
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoenzymatic synthesis of glycopeptides bearing rare N-glycan sequences with or without bisecting GlcNAc†
Weizhun
Yang
a,
Sherif
Ramadan
 ab,
Jared
Orwenyo
c,
Tayeb
Kakeshpour
a,
Thomas
Diaz
a,
Yigitcan
Eken
a,
Miloslav
Sanda
d,
James E.
Jackson
*a,
Angela K.
Wilson
*a and
Xuefei
Huang
*aef
ab,
Jared
Orwenyo
c,
Tayeb
Kakeshpour
a,
Thomas
Diaz
a,
Yigitcan
Eken
a,
Miloslav
Sanda
d,
James E.
Jackson
*a,
Angela K.
Wilson
*a and
Xuefei
Huang
*aef
aDepartment of Chemistry, Michigan State University, 578 South Shaw Lane, East Lansing, MI 48824, USA. E-mail: jackson@chemistry.msu.edu; wilson@chemistry.msu.edu; xuefei@chemistry.msu.edu
bChemistry Department, Faculty of Science, Benha University, Benha, Qaliobiya 13518, Egypt
cDepartment of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA
dDepartment of Oncology, Georgetown University, Washington DC 20057, USA
eDepartment of Biomedical Engineering, Michigan State University, East Lansing, MI 48824, USA
fInstitute for Quantitative Health Science and Engineering, Michigan State University, East Lansing, MI 48824, USA
First published on 31st August 2018
Abstract
N-Linked glycopeptides have highly diverse structures in nature. Herein, we describe the first synthesis of rare multi-antennary N-glycan bearing glycan chains on 6-OH of both α1,6- and α1,3-linked mannose arms. To expedite divergent generation of N-glycan structures, four orthogonal protective groups were installed at the branching points on the core tetrasaccharide, which could be removed individually without affecting one another. In addition, the synthetic route is flexible, allowing a bisecting glucosamine moiety to be introduced at a late stage of the synthesis, further expanding the diversity of sequences that could be achieved. The bisecting glucosamine unit significantly reduced the glycosylation yields of adjacent mannoses, which was attributed to steric hindrance imposed by the glucosamine based on molecular modelling analysis. The N-glycans were then transformed to oxazoline donors and ligated with a glycopeptide acceptor from haptoglobin promoted by the wild type Arthrobacter endo-β-N-acetylglucosaminidase (Endo-A). Endo-A exhibited interesting substrate preferences depending on donor sizes, which was rationalized through molecular dynamics studies. This is the first time that a glycopeptide bearing a bisecting N-acetyl glucosamine (GlcNAc), the rare N-glycan branch, and two LewisX trisaccharide antennae was synthesized, enabling access to this class of complex glycopeptide structures.
Introduction
Protein glycosylation, one of the most common post-translational modifications,1–3 plays critical roles in directing biological functions,4 stabilities5 and conformations of the parent proteins.6 Among glycoproteins, the N-linked structures form a major subset, in which carbohydrate residues are attached to the core protein backbone through amide bonds with asparagine residues.7 N-glycans share a pentasaccharide core, the two mannose units (D and E) at the non-reducing end of which can bear multiple branches (Fig. 1). While the most common points of attachment are C2/C6 hydroxy groups of mannose D (α1,6-arm) and C2/C4 hydroxy groups of mannose E (α1,3-arm), a novel β1,6-N-acetylglucosaminyltransferase (β1,6GnT-IX also designated as β1,6GnT-Vb) has been identified that can add a glycan to 6-OH of mannose E.8 β1,6 branching is known to be important to malignant phenotypes of prostate cancer, neuroblastoma and melanoma.9,10 In addition to modifications of D and E mannoses in the periphery, 4-OH of mannose C can be modified by a glycan such as N-acetyl glucosamine (GlcNAc), bisecting mannoses D and E. The bisecting GlcNAc is important for many biological processes, including tumor development,11 immune response12 and cell–cell communication.13 However, it is very challenging to obtain bisected N-glycans through either isolation from nature or synthesis.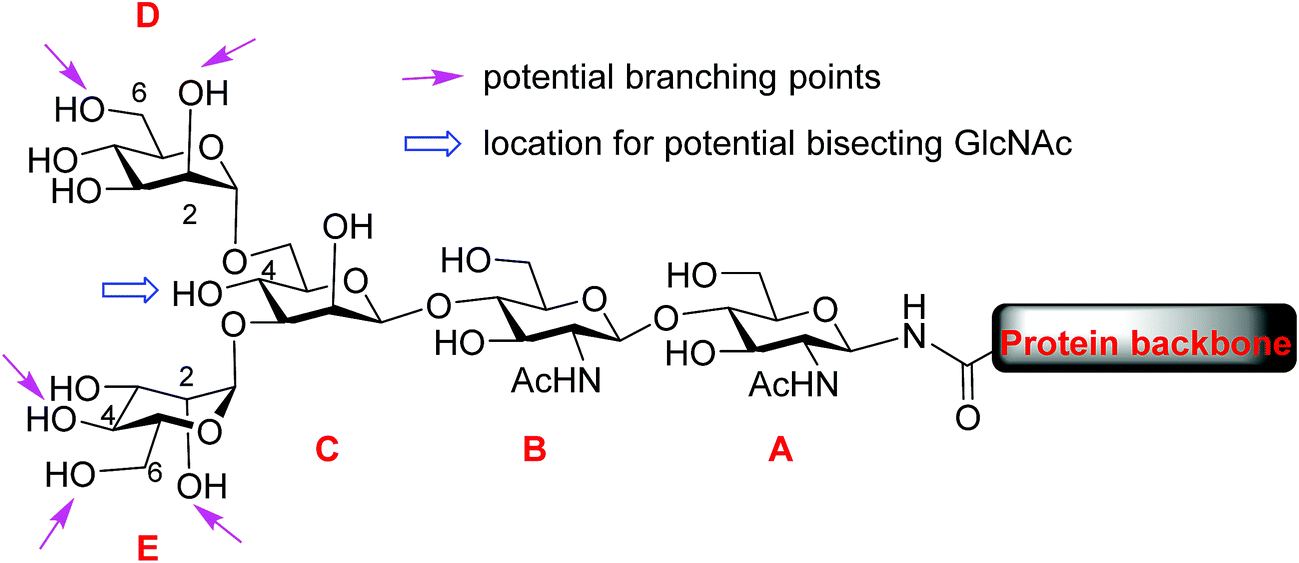 | ||
| Fig. 1 General structure of N-linked glycan. | ||
During the past decade, tremendous advances have been made in synthesis of highly complex N-glycans with many innovative methods developed.14–22 However, to date, N-glycans bearing a branch at 6-OH of mannose E (α1,3-arm) have not been synthesized. Given the structural complexity of N-glycans, synthesis of asymmetric antennae at each branching point remains a highly challenging task. To address this, we report our strategy enabling synthetic access to rare structures of N-glycans bearing antennary glycans on 6-OH of mannose E. To generate diverse glycan structures, a core tetrasaccharide was designed with potential branching points strategically protected with temporary protective groups that can be orthogonally removed. Furthermore, suitable conditions have been identified for the late stage installation of the bisecting GlcNAc moiety. The reducing ends of the synthetic N-glycans were then converted to oxazoline and transferred to a GlcNAc functionalized glycopeptide by endo-β-N-acetylglucosaminidase (Endo-A),23–28 providing access to glycopeptides bearing these rare N-glycan sequences.
To improve the overall efficiency of glyco-assembly, our N-glycan synthesis design hinges on divergent modifications of a key tetrasaccharide 1 bearing four protective groups, i.e., tert-butyl-diphenyl silyl (TBDPS), fluorenylmethyloxycarbonyl (Fmoc), 2,2,2-trichloroethoxycarbonyl (Troc) and levulinoyl (Lev), which are strategically placed at the potential branching points on mannoses D and E and may be removed orthogonally. Orthogonal deprotections have been shown to be powerful strategies for divergent synthesis of a large number of glycans including N-glycans.16,29–31 The free 4-OH of the mannose-C unit in 1 was kept free for late stage installation of the bisecting GlcNAc.
