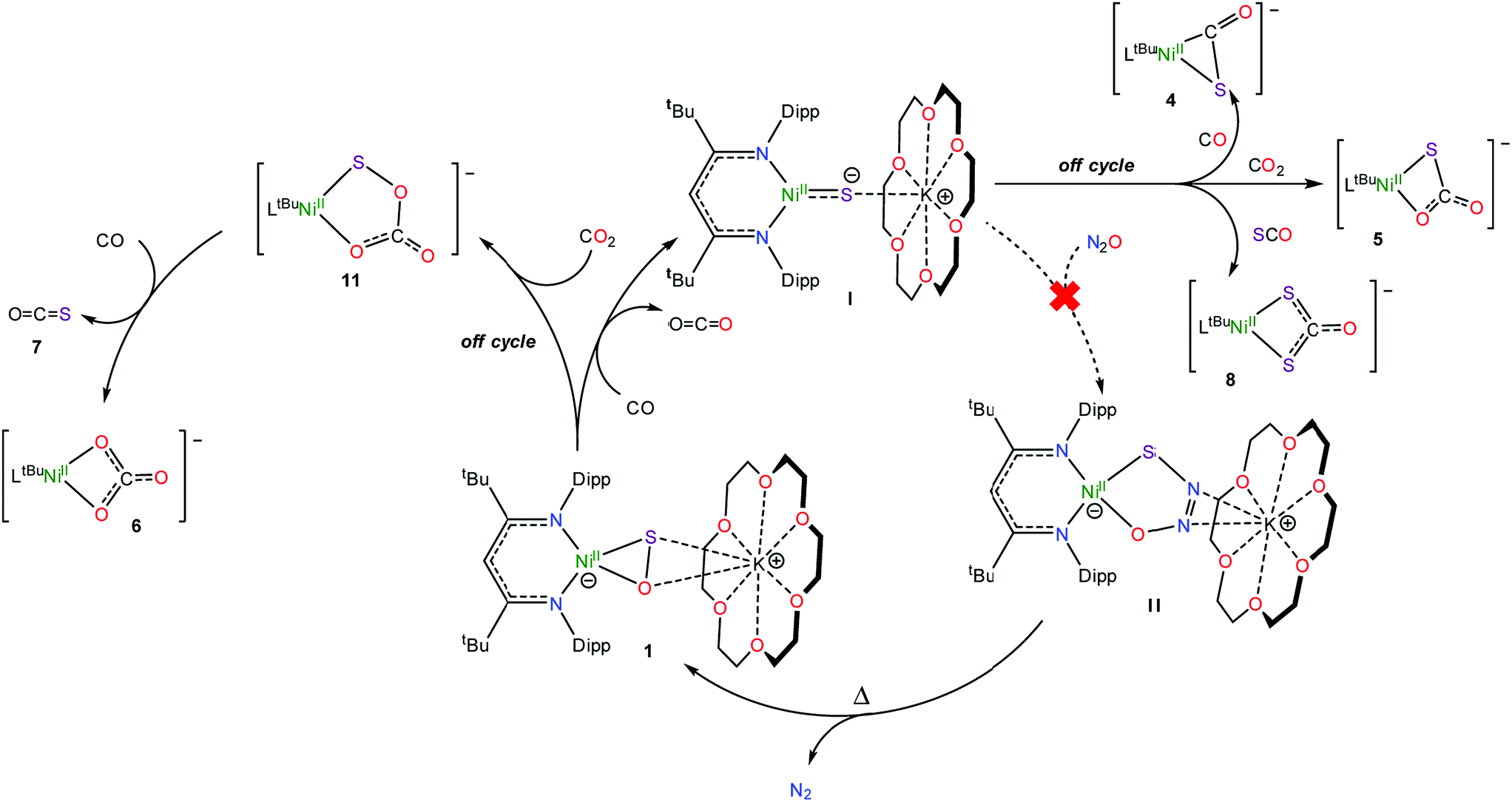 | ||
| Scheme 2 | ||
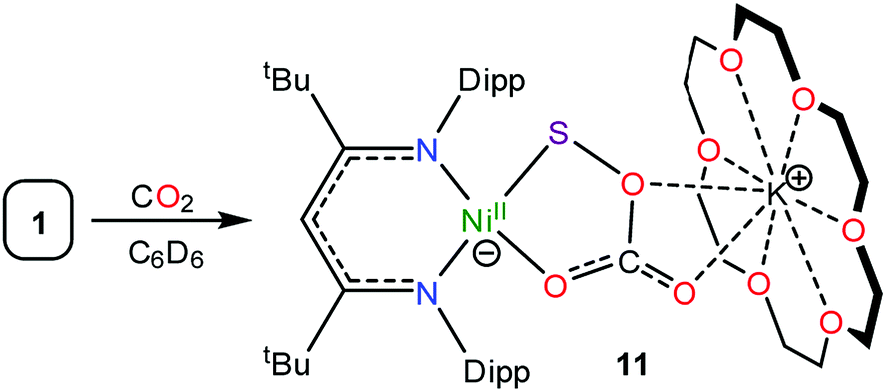
To rationalize the formation of complex 6 and COS, we propose that reaction of the newly formed CO2 with unreacted 1 results in the formation of a transient, unobserved nickel monothiopercarbonate complex, [K(18-crown-6)][LtBuNiII(κ2-SOCO2)] (11). Complex 11 then transfers a sulfur atom to CO to form [K(18-crown-6)][LtBuNiII(κ2-CO3)] (6) and COS (7) (Scheme 2), both of which were confirmed to be present in the in situ reaction mixture. This hypothesis also nicely explains the presence of [K(18-crown-6)][LtBuNiII(κ2-S2CO)] (8), which could be formed via the reaction of 7 with I (Scheme 2). While a monothiopercarbonate complex has not been previously reported, the reaction of metal peroxides (O22−) with CO2 is known to yield peroxocarbonate ([OOCO2]2−) complexes.68–71 Similarly, metal disulfides (S22−) are known to react with CS2 to form perthiocarbonates ([SSCS2]2−).72,73 Moreover, peroxocarbonates are known to be very effective O-atom donors.69,74–77
Consistent with this hypothesis, reaction of [K(18-crown-6)][LtBuNiII(η2-SO)] (1) with CO2 in C6D6 results in the rapid formation of a new diamagnetic NiII complex, as evidenced by the appearance of diagnostic resonances at 4.49 ppm (γ-CH) and 1.20 ppm (tBu) in the in situ1H NMR spectrum of the reaction mixture (Fig. S18†). We have assigned these resonances to the monothiopercarbonate complex [K(18-crown-6)][LtBuNiII(κ2-SOCO2)] (11) (eqn (7)). Complex 11 is the only product observed in the reaction mixture. These results provide further support for the overall reaction mechanism proposed in Scheme 2 and suggest that (SOCO2)2− could function as a very effective a S-atom transfer reagent.78
 | (7) |
Conclusions
Gentle thermolysis of the thiohyponitrite complex, [K(18-crown-6)][LtBuNiII(κ2-SNNO)], results in extrusion of N2 and formation of [K(18-crown-6)][LtBuNiII(η2-SO)], a rare example of a structurally characterized SO complex, along with trace amounts of [K(18-crown-6)][LtBuNiII(η2-OSSO)] and [K(18-crown-6)][LtBuNiII(η2-S2)]. [K(18-crown-6)][LtBuNiII(η2-SO)] reacts