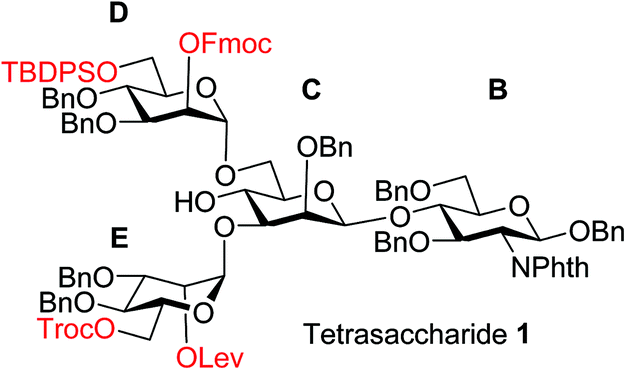
Experimental section
General experimental procedures
All chemical reactions were carried out under nitrogen with anhydrous solvents in flame-dried glassware, unless otherwise noted. Glycosylation reactions were performed in the presence of molecular sieves, which were flame-dried right before the reaction under high vacuum. Glycosylation solvents were dried using a solvent purification system and used directly without further drying. Chemicals used were reagent grade as supplied except where noted. Compounds were visualized by UV light (254 nm) and by staining with a yellow solution containing Ce(NH4)2(NO3)6 (0.5 g) and (NH4)6Mo7O24·4H2O (24.0 g) in 6% H2SO4 (500 mL). Flash column chromatography was performed on silica gel 60 (230–400 mesh). NMR spectra were referenced using residual CHCl3 (δ1H-NMR 7.26 ppm) and CDCl3 (δ13C-NMR 77.0 ppm). Peak and coupling constants assignments are based on 1H-NMR, 1H–1H gCOSY and/or 1H–13C gHMQC and 1H–13C gHMBC experiments.Solid-phase peptide synthesis using the Fmoc-strategy
Most of amino acids and resins were purchased from Chem-impex. Reaction vessels (10 mL, disposable) and the Domino Block Synthesizer were purchased from Torvig. All peptides and glycopeptides were synthesized according to the Fmoc-chemistry based on solid phase peptide synthesis procedure. Resins with pre-loaded amino acid were loaded into a plastic syringe fitted with a filter and swelled in DCM for at least 1 h. For the coupling reactions, the Fmoc-amino acid (5.0 equiv.) was activated by O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) (4.9 equiv.), 1-hydroxybenzotriazole (HOBt) (4.9 equiv.), N,N-diisopropylethylamine (DIPEA) (10.0 equiv.) and anhydrous DMF (5 mL) for 30 min. Then this mixture containing activated Fmoc amino acid was transferred to the syringe containing the resin, which was then rotated on a tube rotator to mix the solvent with the resins. After completion of coupling, the resin was washed with DCM (3 × 5 mL) and DMF (3 × 5 mL) for 1 minute, each time followed by cleavage of the Fmoc group by treatment of the resin with a solution of piperidine (20%) in DMF for at least 2 × 20 min at r.t. After every coupling step, unreacted amino groups were capped by treatment with a mixture of Ac2O (0.5 mL), and DIPEA (1 mL) in DMF (3.5 mL) (capping reagent) for 2 times at 15 min each. For coupling of the glycosylated amino acid building block, (Fmoc-[GlcNAc(OAc)3β1-]Asn-OH) (2 equiv.) was dissolved in DMF and activated with 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (2 equiv.), 1-hydroxy-7-azabenzotriazole (HOAt) (2 equiv.) and DIPEA (4 equiv.). After completion of the glycopeptide chain, the resin was washed and the glycopeptide was cleaved from the resin by treatment with trifluoroacetic acid (TFA)/triisopropylsilyl ether (TIPS)/H2O (95%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5%:2.5%) solution for 2.5 h. After filtration, the resins were washed with trifluoroacetic acid (2 × 10 mL), and the volume of the combined filtrates was concentrated to 1 mL, then absolute Et2O (15 mL) at 0 °C was added dropwise to the residues. The precipitates were separated from the mother liquor by centrifugation and washed with cold Et2O (10 mL). The crude products were dissolved in methanol and treated with NaOMe to cleave the ester protecting group on sugar, then neutralized with AcOH and the solvent was removed. Finally, the crude mixture was dissolved in H2O and subjected to a SUPELCOSILTM LC-18 HPLC column (25 cm × 10 mm, 5 mm) or (25 cm × 4.6 mm, 5 mm) for purification. The solvent systems used were A (0.1% TFA in H2O) and B (acetonitrile (MeCN)) with detection at 220 nm and 254 nm. Mass spectra were obtained by ESI mass spectra (Water Xevo G2-S Q-TOF LC-MS instrument).
:2.5%:2.5%) solution for 2.5 h. After filtration, the resins were washed with trifluoroacetic acid (2 × 10 mL), and the volume of the combined filtrates was concentrated to 1 mL, then absolute Et2O (15 mL) at 0 °C was added dropwise to the residues. The precipitates were separated from the mother liquor by centrifugation and washed with cold Et2O (10 mL). The crude products were dissolved in methanol and treated with NaOMe to cleave the ester protecting group on sugar, then neutralized with AcOH and the solvent was removed. Finally, the crude mixture was dissolved in H2O and subjected to a SUPELCOSILTM LC-18 HPLC column (25 cm × 10 mm, 5 mm) or (25 cm × 4.6 mm, 5 mm) for purification. The solvent systems used were A (0.1% TFA in H2O) and B (acetonitrile (MeCN)) with detection at 220 nm and 254 nm. Mass spectra were obtained by ESI mass spectra (Water Xevo G2-S Q-TOF LC-MS instrument).
General procedure for Fmoc removal
The starting material was dissolved in triethylamine and ethyl acetate (1/1, 0.5 mmol coupling product in 5 mL) and the reaction stirred at room temperature for 6 hours, when TLC indicated the completion of reaction. The solvent was evaporated under reduced pressure, and co-evaporated three times with ethyl acetate. Then the residue was purified by flash column chromatography.General procedure for Lev removal
To a solution of a glycan (0.1 mmol) in DCM:CH3OH (1/1) was added hydrazine acetate (1 mmol), which was prepared in situ by mixing acetic acid and 51% hydrazine (4/1). The reaction was stirred at room temperature for 3 h, after which it was diluted with ethyl acetate and washed with sat. NaHCO3 and brine. The organic phase was dried (Na2SO4), filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography.
General procedure for Troc removal
To a solution of a glycan (0.1 mmol) in THF and acetic acid (4:1, 5 mL) was added zinc dust (200 mg). The reaction was stirred at room temperature for 20 min, after which it was filtered by celite and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography.
General procedure for TBDPS removal
To a solution of a glycan (0.1 mmol) in pyridine (3 mL) was added 70% HF·Py (1 mL) at 0 °C. The reaction was stirred at room temperature for 3 h, after which it was neutralized by sat. NaHCO3. The mixture was extracted with ethyl acetate and washed with 10% HCl, sat. NaHCO3 and brine. The organic phase was dried (Na2SO4), filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure for glycosylation catalyzed by NIS/AgOTf
A mixture of acceptor (20 mM in DCM), thioglycoside donor (3 equiv to each –OH), and 4 Å molecular sieves in DCM was stirred at room temperature for 30 min. Then it was cooled to −30 °C followed by addition of N-iodosuccinimide (1.2 equiv. to donor) and AgOTf (0.1 equiv. to donor). The reaction was stirred at −30 °C for 1.5 hours, after which it was quenched by DIPEA and filtered by celite, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography.General procedure for global deprotection
A mixture of protected oligosaccharide (0.01 mmol) and ethylene diamine:nBuOH (5 mL, 1/4) was stirred at 90 °C overnight. The volatiles were evaporated, and the residue was purified by Sephadex LH-20. The crude product was dissolved in 0.5 mL pyridine followed by the addition of 0.05 mL acetic anhydride-1,1′-13C2 and the reaction was stirred overnight. The residue was purified by Sephadex LH-20. The products were then de-acetylated using sodium methoxide in MeOH (1.5 mL) overnight. The reaction mixture was neutralized by IR-120, filtered and concentrated in vacuum, and purified by Sephadex LH-20. To the products dissolved in MeOH:H2O:DCM (4/1/1, 2 mL), Pd(OH)2 (100% by weight) was added, and the mixture was hydrogenated overnight. The reaction mixture was filtered through cotton and concentrated. The residues were purified by G-15 gel filtration chromatography using water as the eluent. The collected solution containing the product was lyophilized to obtain the product as a white powder.
General procedure for synthesis of glycosyl oxazolines
The free oligosaccharide was dissolved in H2O and cooled in an ice bath. Triethylamine (45 equiv.) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (20 equiv.) were added and the reaction mixture was stirred in the ice bath. The reaction was monitored using High pH Anion Exchange Chromatography (HPAEC). Upon completion, purification was carried out on a Sephadex G10 gel filtration chromatography column eluting with 0.1% aqueous triethylamine. The pooled product fractions were then lyophilized to afford the product glycan oxazolines. The products were characterized using HPAEC and ESI mass spectrometry.Glycopeptide synthesis by Endo-A
Computational modeling of compounds 17 and 20
To obtain conformers covering a wide range of the configuration space, plain MD simulations were performed at 900 K. From the resulting MD frames, 1000 conformers were extracted and further optimized using the AM1 semi-empirical method. Then, the optimized 1000 conformers were sorted by their AM1 total electronic energies and the conformers within 6.0 kcal mol−1 of the lowest energy conformation of each compound were selected for geometrical comparison. The bonds, angles, and dihedral torsion parameters involving Si, that were absent in the amber general force field (gaff), were generated by fitting amber energies to the B3LYP/6-31G* energies of various conformers of TBDPS-OMe obtained from scanning all its dihedral angles. Amber 14 (Tools 15)32 and Gaussian33 were used for MD and quantum chemical simulations, respectively.Computational modeling of Endo-A complexes with sugar oxazolines 39 and 41
Initial coordinates of Endo-A were obtained from the Protein Data Bank34 (PDB ID: 3FHA).35 As the focus of the study was on pocket residue–ligand interaction, missing segments and residues outside the pocket region were capped using Molecular Operating Environment v.2016.08 (MOE).36 Gate-keeper residues, W216 and W244 are positioned parallel to one another during transglycoslyation.35 W244 was rotated from its original perpendicular orientation to parallel with W216. Protein was initially minimized in MOE under the AMBER ff10 force field37 and Extended Hückel Theory.The volumes of N-glycan oxazolines 39 and 41 were calculated using the van der Waals volume QSAR descriptor of MOE, using a connection table approximation to calculate 2D molecular descriptors. The compounds were then non-covalently docked with the docking program within MOE. Binding poses were refined using an induced fit refinement method.