rapidly with CO, forming the “masked” terminal Ni(II) sulfide intermediate, [K(18-crown-6)][LtBuNiII(S)], along with CO2, via O-atom abstraction. This Ni(II) sulfide intermediate then reacts with CO or CO2 to form [K(18-crown-6)][LtBuNiII(η2-SCO)] and [K(18-crown-6)][LtBuNi(S,O:κ2-SCO2)], respectively. [K(18-crown-6)][LtBuNiII(η2-SO)] can also react with the newly formed CO2 to form a putative monothiopercarbonate complex, [K(18-crown-6)][LtBuNiII(κ2-SOCO2)], which can then transfer an S atom to CO, forming COS and [K(18-crown-6)][LtBuNiII(κ2-CO3)].Significantly, the observation of [K(18-crown-6)][LtBuNiII(S)] in the reaction mixture, along with the formation of [K(18-crown-6)][LtBuNiII(η2-SCO)] and [K(18-crown-6)][LtBuNiII(S,O:κ2-SCO2)], confirms that the SO ligand is susceptible to O-atom abstraction by CO, which had not been previously demonstrated. More importantly, these reaction products reveal the stepwise conversion of [K(18-crown-6)][LtBuNiII(κ2-SNNO)] to [K(18-crown-6)][LtBuNiII(η2-SO)] and then [K(18-crown-6)][LtBuNiII(S)], which represents a formal reduction of N2O by CO, forming N2 and CO2. Significantly, this transformation parallels the chemistry mediated by nano-particulate Pt/Rh in catalytic converters. In contrast to the metal-centered redox of the catalytic converter example, however, the redox chemistry in our system occurs at the sulfide ligand, while the nickel center remains in the 2+ oxidation state at every step. The use of ligand-centered redox is an intriguing strategy for N2O reduction and we suggest that the study of model systems, such as the one presented in this manuscript, could inspire the design of a new generation of homogeneous and heterogeneous N2O reduction catalysts.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the National Science Foundation (CHE 1361654) for financial support of this work. We would also like to thank the Scott Group at UCSB for the use of their 13CO. This research made use of the 400 MHz NMR Spectrometer in the Department of Chemistry and Biochemistry, an NIH SIG (1S10OD012077-01A1).References
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef PubMed.
- W. C. Trogler, Coord. Chem. Rev., 1999, 187, 303–327 CrossRef.
- A. V. Leontev, O. g. A. Fomicheva, M. V. Proskurnina and N. S. Zefirov, Russ. Chem. Rev., 2001, 70, 91 CrossRef.
- M. J. Prather, Science, 1998, 279, 1339–1341 CrossRef PubMed.
- E. A. Davidson and D. Kanter, Environ. Res. Lett., 2014, 9, 105012 CrossRef.
- M. Konsolakis, ACS Catal., 2015, 5, 6397–6421 CrossRef.
- B. Neuffer, N. Frank and M. Desai, Available and Emerging Technologies for Reducing Greenhouse Gas Emissions from the Nitric Acid Production Industry, Report of the Office of Air and Radiation, U.S. Environmental Protection Agency, Research Triangle Park, NC, 2010 Search PubMed.
- M. Dameris, Angew. Chem., Int. Ed., 2010, 49, 489–491 CrossRef PubMed.
- P. Granger and V. I. Parvulescu, Chem. Rev., 2011, 111, 3155–3207 CrossRef PubMed.
- J. P. Reeds, B. L. Yonke, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2011, 133, 18602–18605 CrossRef PubMed.
- B. Horn, C. Limberg, C. Herwig, M. Feist and S. Mebs, Chem. Commun., 2012, 48, 8243–8245 RSC.
- A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072–5085 CrossRef.
- J.-H. Lee, M. Pink, J. Tomaszewski, H. Fan and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 8706–8707 CrossRef PubMed.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 CrossRef PubMed.
- R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720–5723 CrossRef PubMed.
- N. J. Hartmann, G. Wu and T. W. Hayton, Angew. Chem., Int. Ed., 2015, 54, 14956–14959 CrossRef PubMed.
- K. Severin, Chem. Soc. Rev., 2015, 44, 6375–6386 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- F. X. Powell and D. R. Lide Jr, J. Chem. Phys., 1964, 41, 1413–1419 CrossRef.
- B. Horn, C. Limberg, C. Herwig and B. Braun, Inorg. Chem., 2014, 53, 6867–6874 CrossRef PubMed.
- C. Bianchini, C. Mealli, A. Meli and M. Sabat, J. Chem. Soc., Chem. Commun., 1985, 1024–1025 RSC.
- A. Neher, O. Heyke and I. P. Lorenz, Z. Anorg. Allg. Chem., 1989, 578, 185–190 CrossRef.
- R. Huang, I. A. Guzei and J. H. Espenson, Organometallics, 1999, 18, 5420–5422 CrossRef.