The geometries of the N-glycan oxazoline compounds were optimized using the Gaussian 16 program package. The optimizations were performed using the AM1 method.38 The obtained Mulliken charges were used with the antechamber module of Amber 16 in the generation of parameters for the N-glycan compounds. The systems were prepared using the Leap module of AmberTools16 (ref. 32) under the AMBER ff14SB and GAFF force fields. Each enzyme complex was solvated in a 14 Å cube of TIP4P-Ew water beyond the solute and 100 mM sodium chloride. The systems were relaxed under NVT conditions over six minimization procedures with decreasing restraints on the protein of 500.0, 200.0, 20.0, 10.0, 5.0 kcal mol−1 Å−2 to no restraints. The systems were then heated to 300 K over 30 ps. Atomistic molecular dynamics simulations were performed for 30 ns at 300 K and 1 atm using AMBER 16. The SHAKE algorithm constrained bonds involving hydrogen.39 The trajectories were produced using Langevin dynamics and the pressure of the system was regulated with isotropic position scaling. Long-range electrostatic effects were modeled using the particle-mesh Ewald method with a 10 Å cutoff.
The produced trajectories were analyzed using AMBER 16 and visualized with MOE and the UCSF Chimera package. Free energy of binding was calculated for every picosecond using the Poisson Boltzmann model form the MMPBSA.py module of AmberTools and AMBER 16. The relative free energy trends between models were compared so solute entropy was neglected.
Results and discussion
The synthesis of key tetrasaccharide 1 commenced from disaccharide 227 (Scheme 1). Upon removal of the p-methoxybenzyl (PMB) group from 2, the glycosylation between the newly generated acceptor 3 and donor 4 went smoothly as promoted by N-iodosuccinimide (NIS) and TfOH to provide trisaccharide 5 in 88% yield. Trisaccharide 5 was treated with 80% AcOH to remove the benzylidene moiety followed by glycosylation with donor 7 to afford tetrasaccharide 1 bearing four different protecting groups at the branching points.
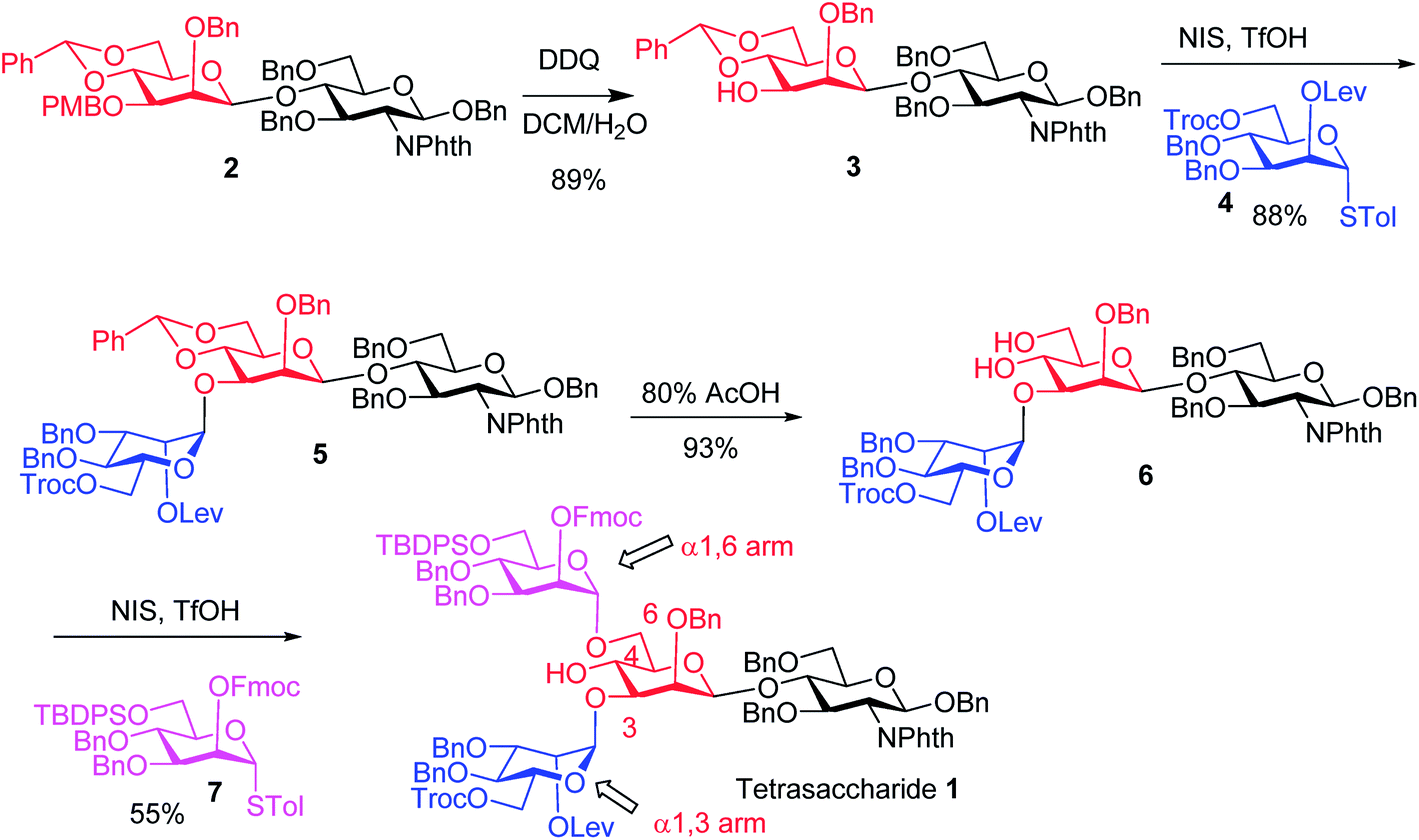 | ||
| Scheme 1 Synthesis of tetrasaccharide 1 bearing different protective groups at the four strategic branching points. | ||
We next focused on the introduction of bisecting GlcNAc to the free 4-OH of tetrasaccharide 1. Glycosylation of the 4-OH group is known to be challenging as it is flanked by two glycans.18,40,41 Prior strategies to synthesize N-glycans with the bisecting glucosamine unit typically introduced the bisecting residue at 4-OH first followed by the removal of the protective group on 6-OH and installation of the α1,6-arm.27,42–47 The direct glycosylation of an acceptor containing free 4-OH with both α1,3- and α1,6-arms is attractive as it reduces protective group manipulations needed. Unverzagt and coworkers successfully developed this approach although a large excess (10 equiv.) of glycosyl donors was needed to overcome the low nucleophilicity of the 4-OH.18,40,41
In our synthesis, we first tested the direct glycosylation of tetrasaccharide 1 with 1.5 equiv. of glucosamine donor 8 (ref. 48) in the presence of NIS/TfOH. Surprisingly, while the desired pentasaccharide was detected from the reaction mixture by mass spectrometry, a pentasaccharide side product containing an iodide group was also formed, which likely resulted from electrophilic substitution of the product by the electron-deficient iodonium ion generated from NIS. The side product could not be separated from the desired pentasaccharide. While NIS/TfOH is a promoter system widely used to activate thioglycosides,49 the side reaction of iodination was not unprecedented as the Yu group reported aglycon iodination during NIS promoted thioglycosylation of a steroid derivative.50 As an alternative to NIS/TfOH, we tested the reaction of 1 and 8 using p-TolSCl/AgOTf51 as the promoter. In this case, however, a pentasaccharide side product containing a p-TolS moiety was found.
We hypothesize that the most likely location in tetrasaccharide 1 that was electrophilically substituted would be the methyl or methylene group α to the ketone carbonyl of the Lev. As electron-withdrawing protective groups, i.e., Troc and Lev, are on the same mannose unit of 1, the acidity of α-H of ketone may be higher. This consideration led us to switch the protective groups of Fmoc and Lev on the mannosyl donors, i.e., using new donors 9 and 12. The coupling between donor 9 and disaccharide acceptor 3 went smoothly, which followed by subsequent benzylidene removal gave the trisaccharide 11 in 77% yield (Scheme 2). Selective glycosylation at the primary hydroxyl group of 11 by mannose donor 12 generated tetrasaccharide 13. The 4-OH of tetrasaccharide 13 was protected as the O-acetate (tetrasaccharide 14) to synthesize N-glycans without the bisecting moiety.