- C. Minh Tuong, W. K. Hammons, A. L. Howarth, K. E. Lutz, A. D. Maduvu, L. B. Haysley, B. R. T. Allred, L. K. Hoyt, M. S. Mashuta and M. E. Noble, Inorg. Chem., 2009, 48, 5027–5038 CrossRef PubMed.
- K. Oya, H. Seino, M. Akiizumi and Y. Mizobe, Organometallics, 2011, 30, 2939–2946 CrossRef.
- R. Wang, M. S. Mashuta, J. F. Richardson and M. E. Noble, Inorg. Chem., 1996, 35, 3022–3030 CrossRef.
- L. E. Longobardi, V. Wolter and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 809–812 CrossRef PubMed.
- L. Markó, B. Markó-Monostory, T. Madach and H. Vahrenkamp, Angew. Chem., Int. Ed., 1980, 19, 226–227 CrossRef.
- J. E. Hoots, D. A. Lesch and T. B. Rauchfuss, Inorg. Chem., 1984, 23, 3130–3136 CrossRef.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2018, 54, 1097–1100 RSC.
- F. J. Lovas, E. Tiemann and D. R. Johnson, J. Chem. Phys., 1974, 60, 5005–5010 CrossRef.
- R. D. Harcourt, J. Mol. Struct.: THEOCHEM, 1989, 186, 131–165 CrossRef.
- R. D. Harcourt, Eur. J. Inorg. Chem., 2000, 2000, 1901–1916 CrossRef.
- M. A. Martin-Drumel, J. van Wijngaarden, O. Zingsheim, F. Lewen, M. E. Harding, S. Schlemmer and S. Thorwirth, J. Mol. Spectrosc., 2015, 307, 33–39 CrossRef.
- C. J. Marsden and B. J. Smith, Chem. Phys., 1990, 141, 335–353 CrossRef.
- G. Schmid, G. Ritter and T. Debaerdemaeker, Chem. Ber., 1975, 108, 3008–3013 CrossRef.
- I.-P. Lorenz and J. Kull, Angew. Chem., Int. Ed., 1986, 25, 261–262 CrossRef.
- The other structurally characterized example, [(diphos)2Ir(OSSO)]Cl (diphos = Ph2PCH2CH2PPh2), features a trans-OSSO ligand (dihedral angle = 68°) with an S–S bond length of 2.041 Å. Curiously, one of its S–O bond lengths (1.21(2) Å) is anomalously short, which may indicate the presence of unresolved disorder in the structure. See ref. 36.
- D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 12683–12685 CrossRef PubMed.
- Z. K. Sweeney, J. L. Polse, R. A. Andersen, R. G. Bergman and M. G. Kubinec, J. Am. Chem. Soc., 1997, 119, 4543–4544 CrossRef.
- Z. K. Sweeney, J. L. Polse, R. G. Bergman and R. A. Andersen, Organometallics, 1999, 18, 5502–5510 CrossRef PubMed.
- C. Mealli and S. Midollini, Inorg. Chem., 1983, 22, 2785–2786 CrossRef.
- R. J. Pleus, H. Waden, W. Saak, D. Haase and S. Pohl, J. Chem. Soc., Dalton Trans., 1999, 2601–2610 RSC.
- J. Cho, K. M. Van Heuvelen, G. P. A. Yap, T. C. Brunold and C. G. Riordan, Inorg. Chem., 2008, 47, 3931–3933 CrossRef PubMed.
- S. Yao, C. Milsmann, E. Bill, K. Wieghardt and M. Driess, J. Am. Chem. Soc., 2008, 130, 13536–13537 CrossRef PubMed.
- M. Inosako, A. Kunishita, M. Kubo, T. Ogura, H. Sugimoto and S. Itoh, Dalton Trans., 2009, 9410–9417 RSC.
- V. M. Iluc, C. A. Laskowski, C. K. Brozek, N. D. Harrold and G. L. Hillhouse, Inorg. Chem., 2010, 49, 6817–6819 CrossRef PubMed.
- F. Olechnowicz, G. L. Hillhouse and R. F. Jordan, Inorg. Chem., 2015, 54, 2705–2712 CrossRef PubMed.
- S. Yao, Y. Xiong, X. Zhang, M. Schlangen, H. Schwarz, C. Milsmann and M. Driess, Angew. Chem., Int. Ed., 2009, 48, 4551–4554 CrossRef PubMed.
- N. J. Hartmann, G. Wu and T. W. Hayton, Dalton Trans., 2016, 45, 14508–14510 RSC.
- N. J. Hartmann, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 12352–12355 CrossRef PubMed.
- W. S. Farrell, P. Y. Zavalij and L. R. Sita, Angew. Chem., Int. Ed., 2015, 54, 4269–4273 CrossRef PubMed.
- J. D. E. T. Wilton-Ely, D. Solanki and G. Hogarth, Inorg. Chem., 2006, 45, 5210–5214 CrossRef PubMed.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC.
- B. Horn, S. Pfirrmann, C. Limberg, C. Herwig, B. Braun, S. Mebs and R. Metzinger, Z. Anorg. Allg. Chem., 2011, 637, 1169–1174 CrossRef.
- R. H. Holm and J. P. Donahue, Polyhedron, 1993, 12, 571–589 CrossRef.
- O. P. Lam, S. M. Franke, F. W. Heinemann and K. Meyer, J. Am. Chem. Soc., 2012, 134, 16877–16881 CrossRef PubMed.
- C. Schoo, S. V. Klementyeva, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2016, 52, 6654–6657 RSC.
- D. E. Smiles, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2014, 136, 96–99 CrossRef PubMed.
- D. E. Smiles, G. Wu, N. Kaltsoyannis and T. W. Hayton, Chem. Sci., 2015, 6, 3891–3899 RSC.
- A. Döhring, P. W. Jolly, C. Krüger and M. J. Romão, Z. Naturforsch. B Chem. Sci., 1985, 40, 484–488 Search PubMed.
- R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461–1475 RSC.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC.
- J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203–10207 CrossRef PubMed.
- S. Pfirrmann, C. Limberg, C. Herwig, R. Stößer and B. Ziemer, Angew. Chem., Int. Ed., 2009, 48, 3357–3361 CrossRef PubMed.
- C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 13802–13803 CrossRef PubMed.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2018, 140, 2179–2185 CrossRef PubMed.
- P. Zimmermann, S. Hoof, B. Braun-Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2018, 57, 7230–7233 CrossRef PubMed.
- K. Hashimoto, S. Nagatomo, S. Fujinami, H. Furutachi, S. Ogo, M. Suzuki, A. Uehara, Y. Maeda, Y. Watanabe and T. Kitagawa, Angew. Chem., Int. Ed., 2002, 41, 1202–1205 CrossRef PubMed.
- G. Meier and T. Braun, Angew. Chem., Int. Ed., 2012, 51, 12564–12569 CrossRef PubMed.
- M. Yamashita, K. Goto and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 7294–7295 CrossRef PubMed.
- D. Coucouvanis and M. Draganjac, J. Am. Chem. Soc., 1982, 104, 6820–6822 CrossRef.
- S.-B. Yu and R. H. Holm, Polyhedron, 1993, 12, 263–266 CrossRef.
- A. McKillop and W. R. Sanderson, Tetrahedron, 1995, 51, 6145–6166 CrossRef.
- T. Tsugawa, H. Furutachi, M. Marunaka, T. Endo, K. Hashimoto, S. Fujinami, S. Akine, Y. Sakata, S. Nagatomo, T. Tosha, T. Nomura, T. Kitagawa, T. Ogura and M. Suzuki, Chem. Lett., 2015, 44, 330–332 CrossRef.
- B. S. Lane, M. Vogt, V. J. DeRose and K. Burgess, J. Am. Chem. Soc., 2002, 124, 11946–11954 CrossRef PubMed.
- H. Yao and D. E. Richardson, J. Am. Chem. Soc., 2000, 122, 3220–3221 CrossRef.
- J. P. Donahue, Chem. Rev., 2006, 106, 4747–4783 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details and spectral data. CCDC 1847162–1847167. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02536c |
| This journal is © The Royal Society of Chemistry 2018 |