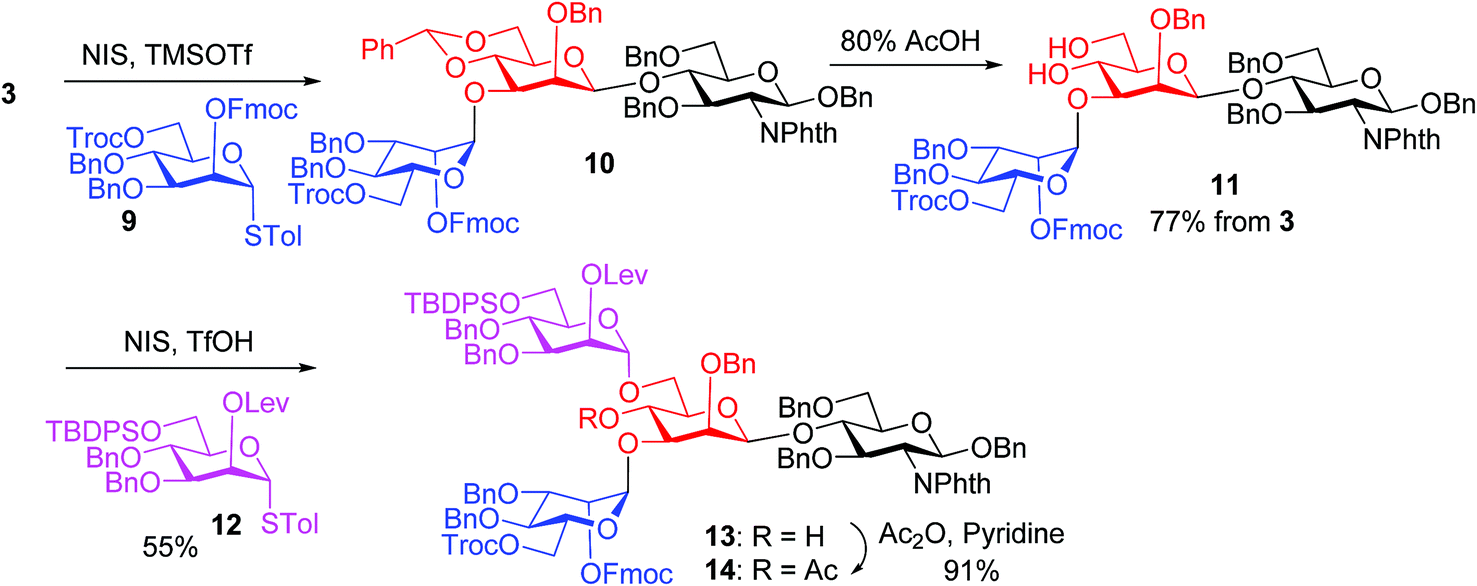 | ||
| Scheme 2 Synthesis of tetrasaccharide 13. | ||
The bisecting glucosamine was successfully introduced via the reaction of 13 with 2 equiv. of donor 8 with NIS/TfOH promoter to form pentasaccharide 15 (Table 1, entry 1). Although the yield was a modest 37%, the undesired iodide substitution products were not detected. When 3 equiv. of donor 8 was used, the yield increased to 53% (Table 1, entry 2). Further increasing the amount of donor did not improve the yield significantly, but rendered purification more complicated. The major side product was due to the reaction of succinimide generated from NIS with the activated glycosyl donor, which competed with the formation of the desired product. The promoter p-TolSCl/AgOTf was utilized next instead of NIS/TfOH. With 1.5 equiv. of donor 8, the desired pentasaccharide 15 was obtained in 44% yield (Table 1, entry 3). Increasing the amount of donor from 1.5 to 3.0 equiv. led to a significant increase of the yield of glycosylation to 81% (Table 1, entries 3–5). This compared favorably with several literature approaches18,40,41 for introduction of the bisecting glycan as good glycosylation yield was obtained without the need for a large excess (10 equiv.) of the glycosyl donor.
|
|
|||
|---|---|---|---|
| Entry | Donor amount | Promoter | Yield |
| 1 | 2.0 equiv. | NIS/TfOH | 37% |
| 2 | 3.0 equiv. | NIS/TfOH | 53% |
| 3 | 1.5 equiv. | p-TolSCl/AgOTf | 44% |
| 4 | 2.2 equiv. | p-TolSCl/AgOTf | 67% |
| 5 | 3.0 equiv. | p-TolSCl/AgOTf | 81% |

With the key pentasaccharide 15 in hand, the possibility of orthogonally removing each of the temporary protective groups was explored. As shown in Scheme 3, treatment of 15 with triethylamine selectively removed the Fmoc group in 95% yield without affecting any other protective groups. The Lev group in 15 could be deprotected by hydrazine acetate, while Troc and TBDPS moieties were cleaved by Zn/AcOH and HF·pyridine respectively in good yields.
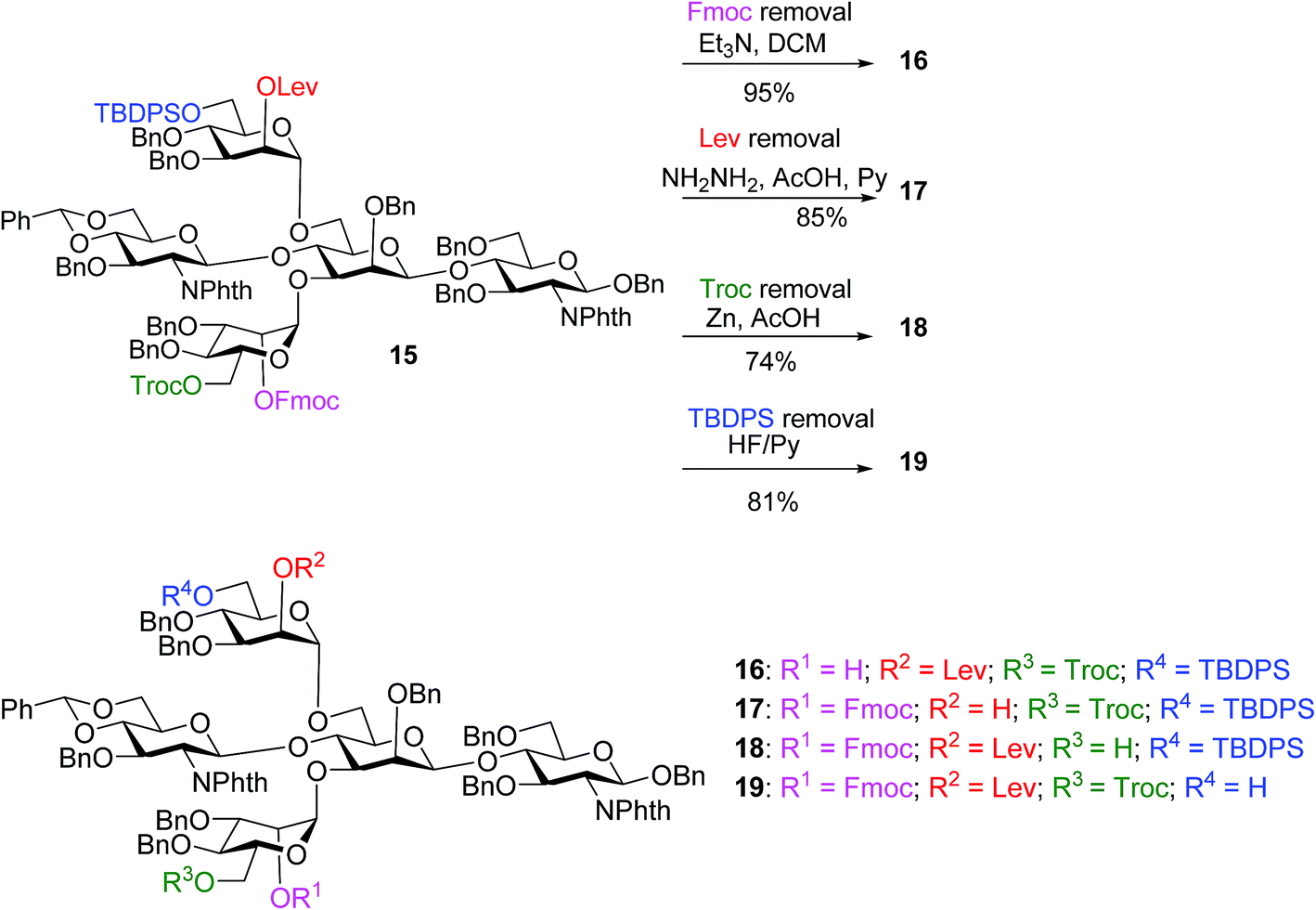 | ||
| Scheme 3 The four temporary protective groups on key pentasaccharide 15 could be orthogonally removed without affecting one another. | ||
With the orthogonality of protective group removal established, glycosylations of the newly liberated hydroxyl groups were tested for the construction of asymmetrically branched N-glycans. To understand the impact of the bisecting glycan on glycosylation, both pentasaccharide 17 and tetrasaccharide 20 obtained from Lev removal of 14 were utilized as acceptors.
Glycosylation of pentasaccharide 17 with donor 8 promoted by NIS/AgOTf gave hexasaccharide 21 in 53% yield (Scheme 4). Other promoter systems such as NIS/TMSOTf, NIS/TfOH and p-TolSCl/AgOTf were also tested, but did not lead to better yields. Compound 21 was treated with Et3N followed by glycosylation by disaccharide 23 and removal of TBDPS, affording octasaccharide 24 in 43% overall yield. LewisX trisaccharide thioglycosyl donor 26 was prepared through a one pot procedure in gram scale.52 Introduction of donor 26 onto the primary OH of octasaccharide 24 proceeded to give undecasaccharide 27 in 33% yield, where 21% of the unreacted acceptor was recovered in the reaction.
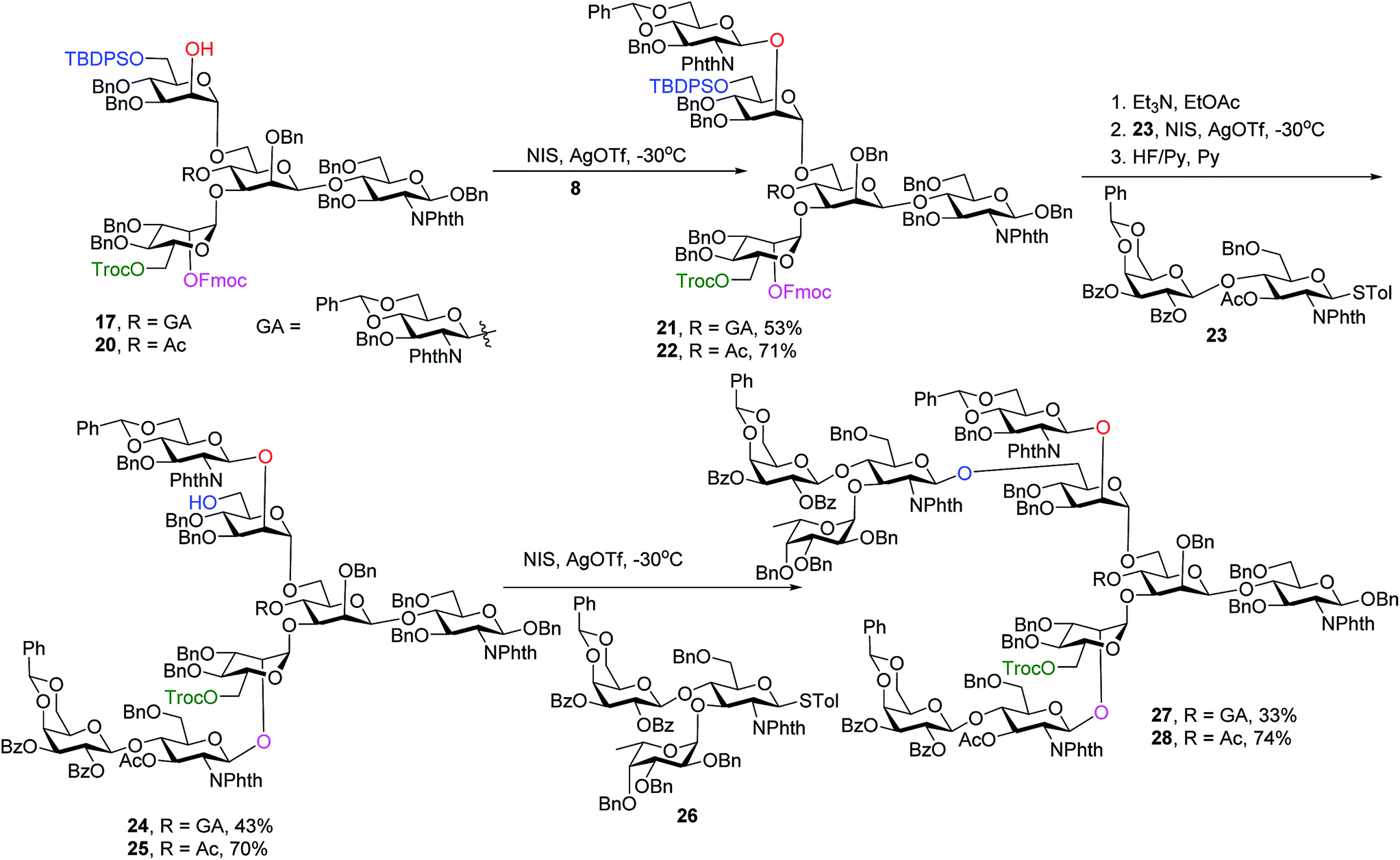 | ||
| Scheme 4 Glycans can be installed at the strategic branching points after orthogonal deprotections. | ||
To synthesize glycans without the bisecting glycan, analogous glycosylations with donors 8, 23, 26 followed by subsequent deprotections were performed on the tetrasaccharide acceptor 20 (Scheme 4). The penta-, hepta- and deca-saccharides 22, 25 and 28 were efficiently synthesized in 71%, 70% and 74% yields respectively, significantly higher than the analogous yields for forming 21, 24 and 27 (53%, 43% and 33%). These results demonstrate that the bisecting glucosamine negatively impacted glycosylation yields at branching points.
To better understand the effect of the bisecting glycan, molecular modeling studies were performed. Both glycans 17 and 20 were subjected to molecular dynamics (MD) simulations at 900 K. From the resulting MD frames, 1000 conformers were extracted and optimized using the AM1 semi-empirical method. The conformers within 6.0 kcal mol−1 from the lowest energy conformation were selected for comparisons. It was noteworthy that the glycan 17 was more rigid than 20, as there were fewer distinct conformers of 17 than 20 in this energy range (Table S1†). Conformational analysis showed that the free 2-OH of compound 17 was much more sterically hindered than that in compound 20. The average number of atoms within 5 Å of the OH group in the low energy conformers of 17 was quantified and found to be significantly higher than that in compound 20 (42.9 vs. 35.9 Table S1†). Furthermore, the OH group was pointing at the bisecting glucosamine unit in many conformers of 17 (Fig. 2). There were no additional hydrogen bond donors interacting with the free OH group in the optimized structures of 17 compared to that in 14. Therefore, the lower yields of glycosylation of 17 were mainly attributed to the increased steric hindrance around the OH group caused by the bisecting glucosamine unit.
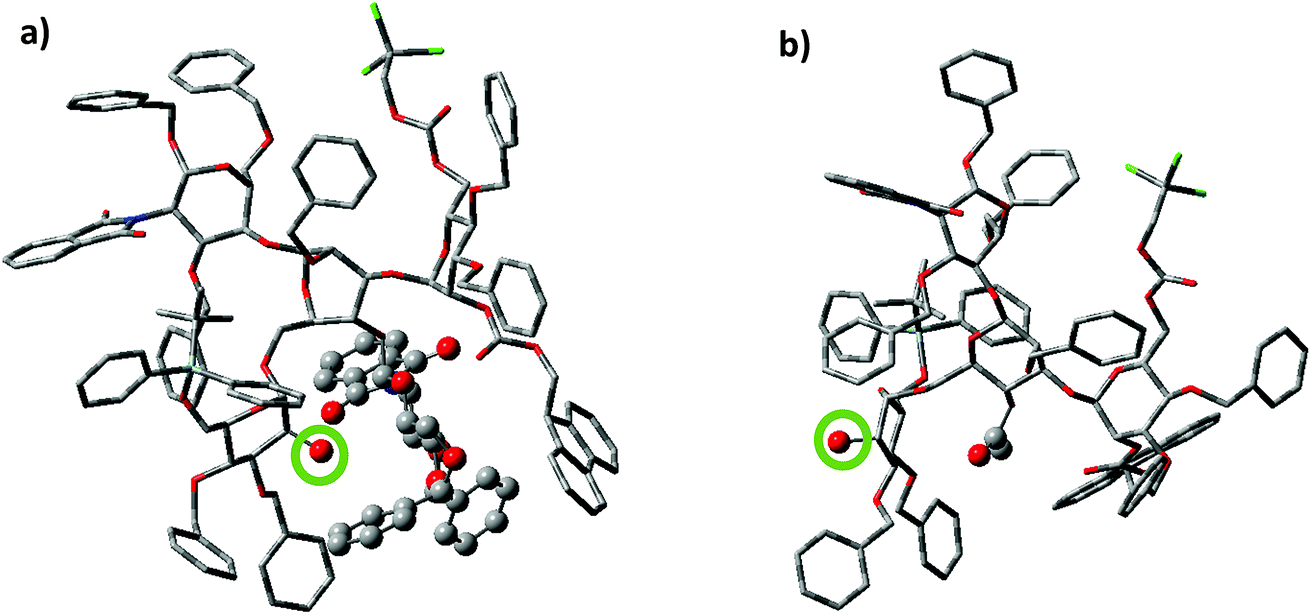 | ||
| Fig. 2 Representative conformations of (a) compound 17 and (b) compound 20 based on MD simulations. The red atom circled by the green circle is the free 2-OH of the non-reducing end mannose. In compound 17, the 2-OH group is pointing towards the bisecting glucosamine moiety potentially hindering its approach towards the activated donor for productive glycosylation. In comparison, the 2-OH in compound 20 is much less hindered, presumably resulting in higher nucleophilicity. The rest of atoms displayed with the ball and stick model are the bisecting glucosamine (compound 17) and the 4-OAc (compound 20) respectively. | ||
As N-glycans are attached to proteins, we next explored the possibility of introducing the rare N-glycan to peptides with haptoglobin peptide (amino acids 233–254)53,54 as a representative acceptor. The transglycosylation strategy by endoglycosidases23–28 pioneered by the Wang lab can enable convergent synthesis of glycopeptides by transferring glycosyl oxazoline donors to peptide acceptors bearing a GlcNAc on an asparagine residue. While the successful transfer of a bisected N-glycan by the endoglycosidase Endo-A has been reported,27 the donor tested did not contain α1,3- or α1,6-glycan branches. Furthermore, it is unclear how the bisecting GlcNAc may impact the transglycosylation reaction.
To synthesize N-glycans for haptoglobin glycopeptides, compounds 15 and 14 were first treated with Zn/AcOH and HF/Py giving diols 29 and 30. LewisX trisaccharide thioglycosyl donor 26 glycosylated diols 29 and 30 using the NIS/AgOTf promoter system followed by subsequent Lev and Fmoc removal producing undeca- and deca-saccharides 31 and 32 in 40% and 51% yields respectively (Scheme 5a). To prepare tri-antennary N-glycan, the Lev group in 29 and 30 was selectively removed. The resulting triols were glycosylated with LewisX trisaccharide thioglycosyl donor 26 leading to tetradeca- and trideca-saccharides 33 and 34 (Scheme 5b).
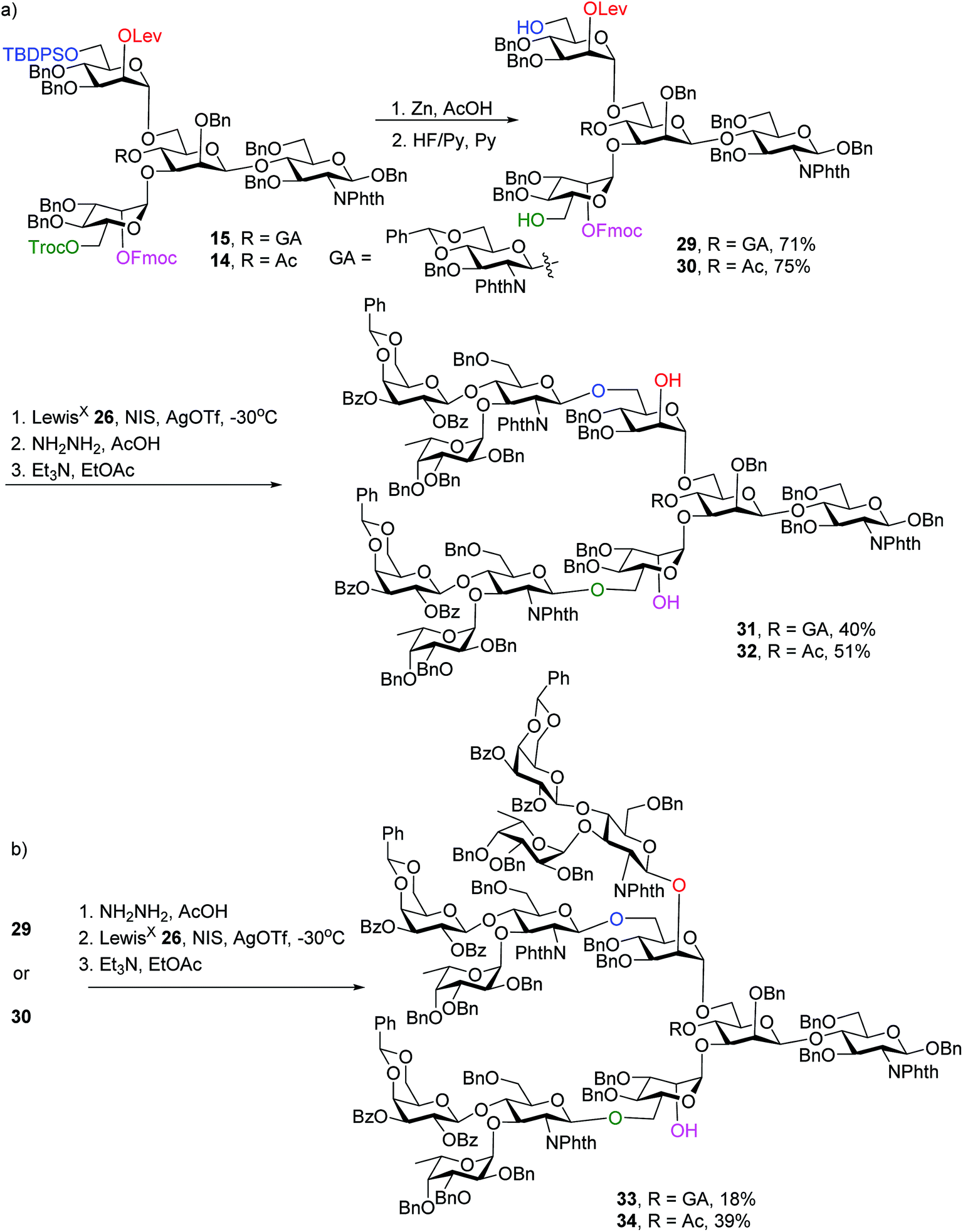 | ||
| Scheme 5 Orthogonal deprotection and glycosylation for the synthesis of complex N-glycans 31–34. | ||
The four oligosaccharides 31–34 were deprotected by first incubating with ethylenediamine in n-butanol at 90 °C,31 which removed all ester and carbonate groups as well as the Phth to generate free amines (Scheme 6). To facilitate future quantification by mass spectrometry,55,56 the free amines were protected with 13C labeled acetic anhydride to introduce 13C labels into the GlcNAcs. Subsequent hydrogenolysis with Pearlman's catalyst under a hydrogen atmosphere57 led to free N-glycans 35–38.
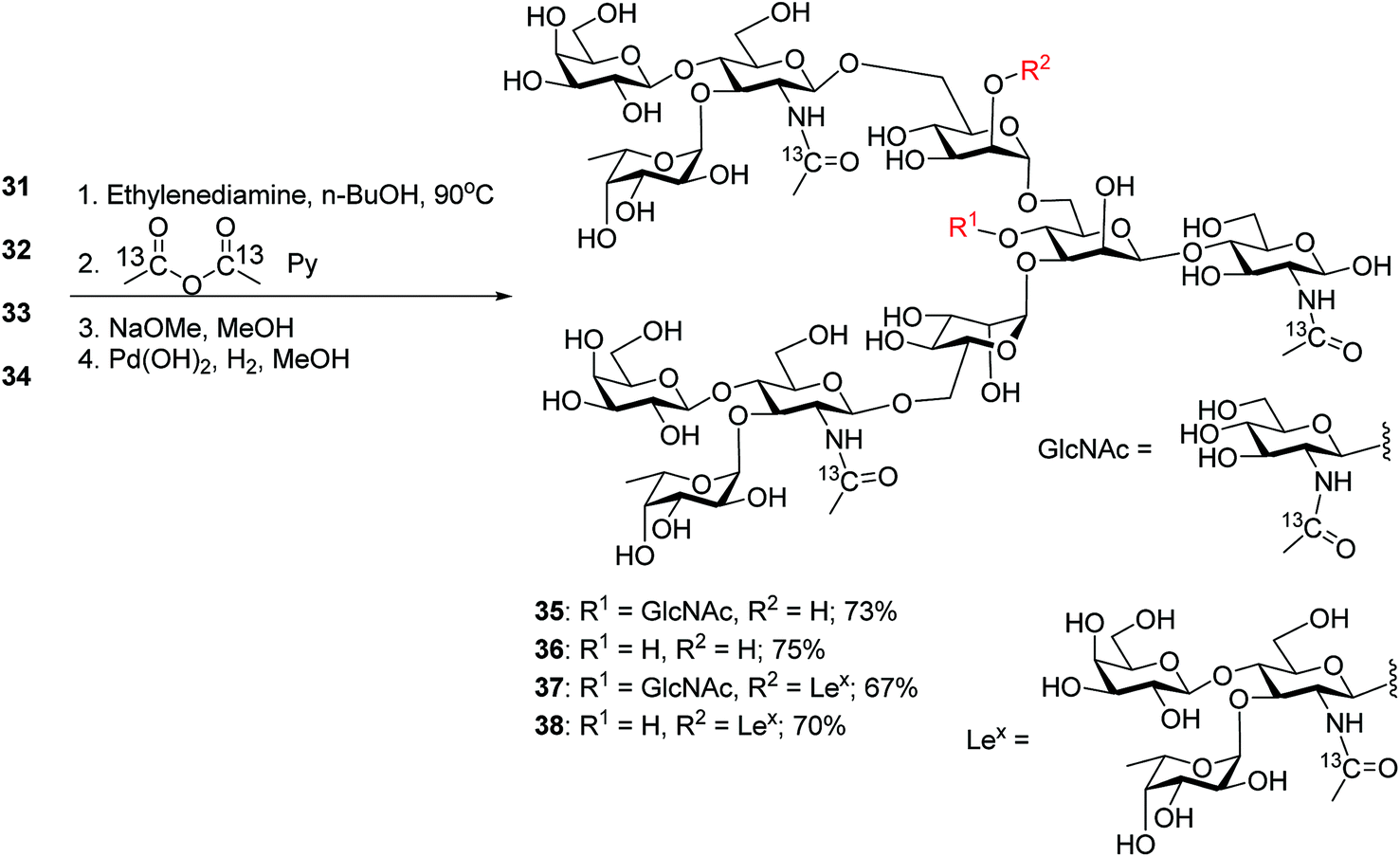 | ||
| Scheme 6 Synthesis of 13C-labeled deprotected N-glycans 35–38. | ||
With the free N-glycans in hand, they were transformed to the respective oligosaccharide oxazolines 39, 40, 41 and 42 using a mild chloroformamidinium dehydrating agent 43 (Scheme 7a), which were directly subjected to the enzymatic transglycosylation reactions with GlcNAc bearing haptoglobin glycopeptide 44 as the acceptor (Scheme 7b). Glycosyl oxazoline 40 was first tested using endoglycosidases Endo-A (wild type) and Endo-M (wild type), and their respective Endo-A N171A, Endo-M N175A and Endo-M N175Q mutants as catalysts. Interestingly, we found that wild type Endo-A was able to transfer the glycan to give glycopeptide 46 in 73% overall yield, while all other enzymes tested failed to generate significant quantities of desired products. Next, the bisecting GlcNAc containing oxazoline 39 was utilized as the donor. Similar to donor 40, Endo-A promoted the transglycosylation reaction efficiently producing glycopeptide 45 in 65% yield. These results suggest that the presence of the bisecting GlcNAc does not negatively affect the donor abilities of 39 and glycosyl oxazolines containing bisecting GlcNAc and complex glycans such as rare α1,3- and α1,6-branches can be suitable donors for transglycosylation to glycopeptides.
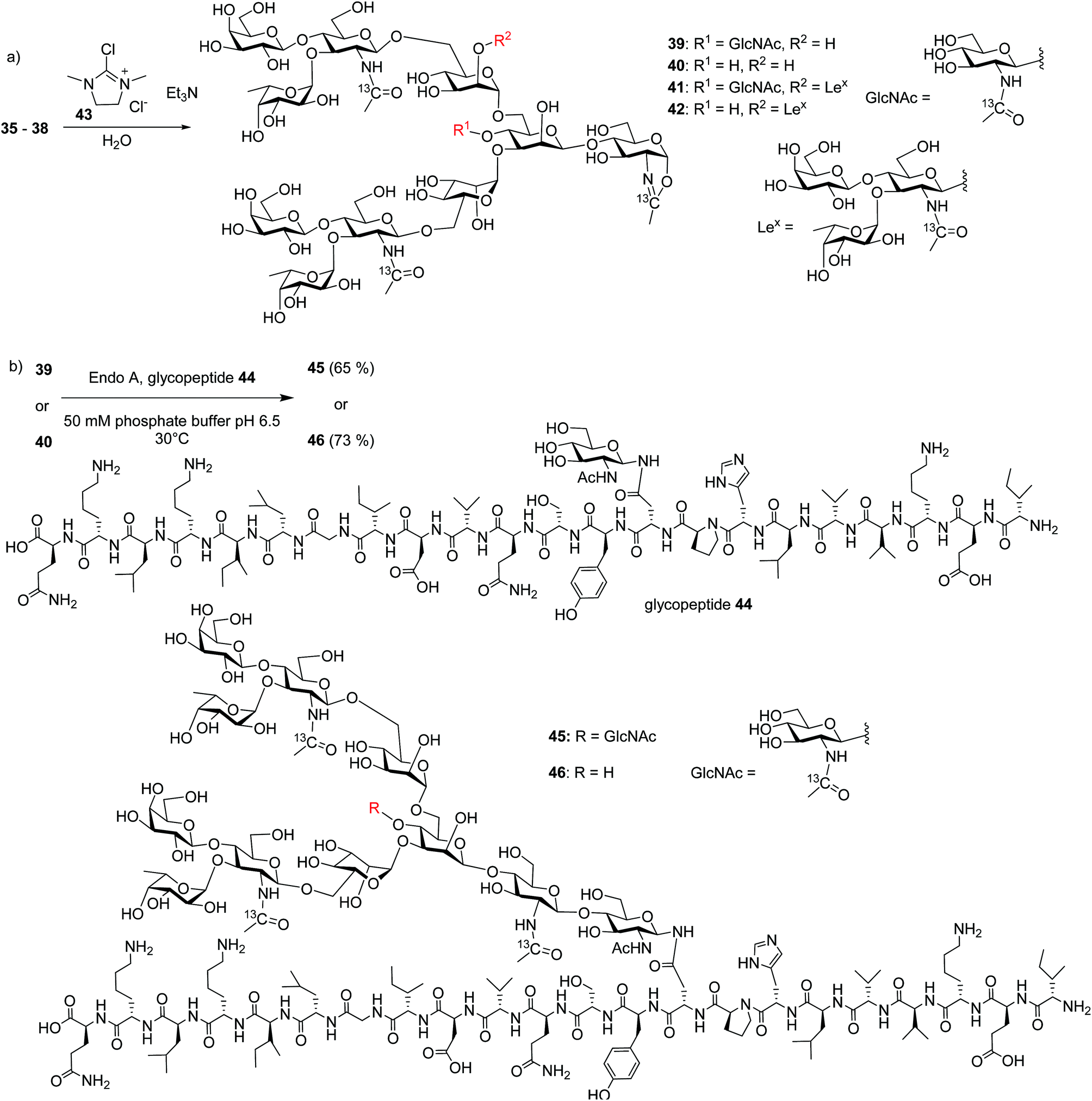 | ||
| Scheme 7 Synthesis of glycopeptides 45 and 46via Endo-A promoted transglycosylations. | ||
To test the limit of the Endo-A enzymes, tri-antennary N-glycan oxazolines 41 and 42 bearing three LeX moieties were subjected to transglycosylation reactions with acceptor peptide 44. However, no desired glycopeptides were observed with Endo-A. Nor did the Endo-M or any of the Endo-A N171A, Endo-M N175A and Endo-M N175Q mutants tested give desired glycopeptides.
To better understand the divergent behavior of glycosyl oxazolines 39vs.41, computational studies were performed on their complexes with Endo-A. The initial coordinates of Endo-A were obtained from the Protein Data Bank34 (PDB ID: 3FHA)35 and N-glycan oxazolines 39 and 41 were docked into the binding pocket of Endo-A using the docking program within Molecular Operating Environment v.2016.08 (MOE).36 Binding poses were scored using the GBVI/WSA ΔG scoring function. It is known that W93, N171, E173, Y205, F125, W216, F243, W244 and Y299 are the active site residues interacting with the donor substrate.35 Poses with the oxazoline ring positioned within range of these residues were simulated using atomistic MD with the AMBER 16 software package.32 The lowest energies poses with the oxazolines remaining in the binding pockets were obtained and analyzed.
MD simulations demonstrate that the binding energy of 39 and Endo-A is on average −81.44 ± 11.10 kcal mol−1 (Table 2). In contrast, the binding energy of 41 is significantly lower at −56.24 ± 9.95 kcal mol−1, suggesting the much weaker binding of 41 by Endo-A. In crystal structures of Endo-A as well as that of Endo-A complexed with a tetrasaccharide oxazoline substrate,35 the indole rings of W216 and W244 are perpendicular to each other, possibly forming a gate sealing off the active site. When oxazoline 39 was docked into Endo-A, the indole of W244 was moved to be parallel to that of W216 to allow the substrate to enter the active site. In all poses generated from subsequent MD simulations, the indole of W244 returned to the original perpendicular orientation to W216 (Fig. 3a). However, in the complex of the tri-antennae bearing donor 41 with Endo-A, the additional antenna of 41 is located between these two tryptophan residues, possibly preventing the rotation of the indole ring. As a result, the two indoles of W244 and W216 remained close to parallel to each other (Fig. 3b). This may have precluded the formation of a closed active site for catalysis, reducing the yields for transglycosylation.
| Compound | Binding pose | Binding energy (kcal mol−1) |
|---|---|---|
| 39 | 1 | −72.97 ± 6.04 |
| 2 | −94.00 ± 9.15 | |
| 3 | −77.36 ± 7.96 | |
| Average | −81.44 ± 11.10 | |
| 41 | 1 | −52.08 ± 11.26 |
| 2 | −60.17 ± 11.56 | |
| 3 | −55.26 ± 7.95 | |
| 4 | −58.80 ± 7.83 | |
| 5 | −54.86 ± 11.17 | |
| Average | −56.24 ± 9.95 | |
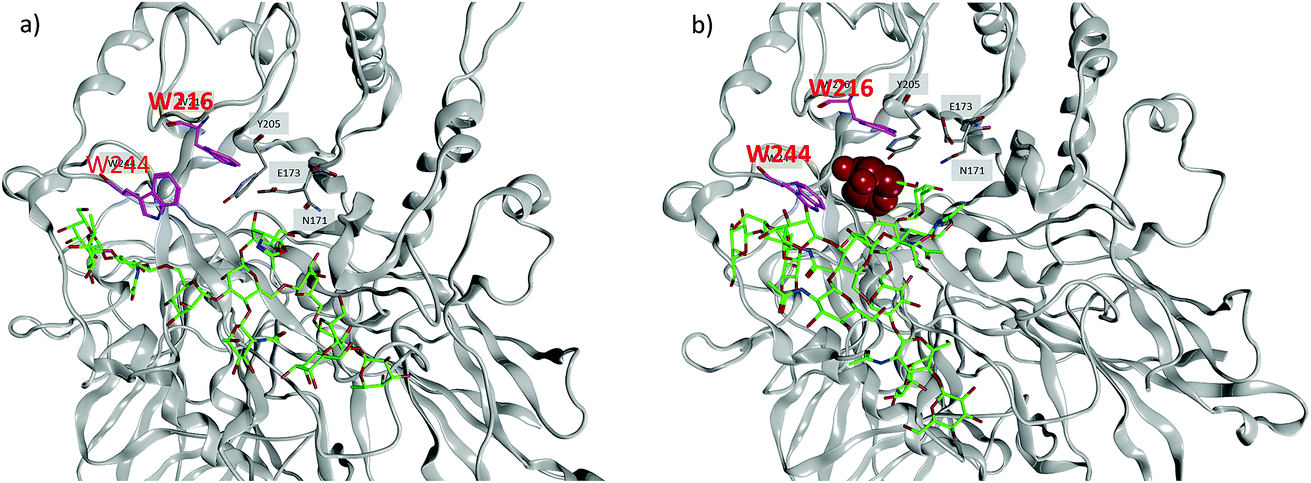 | ||
| Fig. 3 Representation binding pose of (a) N-glycan oxazoline 39 and Endo-A, and (b) 41 with Endo-A. Critical pocket residues are indicated. The indole rings of W216 and W244 are highlighted in magenta. In (a), the two indoles are perpendicular to each other, while in (b), they remain close to parallel. The glycan structures are shown in stick models (green color: carbon; red color: oxygen; blue color: nitrogen). A galactosyl unit (shown in space filling model) at the non-reducing end of 41 is located in between the two indole rings of W216 and W244, which presumably prevents the indoles to become perpendicular to each other. | ||
Conclusion
A flexible synthetic route to rare N-glycan structures has been designed with the core tetrasaccharide 13 bearing four orthogonal protecting groups serving as a linchpin. Through this key intermediate, many N-glycan oligosaccharides could be accessed including asymmetric antennary structures through orthogonal deprotection followed by glycosylations. During the synthesis, it was found that if Troc and Lev groups were on the same mannosyl donor, subsequent glycosylation could not give the desired product in good purity due to the introduction of an iodine into the molecule. This problem was successfully solved by switching the protecting groups of Fmoc and Lev in the two mannosyl donors. Furthermore, the synthesis was designed to enable late stage introduction of bisecting glucosamine. Suitable conditions have been identified to install the bisecting glycan in a high yield without resorting to a large excess of the glycosyl donor. The bisecting glucosamine was found to reduce the glycosylation yields of each branching point. Molecular modeling results indicated that the bisecting glycan sterically hindered the access of the free hydroxyl groups of the mannose branches for glycosylation. Finally, the limit and scope of endoglycosidase promoted transglycosylation reactions were tested. Two homogenous haptoglobin glycopeptides bearing an undeca- and a dodeca-saccharide respectively could be produced via wild type Endo-A catalyzed transglycosylation reactions. Sugar oxazoline donors containing three LeX in the antennae did not undergo productive transfer by the enzymes, which was likely because the bulky glycan substrate reduced binding affinity and prevented the necessary reactive conformations of the enzymes. This is the first time that glycopeptides bearing bisecting GlcNAc and rare branches on α1,3-mannose have been produced, expanding the diversity of N-glycans and glycopeptides that can be accessed.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
We are grateful for the financial support from National Science Foundation (CHE 1507226), the National Institute of General Medical Sciences, NIH (R01GM072667), the National Cancer Institute, NIH (U01 CA168926 and R01 CA135069). We would like to thank Prof. Lai-xi Wang for helpful suggestions.References
- G. W. Hart and R. J. Copeland, Cell, 2010, 143, 672–676 CrossRef PubMed.
- S. V. Kasteren, Biochem. Soc. Trans., 2012, 40, 929–944 CrossRef PubMed.
- G. Walsh and R. Jefferis, Nat. Biotechnol., 2006, 24, 1241–1252 CrossRef PubMed.
- J. N. Arnold, M. R. Wormald, R. B. Sim, P. M. Rudd and R. A. Dwek, Annu. Rev. Immunol., 2007, 25, 21–50 CrossRef PubMed.
- D. Shental-Bechor and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8256–8261 CrossRef PubMed.
- M. R. Wormald, A. J. Petrescu, Y. L. Pao, A. Glithero, T. Elliott and R. A. Dwek, Chem. Rev., 2002, 102, 371–386 CrossRef PubMed.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef PubMed.
- K. i. Inamori, T. Endo, Y. Ide, S. Fujii, J. Gu, K. Honke and N. Taniguchi, J. Biol. Chem., 2003, 278, 43102–43109 CrossRef PubMed.
- T. Lange, S. Ullrich, I. Muller, M. F. Nentwich, K. Stubke, S. Feldhaus, C. Knies, O. J. C. Hellwinkel, R. L. Vessella, C. Abramjuk, M. Anders, J. Schroder-Schwarz, T. Schlomm, H. Huland, G. Sauter and U. Schumacher, Clin. Cancer Res., 2012, 18, 1364–1373 CrossRef PubMed.
- N. Taniguchi and H. Korekane, BMB Rep., 2011, 44, 772–781 CrossRef PubMed.
- Y. Song, J. A. Aglipay, J. D. Bernstein, S. Goswami and P. Stanley, Cancer Res., 2010, 70, 3361–3371 CrossRef PubMed.
- M. Nagae, K. Yamanaka, S. Hanashima, A. IKeda, K. Morita-Matsumoto, T. Satoh, N. Matsumoto, K. Yamamoto and Y. Yamaguchi, J. Biol. Chem., 2013, 288, 33598–33610 CrossRef PubMed.
- M. Takahashi, Y. Kuroki, K. Ohtsubo and N. Taniguchi, Carbohydr. Res., 2009, 344, 1387–1390 CrossRef PubMed.
- L. Li, Y. Liu, C. Ma, J. Qu, A. D. Calderon, B. Wu, N. Wei, X. Wang, Y. Guo, Z. Xiao, J. Song, G. Sugiarto, Y. Li, H. Yu, X. Chen and P. G. Wang, Chem. Sci., 2015, 6, 5652–5661 RSC.
- H. V. Ravi Kumar, K. Naruchi, R. Miyoshi, H. Hinou and S. I. Nishimura, Org. Lett., 2013, 15, 6278–6281 CrossRef PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef PubMed.
- V. Y. Dudkin, J. S. Miller, A. S. Dudkina, C. Antczak, D. A. Scheinberg and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 13598–13607 CrossRef PubMed.
- M. Monnich, S. Eller, T. Karagiannis, L. Perkams, T. Luber, D. Ott, M. Niemietz, J. Hoffman, J. Walcher, L. Berger, M. Pischl, M. Weishaupt, C. Wirkner, R. G. Lichtenstein and C. Unverzagt, Angew. Chem., Int. Ed., 2016, 55, 10487–10492 CrossRef PubMed.
- C. W. Lin, M. H. Tsai, S. T. Li, T. I. Tsai, K. C. Chu, Y. C. Liu, M. Y. Lai, C. Y. Wu, Y. C. Tseng, S. S. Shivatare, C. H. Wang, P. Chao, S. Y. Wang, H. W. Shih, Y. F. Zeng, T. H. You, J. Y. Liao, Y. C. Tu, Y. S. Lin, H. Y. Chuang, C. L. Chen, C. S. Tsai, C. C. Huang, N. H. Lin, C. Ma, C. Y. Wu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10611–10616 CrossRef PubMed.
- S. S. Shivatare, S. H. Chang, C. S. Tsai, S. Y. Tseng, V. S. Shivatare, Y. S. Lin, Y.-Y. Cheng, C.-T. Ren, C. C. D. Lee, S. Pawar, C. S. Tsai, H. W. Shih, Y. F. Zeng, C. H. Liang, P. D. Kwong, D. R. Burton, C. Y. Wu and C. H. Wong, Nat. Chem., 2016, 8, 338–346 CrossRef PubMed.
- C. Unverzagt and Y. Kajihara, Chem. Soc. Rev., 2013, 42, 4408–4420 RSC.
- M. Murakami, T. Kiuchi, M. Nishihara, K. Tezuka, R. Okamoto, M. Izumi and Y. Kajihara, Sci. Adv., 2016, 2, e1500678 Search PubMed.
- B. Li, Y. Zeng, S. Hauser, H. Song and L. X. Wang, J. Am. Chem. Soc., 2005, 127, 9692–9693 CrossRef PubMed.
- W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L.-X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef PubMed.
- M. N. Amin, J. S. McLellan, W. Huang, J. Orwenyo, D. R. Burton, W. C. Koff, P. D. Kwong and L. X. Wang, Nat. Chem. Biol., 2013, 9, 521–526 CrossRef PubMed.
- H. Ochiai, W. Huang and L. X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef PubMed.
- G. Zou, H. Ochiai, W. Huang, Q. Yang, C. Li and L. X. Wang, J. Am. Chem. Soc., 2011, 133, 18975–18991 CrossRef PubMed.
- W. Huang, J. Giddens, S. Q. Fan, C. Toonstra and L. X. Wang, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef PubMed.
- C. H. Wong, X.-S. Ye and Z. Zhang, J. Am. Chem. Soc., 1998, 120, 7137–7138 CrossRef.
- T. Li, M. Huang, L. Liu, S. Wang, K. W. Moremen and G. J. Boons, Chem. - Eur. J., 2016, 22, 18742–18746 CrossRef PubMed.
- I. A. Gagarinov, T. Li, J. S. Torano, T. Caval, A. D. Srivastava, J. A. W. Kruijtzer, A. J. R. Heck and G. J. Boons, J. Am. Chem. Soc., 2017, 139, 1011–1018 CrossRef PubMed.
- D. A. Case, D. S. Cerutti, T. E. Cheatham, T. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, D. Greene, N. Homeyer, S. Izadi, A. V. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, C. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2017, University of California, San Francisco, 2017 Search PubMed.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., 2016 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef PubMed.
- J. Yin, L. Li, N. Shaw, Y. Li, J. K. Song, W. Zhang, C. Xia, R. Zhang, A. Joachimiak, H. C. Zhang, L. X. Wang, Z. J. Liu and P. Wang, PLoS One, 2009, 4, e4658, DOI:10.1371/journal.pone.0004658.
- Molecular Operating Environment (MOE), 2016.08, Chemical Computing Group Inc., 2016 Search PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef.
- C. Unverzagt and J. Seifert, Tetrahedron Lett., 2000, 41, 4549–4553 CrossRef.
- S. Eller, C. Raps, M. Niemietz and C. Unverzagt, Tetrahedron Lett., 2010, 51, 2648–2651 CrossRef.
- S. Eller, R. Schuberth, G. Gundel, J. Seifert and C. Unverzagt, Angew. Chem., Int. Ed., 2007, 46, 4173–4175 CrossRef PubMed.
- H. Paulson, M. Heume and H. Nurnberger, Carbohydr. Res., 1990, 200, 127–166 CrossRef.
- F. Yamazaki, T. Kitajima, T. Nukada and Y. Ito, Carbohydr. Res., 1990, 201, 15–30 CrossRef PubMed.
- H. Weiss and C. Unverzagt, Angew. Chem., Int. Ed., 2003, 42, 4261–4263 CrossRef PubMed.
- R. Schuberth and C. Unverzagt, Tetrahedron Lett., 2005, 46, 4201–4204 CrossRef.
- P. Wang, J. Zhu, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 16669–16671 CrossRef PubMed.
- Y. Wang, X. Huang, L. H. Zhang and X. S. Ye, Org. Lett., 2004, 6, 4415–4417 CrossRef PubMed.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef PubMed.
- B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef PubMed.
- X. Huang, L. Huang, H. Wang and X.-S. Ye, Angew. Chem., Int. Ed., 2004, 43, 5221–5224 CrossRef PubMed.
- A. Miermont, Y. Zeng, Y. Jing, X. S. Ye and X. Huang, J. Org. Chem., 2007, 72, 8958–8961 CrossRef PubMed.
- P. Pompach, Z. Brnakova, M. Sanda, J. Wu, N. Edwards and R. Goldman, Mol. Cell. Proteomics, 2013, 12, 1281–1293 CrossRef PubMed.
- M. R. Langlois and J. R. Delanghe, Clin. Chem., 1996, 42, 1589–1600 Search PubMed.
- B. Echeverria, J. Etxebarria, N. Ruiz, A. Hernandez, J. Calvo, M. Haberger, D. Reusch and N. C. Reichardt, Anal. Chem., 2015, 87, 11460–11467 CrossRef PubMed.
- G. Yang, Z. Tan, W. Lu, J. Guo, H. Yu, J. Yu, C. Sun, X. Qi, Z. Li and F. Guan, J. Proteome Res., 2015, 14, 639–653 CrossRef PubMed.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S. i. Shoda, J. Org. Chem., 2009, 74, 2210–2212 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data, Table S1 as well as NMR spectra. See DOI: 10.1039/c8sc02457j |
| This journal is © The Royal Society of Chemistry 2018 |